
Liposomal Formulations of a Polyleucine–Antigen Conjugate as Therapeutic Vaccines against Cervical Cancer

 , , , , ,
, , , , ,  ,
, 
 ,
,  , ,
, , 

Abstract
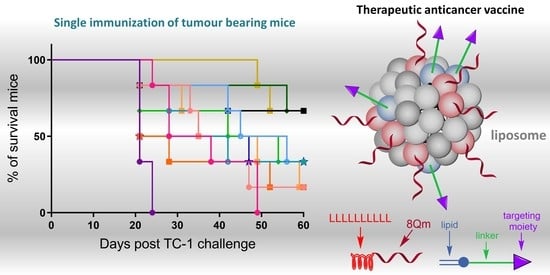
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of Peptides, Conjugates and Targeting Moieties
2.3. Formulation of Vaccine Candidates
2.4. Characterization of Nanoliposomes
2.5. Mice and TC-1 Cell Lines
2.6. In Vivo Tumor Treatment Experiments
2.7. IFN-Gamma ELISPOT Assays
2.8. Statistical Analysis
3. Results and Discussion
3.1. Vaccine Design
3.2. Vaccine Preparation
3.3. Immunological Evaluation of the Vaccine Candidates
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Maine, D.; Hurlburt, S.; Greeson, D. Cervical cancer prevention in the 21st century: Cost is not the only issue. Am. J. Public Health 2011, 101, 1549–1555. [Google Scholar] [CrossRef]
- Alharbi, N.; Skwarczynski, M.; Toth, I. The influence of component structural arrangement on peptide vaccine immunogenicity. Biotechnol. Adv. 2022, 60, 108029. [Google Scholar] [CrossRef]
- Zhang, J.; Fan, J.; Skwarczynski, M.; Stephenson, R.J.; Toth, I.; Hussein, W.M. Peptide-Based Nanovaccines in the Treatment of Cervical Cancer: A Review of Recent Advances. Int. J. Nanomed. 2022, 17, 869–900. [Google Scholar] [CrossRef]
- Reintjens, N.R.M.; Tondini, E.; de Jong, A.R.; Meeuwenoord, N.J.; Chiodo, F.; Peterse, E.; Overkleeft, H.S.; Filippov, D.V.; van der Marel, G.A.; Ossendorp, F.; et al. Self-Adjuvanting Cancer Vaccines from Conjugation-Ready Lipid A Analogues and Synthetic Long Peptides. J. Med. Chem. 2020, 63, 11691–11706. [Google Scholar] [CrossRef]
- Van der Maaden, K.; Heuts, J.; Camps, M.; Pontier, M.; van Scheltinga, A.T.; Jiskoot, W.; Ossendorp, F.; Bouwstra, J. Hollow microneedle-mediated micro-injections of a liposomal HPV E7(43–63) synthetic long peptide vaccine for efficient induction of cytotoxic and T-helper responses. J. Control. Release 2018, 269, 347–354. [Google Scholar] [CrossRef]
- Galliverti, G.; Tichet, M.; Domingos-Pereira, S.; Hauert, S.; Nardelli-Haefliger, D.; Swartz, M.A.; Hanahan, D.; Wullschleger, S. Nanoparticle Conjugation of Human Papillomavirus 16 E7-long Peptides Enhances Therapeutic Vaccine Efficacy against Solid Tumors in Mice. Cancer Immunol. Res. 2018, 6, 1301–1313. [Google Scholar] [CrossRef] [PubMed]
- Mardani, G.; Bolhassani, A.; Agi, E.; Shahbazi, S.; Mehdi Sadat, S. Protein vaccination with HPV16 E7/Pep-1 nanoparticles elicits a protective T-helper cell-mediated immune response. IUBMB Life 2016, 68, 459–467. [Google Scholar] [CrossRef]
- Hussein, W.M.; Liu, T.-Y.; Maruthayanar, P.; Mukaida, S.; Moyle, P.M.; Wells, J.W.; Toth, I.; Skwarczynski, M. Double conjugation strategy to incorporate lipid adjuvants into multiantigenic vaccines. Chem. Sci. 2016, 7, 2308–2321. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.-Y.; Hussein, W.M.; Giddam, A.K.; Jia, Z.; Reiman, J.M.; Zaman, M.; McMillan, N.A.; Good, M.F.; Monteiro, M.J.; Toth, I.; et al. Polyacrylate-Based Delivery System for Self-adjuvanting Anticancer Peptide Vaccine. J. Med. Chem. 2015, 58, 888–896. [Google Scholar] [CrossRef]
- Liu, T.Y.; Hussein, W.M.; Jia, Z.F.; Ziora, Z.M.; McMillan, N.A.J.; Monteiro, M.J.; Toth, I.; Skwarczynski, M. Self-adjuvanting polymer-peptide conjugates as therapeutic vaccine candidates against cervical cancer. Biomacromolecules 2013, 14, 2798–2806. [Google Scholar] [CrossRef]
- Nevagi, R.J.; Toth, I.; Skwarczynski, M. Peptide-based Vaccines. In Peptide Applications in Biomedicine, Biotechnology and Bioengineering; Koutsopoulos, S., Ed.; Woodhead Publishing: Oxford, UK, 2018; pp. 327–358. [Google Scholar]
- Azmi, F.; Ahmad Fuaad, A.A.; Skwarczynski, M.; Toth, I. Recent progress in adjuvant discovery for peptide-based subunit vaccines. Hum. Vaccines Immunother. 2014, 10, 778–796. [Google Scholar] [CrossRef] [PubMed]
- Seder, R.; Reed, S.G.; O’Hagan, D.; Malyala, P.; D’Oro, U.; Laera, D.; Abrignani, S.; Cerundolo, V.; Steinman, L.; Bertholet, S. Gaps in knowledge and prospects for research of adjuvanted vaccines. Vaccine 2015, 33 (Suppl. S2), B40–B43. [Google Scholar] [CrossRef]
- Lambrecht, B.N.; Kool, M.; Willart, M.A.; Hammad, H. Mechanism of action of clinically approved adjuvants. Curr. Opin. Immunol. 2009, 21, 23–29. [Google Scholar] [CrossRef]
- Mougel, A.; Terme, M.; Tanchot, C. Therapeutic Cancer Vaccine and Combinations With Antiangiogenic Therapies and Immune Checkpoint Blockade. Front. Immunol. 2019, 10, 467. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.-Y.; Giddam, A.K.; Hussein, W.; Jia, Z.; McMillan, N.; Monteiro, M.; Toth, I.; Skwarczynski, M. Self-Adjuvanting Therapeutic Peptide-Based Vaccine Induce CD8+ Cytotoxic T Lymphocyte Responses in a Murine Human Papillomavirus Tumor Model. Curr. Drug Deliv. 2015, 12, 3–8. [Google Scholar] [CrossRef]
- Hussein, W.M.; Liu, T.-Y.; Jia, Z.; McMillan, N.A.; Monteiro, M.J.; Toth, I.; Skwarczynski, M. Multiantigenic peptide-polymer conjugates as therapeutic vaccines against cervical cancer. Bioorgan. Med. Chem. 2016, 24, 4372–4380. [Google Scholar] [CrossRef] [PubMed]
- Khongkow, M.; Liu, T.Y.; Bartlett, S.; Hussein, W.M.; Nevagi, R.; Jia, Z.F.; Monteiro, M.J.; Wells, J.W.; Ruktanonchai, U.R.; Skwarczynski, M.; et al. Liposomal formulation of polyacrylate-peptide conjugate as a new vaccine candidate against cervical cancer. Precis. Nanomed. 2018, 1, 183–193. [Google Scholar] [CrossRef]
- Skwarczynski, M.; Zhao, G.; Boer, J.C.; Ozberk, V.; Azuar, A.; Cruz, J.G.; Giddam, A.K.; Khalil, Z.G.; Pandey, M.; Shibu, M.A.; et al. Poly(amino acids) as a potent self-adjuvanting delivery system for peptide-based nanovaccines. Sci. Adv. 2020, 6, eaax2285. [Google Scholar] [CrossRef]
- Azuar, A.; Madge, H.Y.R.; Boer, J.C.; Gonzalez Cruz, J.L.; Wang, J.; Khalil, Z.G.; Deceneux, C.; Goodchild, G.; Yang, J.; Koirala, P.; et al. Poly(hydrophobic Amino Acids) and Liposomes for Delivery of Vaccine against Group A Streptococcus. Vaccines 2022, 10, 1212. [Google Scholar] [CrossRef]
- Nevagi, R.J.; Skwarczynski, M.; Toth, I. Polymers for subunit vaccine delivery. Eur. Polym. J. 2019, 114, 397–410. [Google Scholar] [CrossRef]
- Azuar, A.; Li, Z.; Shibu, M.A.; Zhao, L.; Luo, Y.; Shalash, A.O.; Khalil, Z.G.; Capon, R.J.; Hussein, W.M.; Toth, I.; et al. Poly(hydrophobic amino acid)-Based Self-Adjuvanting Nanoparticles for Group A Streptococcus Vaccine Delivery. J. Med. Chem. 2021, 64, 2648–2658. [Google Scholar] [CrossRef] [PubMed]
- Azuar, A.; Shibu, M.A.; Adilbish, N.; Marasini, N.; Hung, H.; Yang, J.; Luo, Y.; Khalil, Z.G.; Capon, R.J.; Hussein, W.M.; et al. Poly(hydrophobic amino acid) Conjugates for the Delivery of Multiepitope Vaccine against Group A Streptococcus. Bioconj. Chem. 2021, 32, 2307–2317. [Google Scholar] [CrossRef]
- Shalash, A.O.; Becker, L.; Yang, J.; Giacomin, P.; Pearson, M.; Hussein, W.M.; Loukas, A.; Toth, I.; Skwarczynski, M. Development of a peptide vaccine against hookworm infection: Immunogenicity, efficacy, and immune correlates of protection. J. Allergy Clin. Immunol. 2022, 150, 157–169.e110. [Google Scholar] [CrossRef] [PubMed]
- Shalash, A.O.; Becker, L.; Yang, J.; Giacomin, P.; Pearson, M.; Hussein, W.M.; Loukas, A.; Skwarczynski, M.; Toth, I. Oral Peptide Vaccine against Hookworm Infection: Correlation of Antibody Titers with Protective Efficacy. Vaccines 2021, 9, 1034. [Google Scholar] [CrossRef]
- Bartlett, S.; Skwarczynski, M.; Xie, X.; Toth, I.; Loukas, A.; Eichenberger, R.M. Development of natural and unnatural amino acid delivery systems against hookworm infection. Precis. Nanomed. 2020, 3, 471–482. [Google Scholar] [CrossRef]
- Ahmad Fuaad, A.A.H.; Skwarczynski, M.; Toth, I. The use of microwave-assisted solid-phase peptide synthesis and click chemistry for the synthesis of vaccine candidates against hookworm infection. Methods Mol. Biol. 2016, 1403, 639–653. [Google Scholar]
- Hussein, W.M.; Cheong, Y.S.; Liu, C.; Liu, G.; Begum, A.A.; Attallah, M.A.; Moyle, P.M.; Torchilin, V.; Smith, R.; Toth, I. Peptide-based targeted polymeric nanoparticles for siRNA delivery. Nanotechnology 2019, 30, 415604. [Google Scholar] [CrossRef]
- Hussein, W.M.; Toth, I.; Skwarczynski, M. Peptide-Polymer Conjugation Via Copper-Catalyzed Alkyne-Azide 1,3-Dipolar Cycloaddition. In Peptide Conjugation: Methods and Protocols; Hussein, W.M., Stephenson, R.J., Toth, I., Eds.; Springer: New York, NY, USA, 2021; pp. 1–7. [Google Scholar]
- Giddam, A.K.; Reiman, J.M.; Zaman, M.; Skwarczynski, M.; Toth, I.; Good, M.F. A semi-synthetic whole parasite vaccine designed to protect against blood stage malaria. Acta Biomater. 2016, 44, 295–303. [Google Scholar] [CrossRef]
- Yang, J.; Firdaus, F.; Azuar, A.; Khalil, Z.G.; Marasini, N.; Capon, R.J.; Hussein, W.M.; Toth, I.; Skwarczynski, M. Cell-Penetrating Peptides-Based Liposomal Delivery System Enhanced Immunogenicity of Peptide-Based Vaccine against Group A Streptococcus. Vaccines 2021, 9, 499. [Google Scholar] [CrossRef]
- Baldin, A.V.; Savvateeva, L.V.; Bazhin, A.V.; Zamyatnin, A.A., Jr. Dendritic Cells in Anticancer Vaccination: Rationale for Ex Vivo Loading or In Vivo Targeting. Cancers 2020, 12, 590. [Google Scholar] [CrossRef]
- Liu, Y.; Yao, L.; Cao, W.; Liu, Y.; Zhai, W.; Wu, Y.; Wang, B.; Gou, S.; Qin, Y.; Qi, Y.; et al. Dendritic Cell Targeting Peptide-Based Nanovaccines for Enhanced Cancer Immunotherapy. ACS Appl. Bio Mater. 2019, 2, 1241–1254. [Google Scholar] [CrossRef] [PubMed]
- Palucka, K.; Banchereau, J. Cancer immunotherapy via dendritic cells. Nat. Rev. Cancer 2012, 12, 265–277. [Google Scholar] [CrossRef]
- Curiel, T.J.; Morris, C.; Brumlik, M.; Landry, S.J.; Finstad, K.; Nelson, A.; Joshi, V.; Hawkins, C.; Alarez, X.; Lackner, A.; et al. Peptides identified through phage display direct immunogenic antigen to dendritic cells. J. Immunol. 2004, 172, 7425–7431. [Google Scholar] [CrossRef] [PubMed]
- Golani-Armon, A.; Golan, M.; Shamay, Y.; Raviv, L.; David, A. DC3-Decorated Polyplexes for Targeted Gene Delivery into Dendritic Cells. Bioconj. Chem. 2015, 26, 213–224. [Google Scholar] [CrossRef]
- Bobrysheva, I.V. Immunomodulator Imunofan Affects Cell Profile of Morphofunctional Zones of Rat Thymus and Delays Its Age-Related Involution. Bull. Russ. State Med. Univ. 2016, 34–38. [Google Scholar] [CrossRef]
- Vinnitsky, L.; Bunatian, K.; Mironova, E.; Akopdjanova, E.; Komarov, A. Immunomodulator imunofan in treatment of post-traumatic complications. Immunology 1996, 89, Ii366. [Google Scholar]
- Toda, S.; Ishii, N.; Okada, E.; Kusakabe, K.I.; Arai, H.; Hamajima, K.; Gorai, I.; Nishioka, K.; Okuda, K. HIV-1-specific cell-mediated immune responses induced by DNA vaccination were enhanced by mannan-coated liposomes and inhibited by anti-interferon-gamma antibody. Immunology 1997, 92, 111–117. [Google Scholar] [CrossRef]
- Kojima, N.; Ishii, M.; Kawauchi, Y.; Takagi, H. Oligomannose-Coated Liposome as a Novel Adjuvant for the Induction of Cellular Immune Responses to Control Disease Status. Biomed Res. Int. 2013, 2013, 562924 . [Google Scholar] [CrossRef]
- Gu, X.G.; Schmitt, M.; Hiasa, A.; Nagata, Y.; Ikeda, H.; Sasaki, Y.; Akiyoshi, K.; Sunamoto, J.; Nakamura, H.; Kuribayashi, K.; et al. A novel hydrophobized polysaccharide/oncoprotein complex vaccine induces in vitro and in vivo cellular and humoral immune responses against HER2-expressing murine sarcomas. Cancer Res. 1998, 58, 3385–3390. [Google Scholar] [PubMed]
- Wang, F.; Xiao, W.; Elbahnasawy, M.A.; Bao, X.; Zheng, Q.; Gong, L.; Zhou, Y.; Yang, S.; Fang, A.; Farag, M.M.S.; et al. Optimization of the Linker Length of Mannose-Cholesterol Conjugates for Enhanced mRNA Delivery to Dendritic Cells by Liposomes. Front. Pharmacol. 2018, 9, 980. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.S.; Na, K.S.; Hwang, H.; Oh, P.S.; Kim, D.H.; Lim, S.T.; Sohn, M.H.; Jeong, H.J. Effect of space length of mannose ligand on uptake of mannosylated liposome in RAW 264.7 cells: In vitro and in vivo studies. J. Biomed. Mater. Res. Part A 2014, 102, 4545–4553. [Google Scholar] [CrossRef]
- Nahar, U.J.; Toth, I.; Skwarczynski, M. Mannose in vaccine delivery. J. Control. Release 2022, 351, 284–300. [Google Scholar] [CrossRef]
- Sedaghat, B.; Stephenson, R.J.; Giddam, A.K.; Eskandari, S.; Apte, S.H.; Pattinson, D.J.; Doolan, D.L.; Toth, I. Synthesis of Mannosylated Lipopeptides with Receptor Targeting Properties. Bioconj. Chem. 2016, 27, 533–548. [Google Scholar] [CrossRef] [PubMed]
- Stanisic, D.I.; Ho, M.-F.; Nevagi, R.; Cooper, E.; Walton, M.; Islam, M.T.; Hussein, W.M.; Skwarczynski, M.; Toth, I.; Good, M.F. Development and Evaluation of a Cryopreserved Whole-Parasite Vaccine in a Rodent Model of Blood-Stage Malaria. mBio 2021, 12, e0265721. [Google Scholar] [CrossRef]
- Al-Nazal, H.A.; Cooper, E.; Ho, M.F.; Eskandari, S.; Majam, V.; Giddam, A.K.; Hussein, W.M.; Islam, M.T.; Skwarczynski, M.; Toth, I.; et al. Pre-clinical evaluation of a whole-parasite vaccine to control human babesiosis. Cell Host Microbe 2021, 29, 894–903.e895. [Google Scholar] [CrossRef]
- Ssemaganda, A.; Giddam, A.K.; Zaman, M.; Skwarczynski, M.; Toth, I.; Stanisic, D.I.; Good, M.F. Induction of Plasmodium-Specific Immune Responses Using Liposome-Based Vaccines. Front Immunol 2019, 10, 135. [Google Scholar] [CrossRef]
- Yang, J.R.; Luo, Y.C.; Shibu, M.A.; Toth, I.; Skwarczynski, M. Cell-Penetrating Peptides: Efficient Vectors for Vaccine Delivery. Curr. Drug Deliv. 2019, 16, 430–443. [Google Scholar] [CrossRef] [PubMed]
- Skwarczynski, M.; Zaman, M.; Urbani, C.N.; Lin, I.C.; Jia, Z.; Batzloff, M.R.; Good, M.F.; Monteiro, M.J.; Toth, I. Polyacrylate dendrimer nanoparticles: A self-adjuvanting vaccine delivery system. Angew. Chem. Int. Ed. 2010, 49, 5742–5745. [Google Scholar] [CrossRef]
- Zaman, M.; Skwarczynski, M.; Malcolm, J.M.; Urbani, C.N.; Jia, Z.F.; Batzloff, M.R.; Good, M.F.; Monteiro, M.J.; Toth, I. Self-adjuvanting polyacrylic nanoparticulate delivery system for group A streptococcus (GAS) vaccine. Nanomed. Nanotechnol. Biol. Med. 2011, 7, 168–173. [Google Scholar] [CrossRef]
- Guo, Y.; Sakonsinsiri, C.; Nehlmeier, I.; Fascione, M.A.; Zhang, H.; Wang, W.; Pöhlmann, S.; Turnbull, W.B.; Zhou, D. Compact, Polyvalent Mannose Quantum Dots as Sensitive, Ratiometric FRET Probes for Multivalent Protein–Ligand Interactions. Angew. Chem. Int. Ed. 2016, 55, 4738–4742. [Google Scholar] [CrossRef]
- Yang, J.; Azuar, A.; Toth, I.; Skwarczynski, M. Liposomes for the Delivery of Lipopeptide Vaccines. In Vaccine Design: Methods and Protocols, Volume 3. Resources for Vaccine Development; Thomas, S., Ed.; Springer: New York, NY, USA, 2022; pp. 295–307. [Google Scholar]
- Facciola, A.; Visalli, G.; Lagana, P.; La Fauci, V.; Squeri, R.; Pellicano, G.F.; Nunnari, G.; Trovato, M.; Di Pietro, A. The new era of vaccines: The “nanovaccinology”. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 7163–7182. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Duong, V.T.; Shalash, A.O.; Skwarczynski, M.; Toth, I. Chemical Conjugation Strategies for the Development of Protein-Based Subunit Nanovaccines. Vaccines 2021, 9, 563. [Google Scholar] [CrossRef]
- Skwarczynski, M.; Toth, I. Recent advances in peptide-based subunit nanovaccines. Nanomedicine 2014, 9, 2657–2669. [Google Scholar] [CrossRef] [PubMed]
- Ghaffar, K.A.; Giddam, A.K.; Zaman, M.; Skwarczynski, M.; Toth, I. Liposomes as nanovaccine delivery systems. Curr. Top. Med. Chem. 2014, 14, 1194–1208. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Su, Q.; Song, H.; Shi, X.; Li, M.; Song, N.; Lou, S.; Wang, W.; Yu, Z. Precisely Shaped Self-Adjuvanting Peptide Vaccines with Enhanced Immune Responses for HPV-Associated Cancer Therapy. ACS Appl. Mater. Interfaces 2021, 13, 49737–49753. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wu, L.; Tong, R.; Yang, F.; Yin, L.; Li, M.; You, L.; Xue, J.; Lu, Y. PD-1/PD-L1 Inhibitors in Cervical Cancer. Front. Pharmacol. 2019, 10. [Google Scholar] [CrossRef]
- Domingos-Pereira, S.; Galliverti, G.; Hanahan, D.; Nardelli-Haefliger, D. Carboplatin/paclitaxel, E7-vaccination and intravaginal CpG as tri-therapy towards efficient regression of genital HPV16 tumors. J. Immunother. Cancer 2019, 7, 122. [Google Scholar] [CrossRef]
- Kokate, R.A.; Thamake, S.I.; Chaudhary, P.; Mott, B.; Raut, S.; Vishwanatha, J.K.; Jones, H.P. Enhancement of anti-tumor effect of particulate vaccine delivery system by ‘Bacteriomimetic’ CpG functionalization of poly-lactic-co-glycolic acid nanoparticles. Nanomedicine 2015, 10, 915–929. [Google Scholar] [CrossRef]
- Liu, C.; Chu, X.; Sun, P.; Feng, X.; Huang, W.; Liu, H.; Ma, Y. Synergy effects of Polyinosinic-polycytidylic acid, CpG oligodeoxynucleotide, and cationic peptides to adjuvant HPV E7 epitope vaccine through preventive and therapeutic immunization in a TC-1 grafted mouse model. Hum. Vaccines Immunother. 2018, 14, 931–940. [Google Scholar] [CrossRef]
- Kim, T.G.; Kim, C.H.; Won, E.H.; Bae, S.M.; Ahn, W.S.; Park, J.B.; Sin, J.I. CpG-ODN-stimulated dendritic cells act as a potent adjuvant for E7 protein delivery to induce antigen-specific antitumour immunity in a HPV 16 E7-associated animal tumour model. Immunology 2004, 112, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, D.M.; Skeate, J.G.; Chavez-Juan, E.; Lühen, K.P.; Wu, J.-M.; Wu, C.-M.; Kast, W.M.; Hwang, K. Therapeutic efficacy of a human papillomavirus type 16 E7 bacterial exotoxin fusion protein adjuvanted with CpG or GPI-0100 in a preclinical mouse model for HPV-associated disease. Vaccine 2019, 37, 2915–2924. [Google Scholar] [CrossRef] [PubMed]
- Rad-Malekshahi, M.; Fransen, M.F.; Krawczyk, M.; Mansourian, M.; Bourajjaj, M.; Chen, J.; Ossendorp, F.; Hennink, W.E.; Mastrobattista, E.; Amidi, M. Self-Assembling Peptide Epitopes as Novel Platform for Anticancer Vaccination. Mol. Pharm. 2017, 14, 1482–1493. [Google Scholar] [CrossRef]
- Rahimian, S.; Fransen, M.F.; Kleinovink, J.W.; Christensen, J.R.; Amidi, M.; Hennink, W.E.; Ossendorp, F. Polymeric nanoparticles for co-delivery of synthetic long peptide antigen and poly IC as therapeutic cancer vaccine formulation. J. Control. Release 2015, 203, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Zwaveling, S.; Ferreira Mota, S.C.; Nouta, J.; Johnson, M.; Lipford, G.B.; Offringa, R.; van der Burg, S.H.; Melief, C.J. Established human papillomavirus type 16-expressing tumors are effectively eradicated following vaccination with long peptides. J. Immunol. 2002, 169, 350–358. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, L.M.; Morale, M.G.; Chaves, A.A.; Cavalher, A.M.; Lopes, A.S.; Diniz Mde, O.; Schanoski, A.S.; de Melo, R.L.; Ferreira, L.C.; de Oliveira, M.L.; et al. Design, Immune Responses and Anti-Tumor Potential of an HPV16 E6E7 Multi-Epitope Vaccine. PLoS ONE 2015, 10, e0138686. [Google Scholar] [CrossRef]
- Nguyen, C.T.; Hong, S.H.; Sin, J.-I.; Vu, H.V.D.; Jeong, K.; Cho, K.O.; Uematsu, S.; Akira, S.; Lee, S.E.; Rhee, J.H. Flagellin enhances tumor-specific CD8+ T cell immune responses through TLR5 stimulation in a therapeutic cancer vaccine model. Vaccine 2013, 31, 3879–3887. [Google Scholar] [CrossRef]
- Namvar, A.; Panahi, H.A.; Agi, E.; Bolhassani, A. Development of HPV(16,18,31,45) E5 and E7 peptides-based vaccines predicted by immunoinformatics tools. Biotechnol. Lett. 2020, 42, 403–418. [Google Scholar] [CrossRef]
- Gan, L.; Jia, R.; Zhou, L.; Guo, J.; Fan, M. Fusion of CTLA-4 with HPV16 E7 and E6 enhanced the potency of therapeutic HPV DNA vaccine. PLoS ONE 2014, 9, e108892. [Google Scholar] [CrossRef]
- Peng, S.; Tan, M.; Li, Y.D.; Cheng, M.A.; Farmer, E.; Ferrall, L.; Gaillard, S.; Roden, R.B.S.; Hung, C.F.; Wu, T.C. PD-1 blockade synergizes with intratumoral vaccination of a therapeutic HPV protein vaccine and elicits regression of tumor in a preclinical model. Cancer Immunol. Immunother. 2021, 70, 1049–1062. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vaccine | Targeting Moiety | Size (nm ± SD) | PDI | Charge (mV ± SD) |
|---|---|---|---|---|
| Blank liposomes | - | 165 ± 4 | 0.06 ± 0.01 | 61 ± 2 |
| D-8Qm | - | 250 ± 50; 1500 ± 100 | 0.70 ± 0.06 | −14 ± 1 |
| pLeu-8Qm | - | 750 ± 350 | 0.60 ± 0.10 | −10 ± 5 |
| L1 | - | 320 ± 20; 4500 ± 600 | 0.50 ± 0.05 | 28 ± 2 |
| L2 | - | 157 ± 2 | 0.10 ± 0.01 | 24 ± 2 |
| L2DC1 | DC1 | 200 ± 10; 3200 ± 1100 | 0.30 ± 0.01 | 17 ± 2 |
| L2DC2 | DC2 | 160 ± 10 | 0.05 ± 0.01 | 23 ± 2 |
| L2M1 | M1 | 162 ± 6 | 0.10 ± 0.01 | 13 ± 1 |
| L2M2 | M2 | 174 ± 2; 4800 ± 300 | 0.20 ± 0.01 | 20 ± 1 |
| L2M3 | M3 | 143 ± 2 | 0.05 ± 0.01 | 23 ± 2 |
| L2CPP 1 | CPP | 610 ± 60 | 0.40 ± 0.03 | 54 ± 5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Firdaus, F.Z.; Bartlett, S.; Hussein, W.M.; Lu, L.; Wright, Q.; Huang, W.; Nahar, U.J.; Yang, J.; Khongkow, M.; Veitch, M.; et al. Liposomal Formulations of a Polyleucine–Antigen Conjugate as Therapeutic Vaccines against Cervical Cancer. Pharmaceutics 2023, 15, 602. https://doi.org/10.3390/pharmaceutics15020602
Firdaus FZ, Bartlett S, Hussein WM, Lu L, Wright Q, Huang W, Nahar UJ, Yang J, Khongkow M, Veitch M, et al. Liposomal Formulations of a Polyleucine–Antigen Conjugate as Therapeutic Vaccines against Cervical Cancer. Pharmaceutics. 2023; 15(2):602. https://doi.org/10.3390/pharmaceutics15020602
Chicago/Turabian StyleFirdaus, Farrhana Z., Stacey Bartlett, Waleed M. Hussein, Lantian Lu, Quentin Wright, Wenbin Huang, Ummey J. Nahar, Jieru Yang, Mattaka Khongkow, Margaret Veitch, and et al. 2023. "Liposomal Formulations of a Polyleucine–Antigen Conjugate as Therapeutic Vaccines against Cervical Cancer" Pharmaceutics 15, no. 2: 602. https://doi.org/10.3390/pharmaceutics15020602
APA StyleFirdaus, F. Z., Bartlett, S., Hussein, W. M., Lu, L., Wright, Q., Huang, W., Nahar, U. J., Yang, J., Khongkow, M., Veitch, M., Koirala, P., Ruktanonchai, U. R., Monteiro, M. J., Gonzalez Cruz, J. L., Stephenson, R. J., Wells, J. W., Toth, I., & Skwarczynski, M. (2023). Liposomal Formulations of a Polyleucine–Antigen Conjugate as Therapeutic Vaccines against Cervical Cancer. Pharmaceutics, 15(2), 602. https://doi.org/10.3390/pharmaceutics15020602