
Nanosized Drug Delivery Systems to Fight Tuberculosis


Abstract
1. Introduction
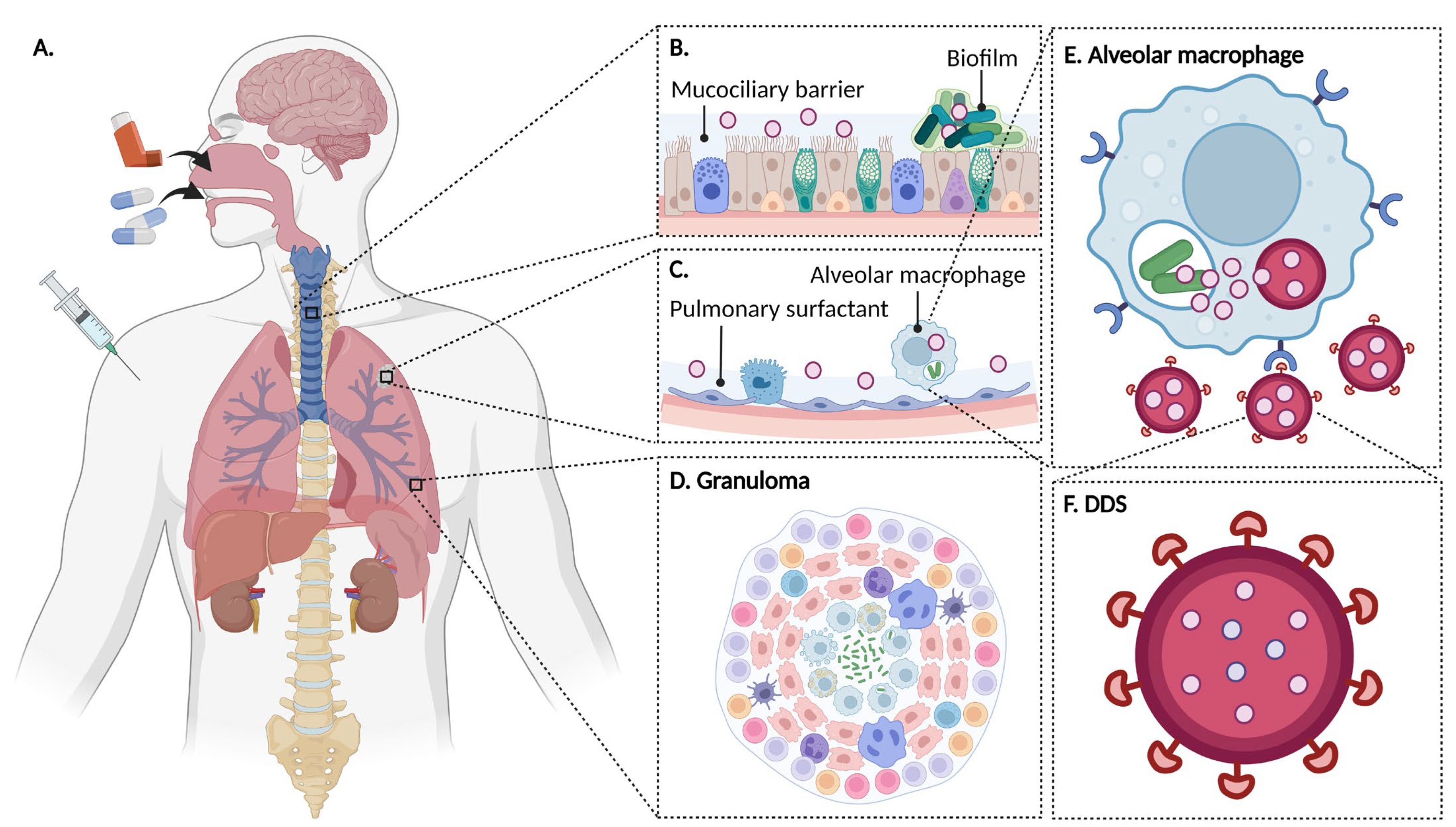
2. Tuberculosis
2.1. Physiopathology
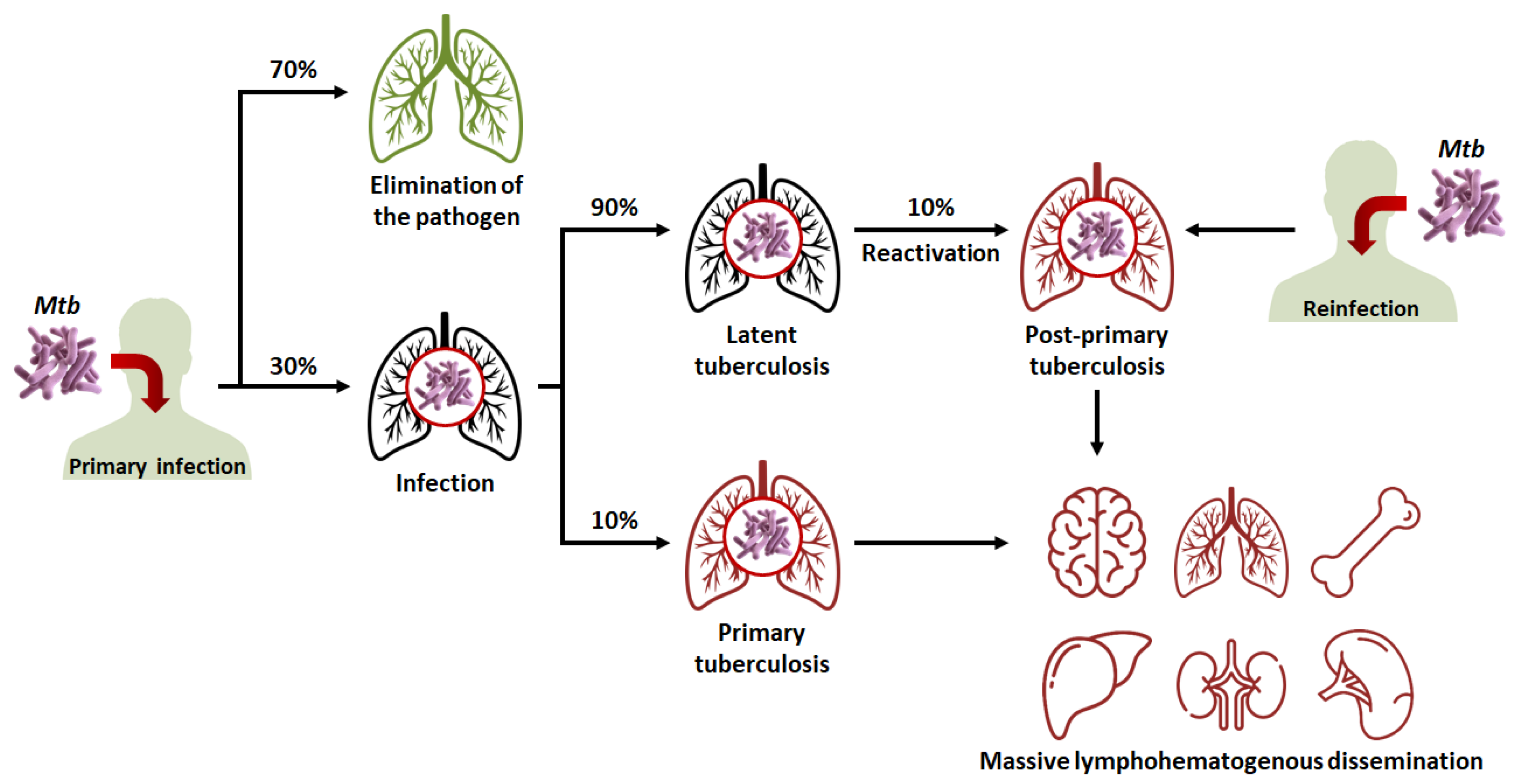
- In 90% of cases, the granuloma, acting as a physical and immunological barrier will succeed in stemming the infection. The pathogen will be contained in necrotic areas within granulomas located in the lungs [25]. Fibrous lesions will develop, and TB will evolve towards a latent form. This explains the fact that almost a third of the world’s population carries the pathogen, but that not more than ten million cases of active TB are diagnosed every year. Thus, most subjects that are latent cases (90%) will carry Mtb for decades, but will not show symptoms or infect other individuals [22].
- In 10% of cases, most often in very young, elderly, or immunocompromised subjects, the granuloma will fail to contain Mtb. This phenomenon can also occur after the reactivation of dormant bacilli (which happens in 10% of latent cases) or with a novel inhalation of bacilli. The adaptive immune response will attempt to eliminate bacteria multiplying and escaping macrophages, but in doing so, will cause the destruction of lung tissue [1,26]. This will lead to the formation of caseous lesions and cavities, within which the growth of Mtb will no longer be controlled. TB will then evolve towards an active form. In certain cases, the pathogen will spread to other organs (brain, bones, liver, spleen, kidneys), and, in the most serious cases, the disease will evolve towards a miliary form, involving the massive lymphohematogenous dissemination of bacteria [27].
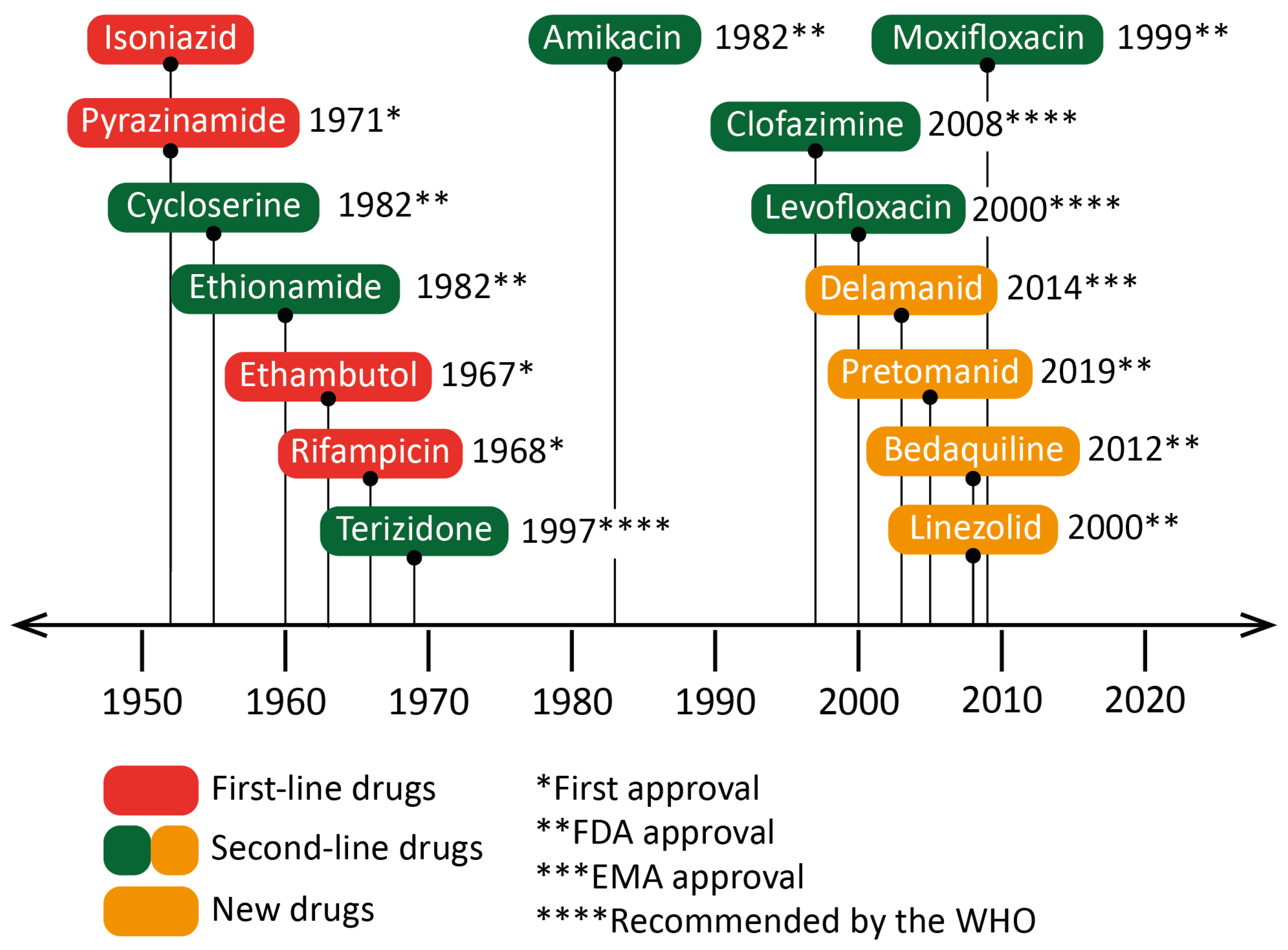
2.2. Treatment
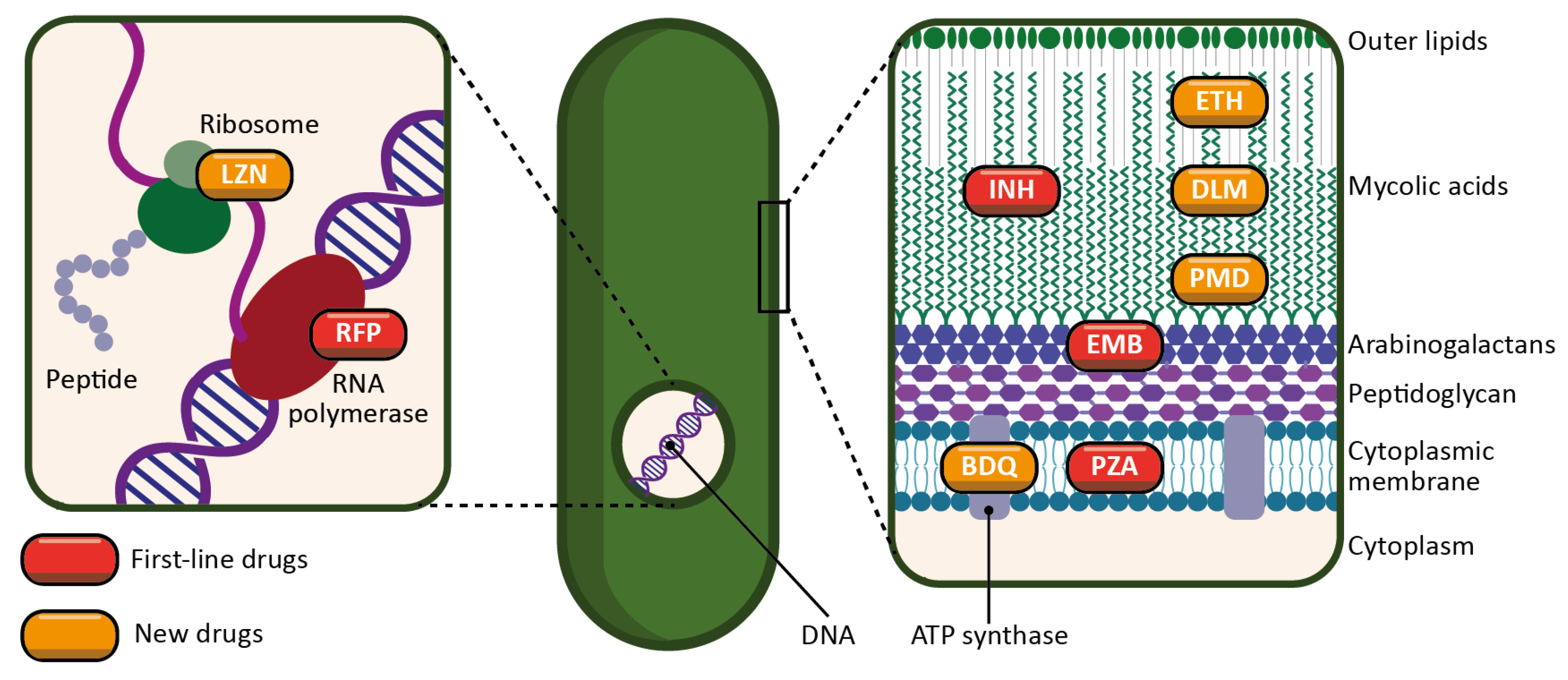
- INH is a prodrug activated by a bacterial enzyme (KatG) [32]. The activation of the molecule produces an inhibitor of another bacterial enzyme, InhA, which results in the inhibition of mycolic acid synthesis, and therefore of the bacterial wall.
- RFP is an inhibitor of the bacterial RNA polymerase, and thus acts by preventing protein synthesis [33]. It inhibits the elongation of bacterial RNA once it reaches two to three nucleotides in length.
- PZA is a prodrug metabolized by a bacterial enzyme (pyrazinamidase) to become pyrazinoic acid [34]. The exact mechanism of action of pyrazinoic acid is still only partially elucidated, but the molecule is thought to act simultaneously on membrane energy production, the ribosomal protein RpsA, and other yet unidentified bacterial targets.
- EMB targets arabinosyl transferase (a bacterial enzyme), thereby inhibiting arabinogalactan and bacterial wall synthesis [35]. EMB is also thought to exert a synergistic effect on INH activity.
- Ethionamide (ETH) is a thioisonicotinamide with a structure similar to that of INH [42]. ETH is a prodrug that, like INH, must be activated in order to inhibit mycobacterial fatty acid synthesis (by inhibiting enoyl-ACP reductase), which is essential for the production and repair of the bacterial cell wall.
- LZN is a synthetic antimicrobial drug of the oxazolidinone class [43]. By binding to the rRNA on the 50S and 30S ribosomal subunits, it blocks the synthesis of bacterial proteins.
- BDQ is the only FDA-approved antitubercular drug that targets the production of ATP [44]. BDQ inhibits the proton pumping mechanism by binding to the c subunit of the ATP synthase complex. It has also been observed that BDQ is able to act on the ε subunit of the enzyme.
- DLM is a prodrug that, like INH, prevents the synthesis of mycolic acid in the bacterial cell wall [45]. DLM inhibits the synthesis of methoxy- and keto-mycolic acid by acting on the mycobacterial F420 system.
- PMD is also a prodrug that acts under different mechanisms [46]. Under aerobic conditions, PMD inhibits protein and lipid synthesis by decreasing the availability of keto-mycolic acids through the inadequate oxidative transformation of the hydroxymycolate precursor. Under anaerobic conditions, PMD generates desnitro metabolites and provokes the release of nitric oxide, which inhibits cytochrome c oxidase and leads to a significant reduction in the amount of ATP present in bacteria.
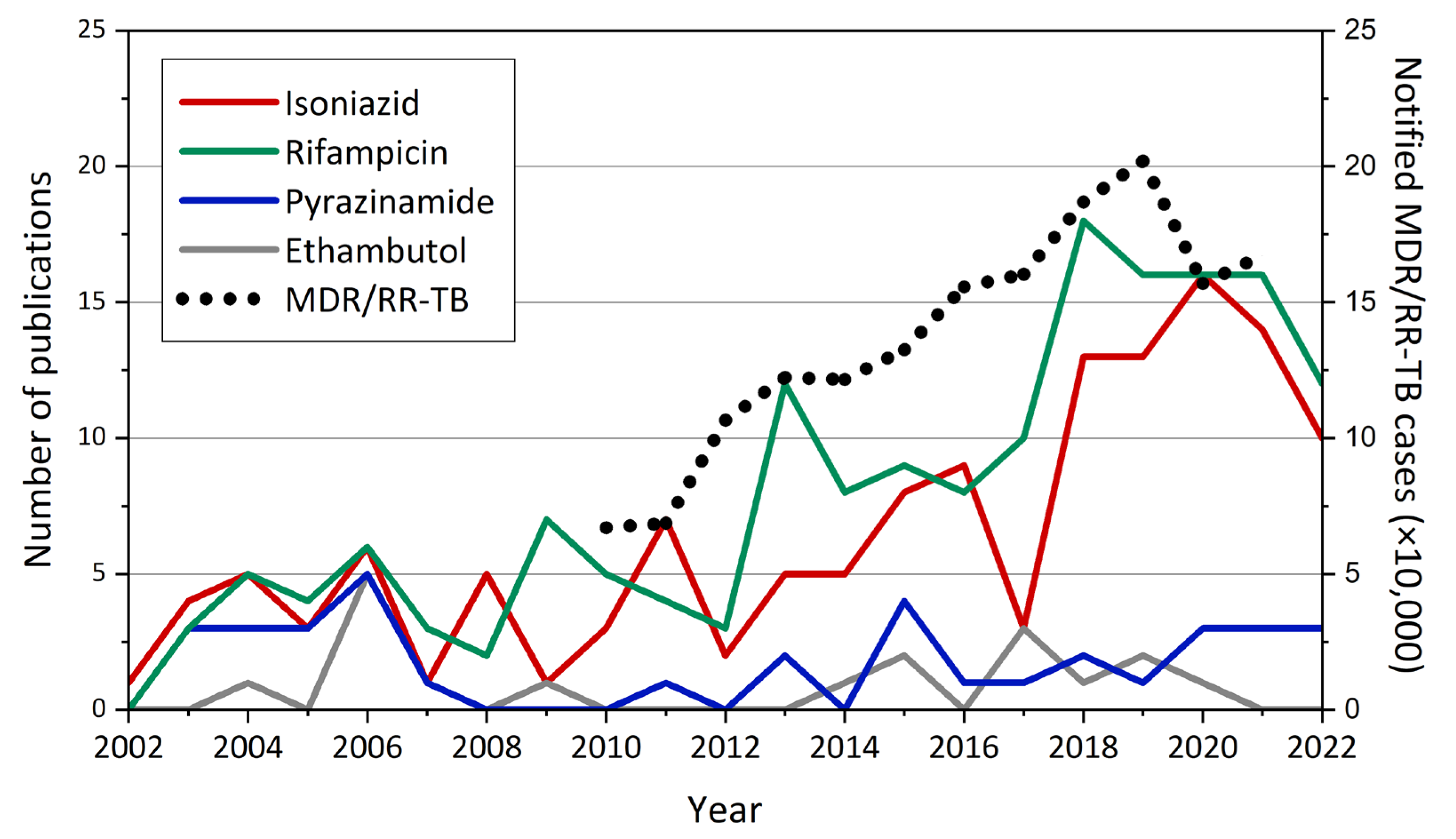
- Multidrug-resistant TB (MDR-TB) is defined as TB with an RFP-resistant (RR-TB) and INH-resistant strain.
- MDR/RR-TB stands for either MDR-TB or RR-TB.
- Pre-extensively drug-resistant TB (pre-XDR-TB) is defined as MDR/RR-TB with resistance to at least one fluoroquinolone (either levofloxacin (LVX) or moxifloxacin (MOX)).
- Extensively drug-resistant TB (XDR-TB) is defined as MDR/RR-TB with resistance to at least one fluoroquinolone (either LVX or MOX) and to at least one of the following two drugs: LZN and BDQ.
- Thus, the updated recommendations are as follows:
- For an INH-resistant only strain, the treatment is continued with RFP, PZA and EMB for a period of six months. INH is replaced by LVX.
- For MDR/RR-TB and pre-XDR-TB:
- Firstly, the WHO suggests adopting a six-month regimen (BPaLM) comprising LZN, BDQ, PMD and MOX (in the absence of a MOX-resistant strain). It is urged to use this new regimen instead of the nine-month or longer regimens for MDR/RR-TB, since BPaLM provides superior results in a shorter period.
- Secondly, for MDR/RR-TB without a resistance to fluoroquinolones, the WHO recommends using a nine-month regimen rather than longer (eighteen-month) regimen. This regimen consists of BDQ (for six months) in combination with a fluoroquinolone (LVX or MOX), INH, PZA, EMB, ETH and clofazimine (CFZ) (for four months, with the possibility of extending this period to six months if the patient remains sputum smear-positive after four months), and then, a fluoroquinolone (LVX or MOX), PZA, EMB and CFZ (for five months). Two months of LZN might be used as an alternative to ETH.
- For XDR-TB, the complementary molecules mentioned above constitute the core of the treatment.
3. Nanoparticles
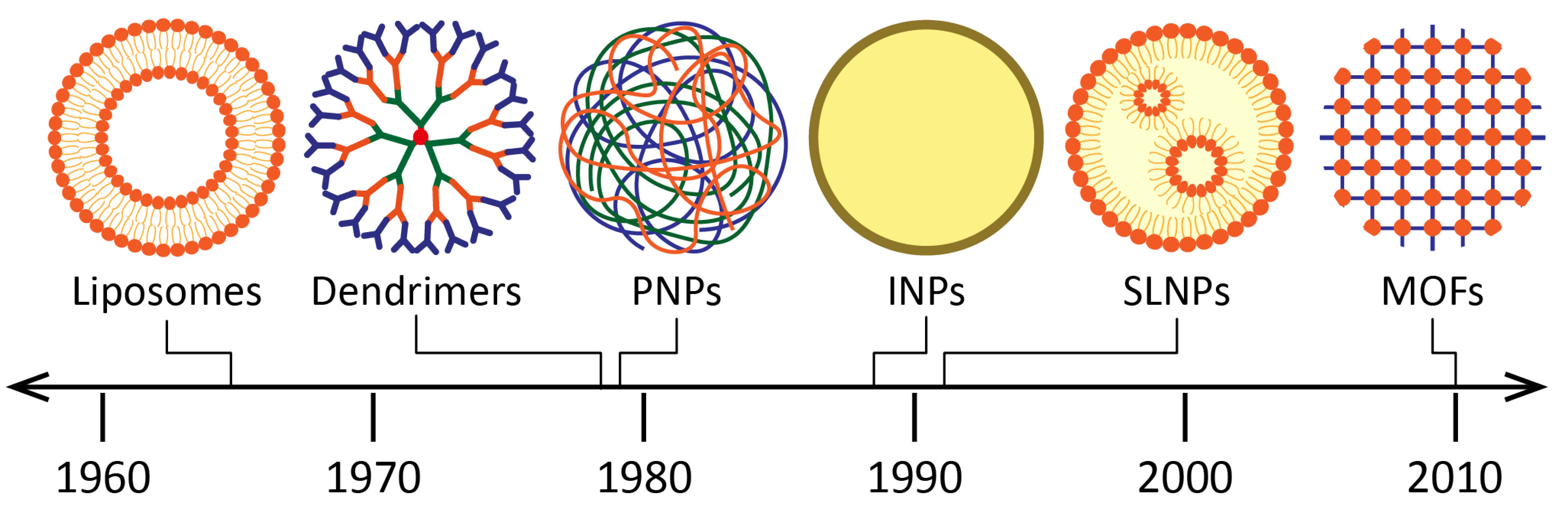
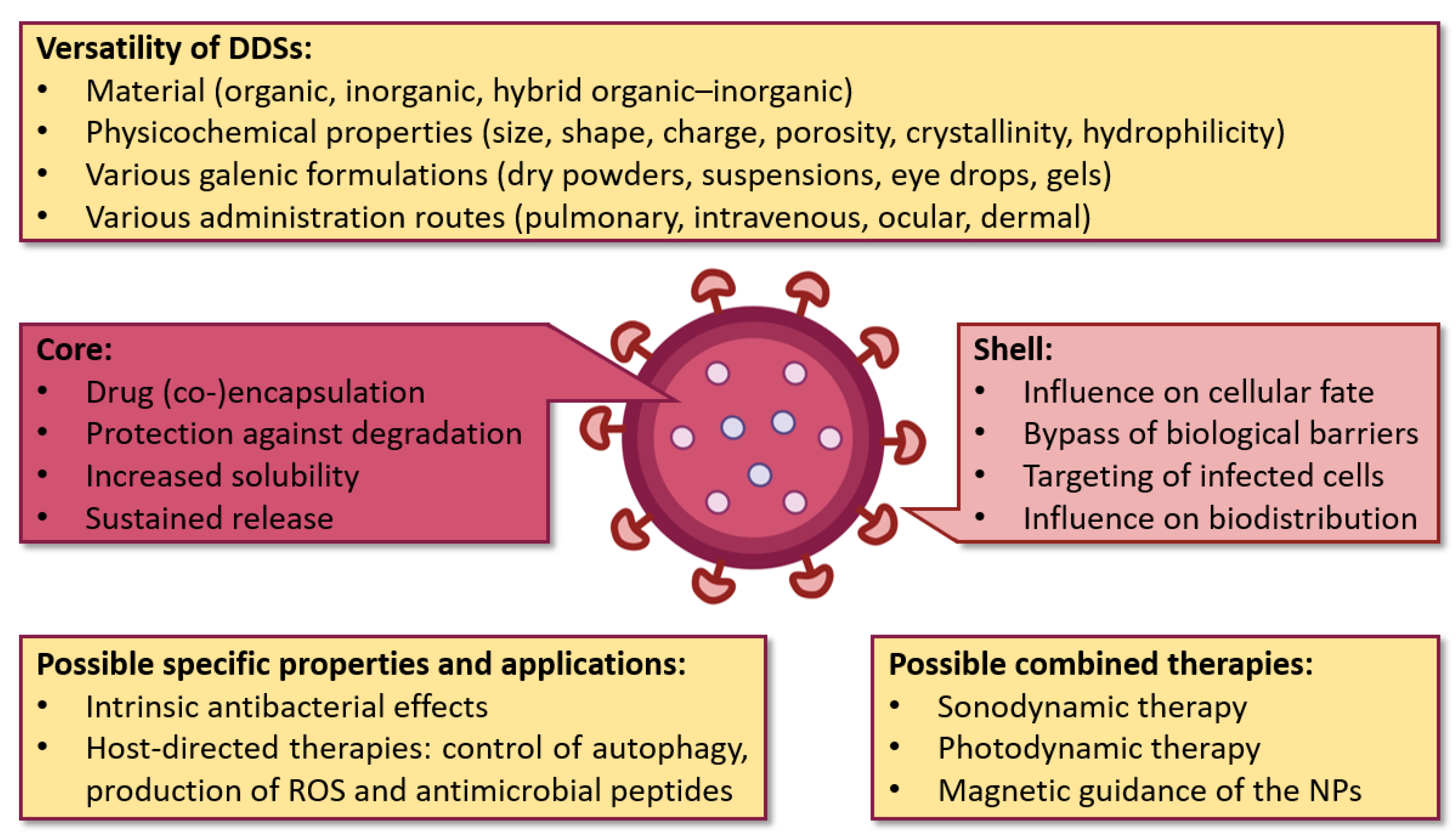
3.1. Diversity and Versatility of Drug Nanocarriers
- Liposomes were introduced as drug carriers as early as 1965. They are vesicles made of at least one lipid bilayer, which is itself made of phospholipids [55]. The amphiphilic character of these molecules (hydrophilic head, hydrophobic tail) enables the simultaneous encapsulation of active molecules with different solubilities. The applications of liposomes are vast (among others, in the food and cosmetics industries), especially in the biomedical field: the ability of liposomes to encapsulate nucleic acids, enzymes, hormones as well as blood factors, makes them suitable carriers for the treatment of infectious diseases, cancers, gene therapy and for molecular imaging [56]. Other structures such as niosomes [57], phytosomes [58] and transferosomes [59] are also vesicular DDSs of interest.
- Nano(micro)emulsions, first introduced in 1943 [60], are another type of DDS that have gained much attention because of their high loading capacity, ease of preparation, and thermodynamic stability [61,62]. They are defined as a system of water, oil and an amphiphile (surfactant and co-surfactant) which is optically isotropic and thermodynamically stable [63].
- Dendrimers were discovered later, at the end of the 1970s [64]. Their structure consists of a hydrophobic core, with chains of repeating units grafted onto it, branching off each other in a dendritic manner [65]. Functional groups can also be grafted at the periphery. Thus, hydrophilic molecules can be integrated into dendrimers (using the large specific surface conferred by the chains of repeated units) as well as hydrophobic ones (using the core cavity). The major advantages of dendrimers are their homogeneity and small size. Their biomedical applications include infectious diseases, cancers, and gene therapy [66]. INH and RFP, mainly, were incorporated in dendrimers to treat TB [67].
- Inorganic nanoparticles (INPs) are nanocarriers which have also been widely studied for drug delivery [68]. In this group, nanomaterials derived from gold, silica, carbon nanotubes and iron oxides can be found. Gold nanoparticles (AuNPs) have aroused great interest for drug delivery. Indeed, they are chemically inert and non-toxic, and they can be used as contrast agents for medical imaging applications [69]. Unlike other nanocarriers, drugs are usually immobilized on the AuNP surface for their loading, while other ligands and chemical moieties can also be added for their protection and targeting. Silica nanoparticles (SiNPs) are biocompatible nanocarriers that have been used as excipients and food additives for years [70]. They stand out due to their high load capacity, mechanical stability, simplicity of functionalization, and customizable release profiles. Mesoporous SiNPs are of particular interest due to their large surface area. Moreover, iron oxide NPs have also been studied in the medical field, although only imaging applications have reached the market [71]. They have a magnetic behavior which can be useful to guide them (with the help of an external magnetic field) towards the target, thus enhancing the drug release [72].
- Metal–organic frameworks (MOFs) are promising porous nanocarriers which have generated growing interest over the past twenty years. The originality of these structures resides in the combination of metal ions and organic ligands, which assemble to form highly porous networks. This feature enables specific surfaces ranging up to almost 2000 m2/g for biocompatible formulations, favorable for drug entrapment [73,74]. The diversity of MOFs is such that nearly one hundred thousand different models are currently deposited in the Cambridge Structural Database. Nanosized MOFs are studied for biomedical applications (infectious diseases and cancers), as well as for industrial uses (gas storage and separation, catalysis, water treatment).
- Solid lipid nanoparticles (SLNPs) were discovered in the early 1990s [75]. They consist of phospholipids whose tails form a solid hydrophobic core surrounded by a surfactant layer. Compared to liposomes, SLNPs offer increased stability for the active molecule (thanks to the solid core), higher encapsulation rates for hydrophobic molecules (since SLNPs do not possess an aqueous core) and the possibility of targeting and sustained release, thanks to the grafting of ligands of interest [76]. In addition, they can be stored for extended periods (up to three years). They are interesting candidates for various administration routes (mainly intravenous and pulmonary), and, as the rest of this review will highlight, they have been widely studied for the treatment of TB.
- Finally, polymeric nanoparticles (PNPs), such as SLNPs, occupy a prominent place in the therapeutic arsenal for the treatment of TB. Polymers are macromolecules formed by repeating covalently linked units (monomers), whose applications for drug delivery have recently been listed in another review [77]. There is a wide variety of polymers, with biodegradable ones being the most widely used: among others, chitosan, PLA (poly(lactic acid)), PLGA (poly(lactic-co-glycolic acid)), and PCL (poly(ε-caprolactone). During the preparation of PNPs, the polymeric chains assemble, often with the help of surfactants for the stabilization of the system. This results in structures suitable for the incorporation of a wide variety of molecules, both hydrophilic and hydrophobic, depending on the properties of the used material. The physicochemical properties of PNPs can be easily modified to design the appropriate nanocarrier for a given pathology, an aspect that will be developed in the following paragraphs.
3.2. Influence of Physicochemical Properties on the Fate of Drug Nanocarriers
- Size plays a major role in the mode of internalization of NPs. It is one of the main parameters studied during their characterization. Thus, NPs of 120 nm–200 nm mainly penetrate inside cells using the clathrin-dependent and caveolin-dependent pathways, while those of more than 200 nm are preferably internalized by macropinocytosis [83]. Those of 250 nm–1 µm are rather taken by phagocytosis. Not all pathways lead to the same intracellular compartments. Indeed, phagocytosis and the clathrin-dependent pathways lead to endosomes, while macropinocytosis leads to lysosomes and the caveolin-dependent pathway leads to caveosomes [84]. Therefore, in the case of TB, adapting the size of NPs so that they target one pathway (and one intracellular compartment) rather than another is a part of the therapeutic strategy itself.
- Depending on the formulation parameters, the shape of NPs can be varied (spheres, cubes, rods and cones), which in turn impact the NP’s intracellular fate [85]. For example, as early as 2006, Chithrani et al. studied the effect of the shape of AuNPs upon their internalization within HeLa cells. They showed that spherical NPs were internalized five times more than rod-shaped ones and hypothesized that this was due to more complex plasma membrane movements for rods than for spheres [86].
- The surface charge is another parameter to consider. Since the plasma membrane is negatively charged, positively charged NPs are more internalized than neutral or negatively charged objects [87]. Moreover, charge can also be used to specifically target an intracellular compartment [85,88]. Indeed, positively charged NPs tend to be internalized by macropinocytosis, while negatively charged NPs rather use the clathrin/caveolin-independent pathway [83], thus leading to different cellular locations.
- Finally, one of the main parameters to consider for the preparation of NPs is surface modification. Indeed, an appropriate surface modification can determine the internalization of NPs within a given cell type, as well as the NP’s fate at the scale of the entire organism [89]. For example, the grafting of amine or carboxyl groups at the surface of nanocarriers gives the latter a more positive or negative charge, respectively, which leads to the consequences explained in the previous paragraph. In addition, the grafting of polyethylene glycol (PEG) at the surface of NPs enables one to prevent the adsorption of opsonins, which makes it possible for NPs to escape the immune system and to prolong their circulation time in the organism.
4. The Potential of Nanoparticles Regarding the Treatment of Tuberculosis
4.1. Preamble
4.2. Delivering Antibiotics to the Site of Infection and Increasing Their Bioavailability
4.3. Potentiating the Antibacterial Effect While Reducing the Posology
4.4. Maximizing Patient Compliance by Reducing Treatment Toxicity
- For the untreated mice, the concentrations of total bilirubin, SGPT and ALP, were, respectively, 0.24 mg/100 mL, 32.2 IU/L and 33.3 IU/L.
- For the mice treated with free INH, these concentrations were, respectively, 0.63 mg/100 mL, 57.5 IU/L and 47.6 IU/L.
- For the mice treated with INH-loaded NPs, these concentrations were, respectively, 0.23 mg/100 mL, 33.45 IU/L and 32.61 IU/L.
4.5. Maximizing Patient Compliance by Reducing Treatment Complexity
4.6. Exploiting the Antibacterial Effect of the Nanocarrier Itself

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Physicochemical Properties | Biological Data | |
|---|---|---|
| Isoniazid (INH) | ||
| Chitosan–tripolyphosphate NPs [94] | ||
| Preparation: ionic gelation Size: 249 nm and 449 nm PdI: For 249 nm NPs: 0.191 For 449 nm: 0.240 ζ potential: For 249 nm NPs: 37.7 mV For 449 nm NPs: 38.9 mV Drug encapsulation: For 249 nm NPs: 13% For 449 nm NPs: 17% Drug loading: For 249 nm NPs: 4% For 449 nm NPs: 6% | In vitro | Drug release: For 249 nm NPs: 50% in 4 h, 95% in 6 days For 449 nm NPs: 40% in 4 h, 80% in 6 days Effect against Staphylococcus aureus and Pseudomonas aeruginosa: Empty NPs: 8-fold reduction in the MIC compared to free INH INH-loaded NPs: 64-fold reduction in the MIC compared to free INH Effect against Mycobacterium avium: Empty NPs: no reduction in the MIC compared to free INH INH-loaded NPs: 16-fold reduction in the MIC compared to free INH |
| In vivo | - | |
| Gelatin NPs [113] | ||
| Preparation: two-step desolvation Mannose-conjugated NPs: Size: 387 nm PdI: 0.262 ζ potential: 10.21 mV Drug encapsulation: 43% | In vitro | Drug release, for a pH of 7.4: INH-loaded NPs: 40% in 4 h, 92% in 120 h Maximum cell uptake in 6 h for J774 cells |
| In vivo | Animal model: mouse INH-loaded NPs: 4-fold higher concentration in the plasma, 9-fold higher concentration in the lungs, and 10-fold lower concentration in the kidney compared to free INH Sustained release compared to free INH Effect against Mtb: INH-loaded NPs: 2.5-fold reduction in CFUs in the lung and in the spleen compared to free INH No hepatotoxicity | |
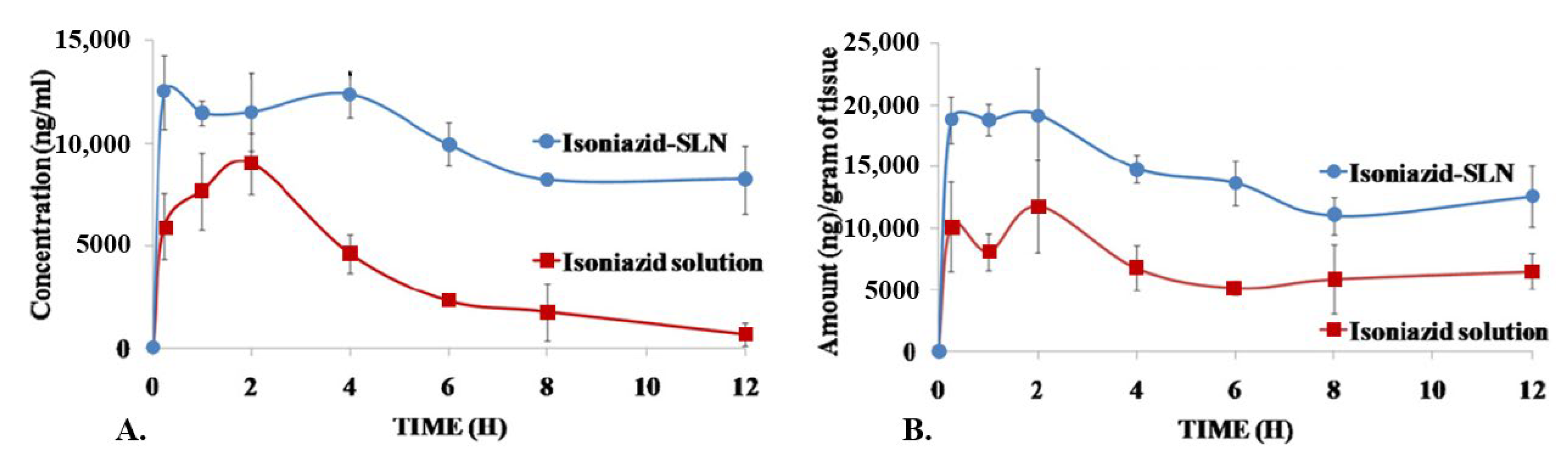
| SLNPs [107] | ||
| Preparation: microemulsification Size: 48 nm PdI: 0.266 ζ potential: −0.101 mV Drug encapsulation: 69% | In vitro | Drug release, for a pH of 6.8: Free INH: 100% in 5 h INH-loaded NPs: 65% in 24 h |
| In vivo | Animal model: rat INH-loaded NPs: significantly higher concentrations in the plasma and in the brain, but not in the liver and in the kidney, compared to free INH | |
| PLGA-PEG-PLGA NPs [133] | ||
| Preparation: double emulsification Size: 250 nm to 400 nm Drug encapsulation: 13%–19% Drug loading: 6%–9% | In vitro | INH-loaded NPs: initial burst release followed by sustained release compared to free INH |
| In vivo | Animal model: rat INH-loaded NPs: sustained release and 28-fold higher bioavailability compared to free INH | |
| Mesoporous SiNPs [118] | ||
| Preparation: formation of liquid-crystalline mesophases of surfactant, in situ polymerization of orthosilicic acid Size: 50 nm and 100 nm Drug loading:50 nm NPs: 3% 100 nm NPs: 6% | In vitro | Effect against Mtb: INH-loaded NPs: similar antibacterial effect compared to free INH |
| In vivo | Animal model: mouse Effect against Mtb: 50 nm NPs: 2-fold higher antibacterial effect compared to free INH 100 nm NPs: 4-fold higher antibacterial effect compared to free INH | |
| Selenium NPs [112] | ||
| Preparation: sodium selenite reduction and chitosan stabilization Mannose-conjugated NPs: Size: 45 nm | In vitro | Drug release: For a pH of 7.4: 45% in 48 h For a pH of 5.3: 80% in 48 h Effect against Mtb: Empty NPs: intrinsic antibacterial effect INH-loaded NPs: synergistic antibacterial effect against intracellular bacteria Promotion of Mtb localization into lysosomes No toxicity towards THP-1 cells |
| In vivo | - | |
| SLNPs [105] | ||
| Preparation: microemulsification Size: 149 nm PdI: 0.15 ζ potential: −0.35 mV Drug encapsulation: 65% Drug loading: 40% | In vitro | Drug release, for a pH of 7.2: Free INH: 100% in 7 h INH-loaded NPs: 28% in 4 h, 45% in 6 h, 94% in 48 h Ex vivo corneal permeation: INH-loaded NPs: 2.5-fold higher compared to free INH Effect against Mtb: INH-loaded NPs: 7.1-fold reduction in the MIC compared to free INH |
| In vivo | Animal model: rat and rabbit INH-loaded NPs: 428% higher bioavailability compared to free INH Drug release:Free INH: detection for up to 12 h INH-loaded NPs: detection for up to 24 h No ocular toxicity | |
| Magnetic NPs [120] | ||
| Preparation: coprecipitation Lipoaminoacid-modified NPs: Size: 13 nm ζ potential: −19.8 mV Drug loading: 3% | In vitro | Effect against Staphylococcus aureus, Escherichia coli and Pseudomonas aeruginosa: Free INH: MIC90 of >500 µg/mL INH-loaded NPs: MIC90 of 38 µg/mL Effect against Mtb: Free INH: MIC90 of 1.26 µg/mL INH-loaded NPs: MIC90 of 1.08 µg/mL |
| In vivo | - | |
| MIL-100 MOFs in mannitol microspheres [95] | ||
| Preparation: spray-drying Size: 137 nm ζ potential: −18 mV Drug loading: 30% | In vitro | Drug release, for a pH of 7.4: In milli-Q water: 21% in 0 h, 27% in 48 h In PBS: 44% in 0 h, 84% in 120 h No toxicity towards A549 cells |
| In vivo | - | |
| SLNPs [134] | ||
| Preparation: ultrasonication of crude emulsion Mannose-conjugated NPs: Size: 236 nm PdI: 0.24 ζ potential: −19 mV Drug encapsulation: 75% Drug loading: 10% | In vitro | Drug release: For a pH of 7.4: 59% in 9 h For a pH of 5.5: 83% in 9 h Mannose-conjugated NPs: higher cell uptake in macrophages (97%) compared to non-modified NPs (42%) No toxicity towards RAW264.7 cells and A549 cells |
| In vivo | Animal model: rat Effect against Mycobacterium smegmatis: Empty NPs: decrease in CFUs of 60% INH-loaded NPs: decrease in CFUs of 83% | |
| Rifampicin (RFP) | ||
| PLA microspheres [116] | ||
| Preparation: modified solvent evaporation Size: 800 nm to 8 µm Drug loading: 19% | In vitro | Drug release: For a pH of 9.8: 10% in 14 h For a pH of 7.4: 20% in 14 h For a pH of 3.0: 55% in 14 h |
| In vivo | - | |
| PLGA NPs in porous NP-aggregate particles [97] | ||
| Preparation: solvent evaporation and spray-drying Size: 195 nm PdI: 0.06 ζ potential: −33 mV Drug loading: 14% | In vitro | RFP-loaded NPs: burst release (80%) followed by slower release for 8 h |
| In vivo | Animal model: guinea pig Free RFP: low or no levels in the lungs 8 h post-treatment RFP-loaded NPs: higher levels in the lungs 8 h post-treatment | |
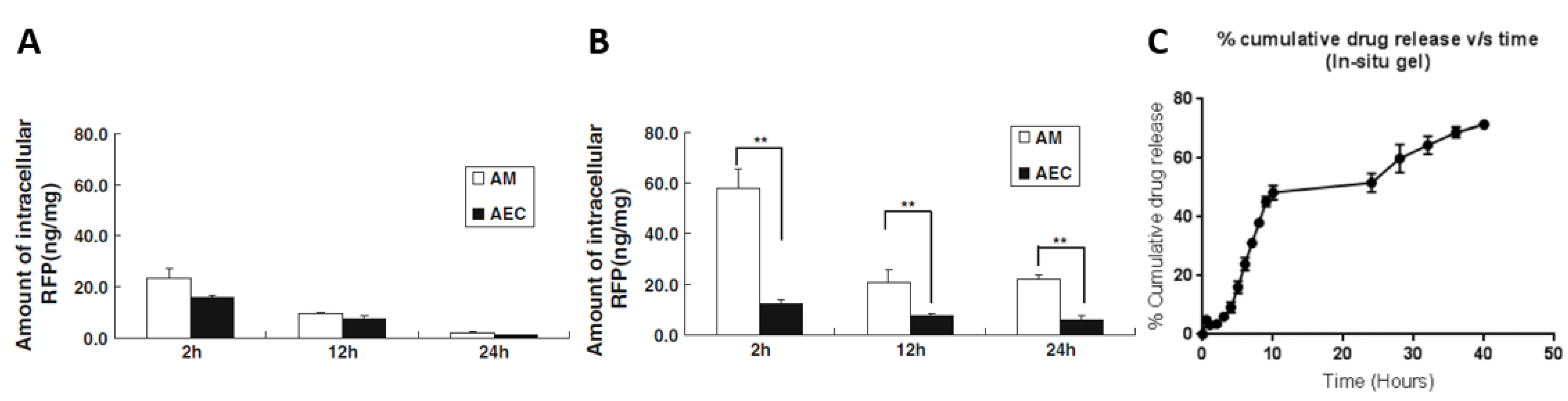
| SLNPs [98] | ||
| Preparation: modified lipid film hydration Size: 830 nm | In vitro | RFP-loaded NPs: significantly higher intracellular amounts in alveolar macrophages than in alveolar epithelial type II cells compared to free RFP No toxicity towards A549 and NR8383 cells |
| In vivo | Animal model: rat RFP-loaded NPs: significantly higher intracellular amounts in alveolar macrophages than in alveolar epithelial type II cells compared to free RFP Significantly higher intracellular concentrations (and for a longer time) in alveolar macrophages compared to free RFP | |
| Chitosan NPs [117] | ||
| Preparation: modified emulsion ionic gelation Size: 222 nm Drug encapsulation: 44% Drug loading: 43% | In vitro | Drug release: For a pH of 7.4: 5% to 8% in 1 h For a pH of 6.8 or 5.2: 8% to 13% in 1 h After 1 h, constant drug release, up to 90% in the range of 28 h–34 h |
| In vivo | - | |
| PLGA-lipid hybrid microparticles [135] | ||
| Preparation: spray-drying Hybrid system of lipid NPs encapsulated within a PLGA NP matrix: Size: 110 nm PdI: 0.15 ζ potential: −7.12 mV Drug encapsulation: 100% Drug loading: 12% | In vitro | Drug release, for a pH of 7.4: RFP-loaded NPs: 8% in 1 h in simulated lung fluid (protection of the drug before phagocytosis), 41% in 48 h in artificial lysosomal fluid Effect against intracellular Staphylococcus aureus: Free RFP: no reduction in CFUs until 5 µg/mL RFP-loaded NPs: 4-fold reduction in CFUs for 0.5 µg/mL |
| In vivo | - | |
| ZnO NPs [132] | ||
| Preparation: precipitation in liquid media Size: 11 nm ζ potential: 19.1 mV | In vitro | Effect against Mycobacterium smegmatis: Free RFP: reduction in CFUs, but not after 36 h Empty NPs: no reduction in CFUs up to 60 h RFP-loaded NPs: significant reduction in CFUs compared to free RFP, up to 60 h; irreversible bacterial membrane damage |
| In vivo | - | |
| Alginate-chitosan NPs [99] | ||
| Preparation: ionic gelation Encapsulation of RFP and ascorbic acid: RFP: Drug encapsulation: 50% Drug loading: 24% Ascorbic acid: Drug encapsulation: 16% Drug loading: 38% | In vitro | Effect against Staphylococcus aureus: Free RFP: MIC of 0.2 µg/mL RFP-loaded NPs: MIC of <0.025 µg/mL Effect against methicillin-resistant Staphylococcus aureus: Free RFP: 3.125 µg/mL RFP-loaded NPs: 1.6 µg/mL Effect against Mtb: Free RFP: MIC of 0.78 µg/mL–1.25 µg/mL RFP-loaded NPs: MIC of 0.039 µg/mL –0.31 µg/mL |
| In vivo | Animal model: rat Intratracheal administration, efficient penetration of the airway mucus, distribution throughout the lung tissues | |
| Chitosan NPs [104] | ||
| Preparation: ionic gelation Mannose-conjugated NPs: Size: 142 nm PdI: 0.154 ζ potential: 38.5 mV Drug encapsulation: 71% Non-conjugated NPs: Size: 138 nm PdI: 0.173 ζ potential: 42.6 mV Drug encapsulation: 74% | In vitro | Mannose-conjugated NPs: Drug release: For a pH of 7.4: 71% in 12 h For a pH of 5.2: 89% in 12 h Incorporation in in situ gelling system: 70% in 40 h Effect against Mtb: RFP-loaded NPs: MIC of 0.009 µg/mL |
| In vivo | - | |
| Chitosan NPs [114] | ||
| Preparation: ionic gelation Mannose-conjugated NPs: Size: 300 nm ζ potential: 18 mV Drug encapsulation: 73% Drug loading: 40% | In vitro | - |
| In vivo | Animal model: rat and rabbit RFP-loaded NPs: 19-fold higher permeation across everted rat intestines compared to free RFP 16-fold higher oral bioavailability in rabbits compared to free RFP Hepatoprotective effect | |
| SLNPs [136] | ||
| Preparation: hot ultrasonication Chitosan-coated NPs: Size: 524 nm ζ potential: 30 mV Drug encapsulation: 90% Drug loading: 5% Non-coated NPs: Size: 245 nm ζ potential: −30 mV Drug encapsulation: 89% Drug loading: 5% | In vitro | Drug release: Chitosan-coated NPs: For a pH of 7.4: 34% in 8 h For a pH of 4.5: 25% in 8 h Non-coated NPs: For a pH of 7.4: 50% in 8 h For a pH of 4.5: 50% in 8 h No toxicity towards A549 cells |
| In vivo | - | |
| Pyrazinamide (PZA) | ||
| PLGA NPs [100] | ||
| Preparation: double emulsion-solvent evaporation Size: 173 nm PdI: 0.05 ζ potential: −1 mV Drug encapsulation: 8% Drug loading: 3% | In vitro | - |
| In vivo | - | |
| Eudragit RS-100 NPs [101] | ||
| Preparation: double emulsion-solvent evaporation Size: 46 nm–300 nm PdI: 0.237–0.823 ζ potential: 3.23 mV–25.2 mV Drug encapsulation: 61%–81% Drug loading: 13%–43% | In vitro | Drug release, for a pH of 6.8: Free PZA: 90% in 6 h, no further release PZA-loaded NPs: rapid release phase up to 11 h, slower release phase over 24 h (approximately 80%) Important uptake in alveolar macrophages 2 h after administration |
| In vivo | - | |
| Ethambutol (EMB) | ||
| Graphene oxide with iron oxide magnetite NPs [137] | ||
| Size: 9 nm Drug loading: 34% | In vitro | Drug release, for a pH of 7.4 or 4.8: Free EMB: 100% in 10 min EMB-loaded NPs: 100% in 50 h Effect against Mycobacterium smegmatis: EMB-loaded NPs: MIC of 6.25 µg/mL No toxicity towards 3T3 cells |
| In vivo | - | |
| PCL NPs [138] | ||
| Preparation: double emulsification Size: 270 nm | In vitro | Effect against BCG: J774A.1 cells, free EMB or EMB-loaded NPs: decrease in percentage of infected cells from 85% to 30% |
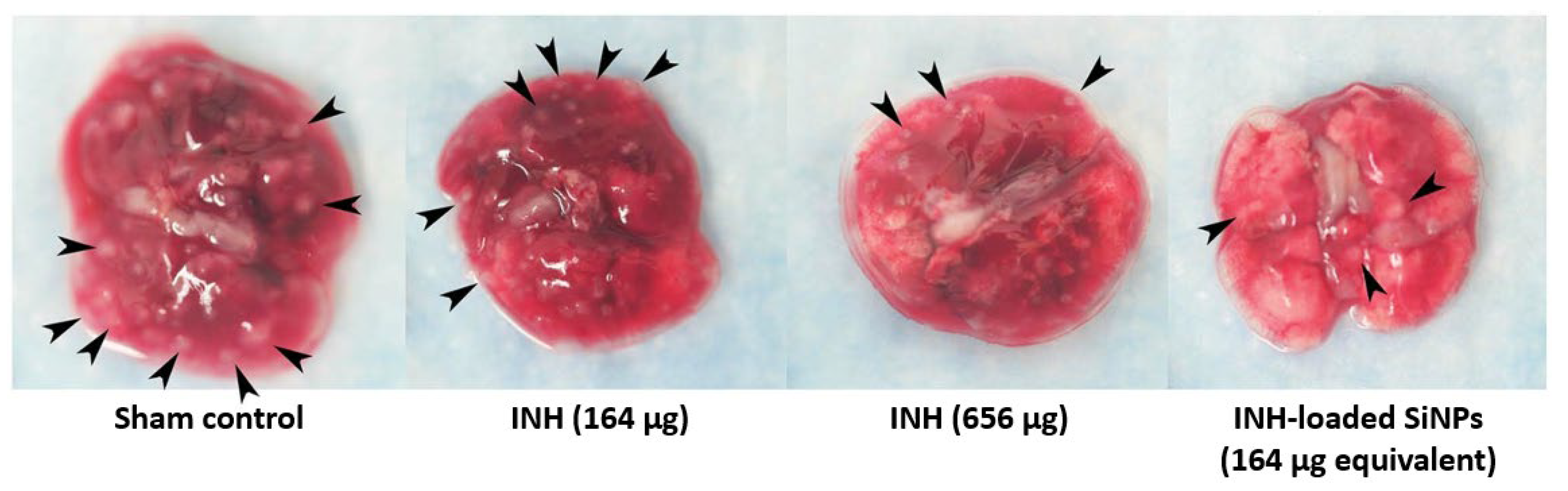
| In vivo | Animal model: mouse Effect against BCG: 18% of EMB-loaded NPs taken up by the lungs | |
| SLNPs [102] | ||
| Preparation: hot homogenization and ultrasonication Size: 58 nm PdI: 0.253 Drug encapsulation: 99% Drug loading: 30% | In vitro | Drug release: Free EMB: 47% in 8 h EMB-loaded NPs: 34% in 8 h No toxicity towards A549 cells compared to free EMB |
| In vivo | - | |
| Combinations | ||
| SLNPs: INH + RFP + PZA [124] | ||
| Preparation: emulsion-solvent diffusion Drug encapsulation: INH: 45% RFP: 51% PZA: 41% | In vitro | - |
| In vivo | Animal model: mouse Bioavailability: Free drugs: detection in the plasma for up to 12 h Loaded NPs: detection in the plasma for up to 8 days Effect against Mtb: Free drugs: 46 doses Loaded NPs: 5 doses In both cases, undetectable CFUs in the lungs and in the spleen | |
| Alginate NPs: INH + RFP + PZA [123] | ||
| Preparation: cation-induced gelification Size: 236 nm PdI: 0.439 Drug encapsulation: INH: 70% to 90% RFP: 80% to 90% PZA: 70% to 90% | In vitro | - |
| In vivo | Animal model: guinea pig Bioavailability: Free drugs: detection in the plasma for up to 14 h Loaded NPs: detection in the plasma for up to 14 days Effect against Mtb: Free drugs: 45 doses Loaded NPs: 3 doses In both cases, undetectable CFUs in the lungs and in the spleen | |
| PLGA NPs: INH + RFP + PZA; EMB [125] | ||
| Preparation: emulsion-solvent evaporation EMB encapsulated separately: drug encapsulation: INH: 67% RFP: 56% PZA: 69% EMB: 43% | In vitro | - |
| In vivo | Animal model: mouse Bioavailability: Free drugs: detection in the plasma for up to 12 h; detection in the brain for up to 1 day, except for EMB (6 days) Loaded NPs: detection in the plasma for up to 8 days for INH and PZA, 5 days for RFP, and 3 days for EMB; from 15- to 30-fold higher bioavailability; detection in the brain for up to 9 days Effect against Mtb: Free drugs: 46 doses Loaded NPs: 10 doses In both cases, undetectable CFUs in the brain | |
| SLNPs: INH + RFP + PZA [121] | ||
| Preparation: microemulsion Size: 188 nm PdI: 0.568 ζ potential: −47.4 mV Drug encapsulation: INH: 84% RFP: 86% PZA: 81% | In vitro | Drug release: Free drugs: For a pH of 6.8: INH: 95% in 1 h RFP: 92% in 1 h PZA: 96% in 1 h For a pH of 1.2: INH: 92% in 1 h RFP: 87% in 1 h PZA: 89% in 1 h Loaded NPs: For a pH of 6.8: INH: 6% in 1 h RFP: 12% in 1 h PZA: 10% in 1 h For a pH of 1.2: INH: 8% in 1 h RFP: 9% in 1 h PZA: 10% in 1 h Effect against Mycobacterium marinum: Loaded NPs: 2-fold reduction in bacterial load compared to free drugs |
| In vivo | - | |
| Chitosan NPs: INH + PZA [122] | ||
| Preparation: ionic gelation Size: 250 nm–576 nm PdI: 0.3–0.4 ζ potential: 25.92 mV–37.44 mV Drug encapsulation: INH: 25%–30% PZA: 25%–30% | In vitro | No toxicity towards NCI-H358, A549 and NR8383 cells Low levels of IL-1β, TNF-α and NO after administration |
| In vivo | - | |
4.7. Encapsulating New Antitubercular Drugs
| Physicochemical Properties | Biological Data | |
|---|---|---|
| Bedaquiline (BDQ) | ||
| Lipid NPs [139] | ||
| Preparation: ultrasonication Trimannose-conjugated NPs: Size: 83 nm to 86 nm PdI: <0.15 ζ potential: −10 mV or 28 mV Drug encapsulation: 93% Drug loading: 3% | In vitro | Drug release:BDQ-loaded NPs (28 mV): 75% in 14 h BDQ-loaded NPs (−10 mV): 95% in 14 h <10% of drug release after 7 days in PBS, RPMI and 7H9Effect against Mtb: Free BDQ, BDQ-loaded NPs (28 mV) and BDQ-loaded NPs (−10 mV): MIC of 0.03 µg/mL No toxicity towards THP-1 cells (below 1 mg/mL), HepG2 cells (below 1 mg/mL) and A549 cells (below 600 µg/mL) |
| In vivo | Animal model: mouse Effect against Mtb: BDQ-loaded NPs: decrease in bacterial load after 13 days Strong accumulation in the lungs | |
| Chitosan NPs [140] | ||
| Preparation: nanoemulsion PEG-coated NPs: Size: 328 nm–456 nm PdI: 0.151–0.204 ζ potential: −9 mV Drug loading: 25% Non-coated NPs: Size: 328 nm–456 nm PdI: 0.151–0.204 ζ potential: 26 mV Drug encapsulation: 70% Drug loading: 28% | In vitro | Drug release: Coated NPs: >40% after 7 days in RPMI, <30% after 7 days in milli-Q water Non-coated NPs: 5% after 7 days in RPMI and in milli-Q water |
| In vivo | - | |
| PLGA NPs [145] | ||
| Preparation: single emulsion Encapsulation of BDQ and Q203: Size: 480 nm PdI: 0.51 Drug encapsulation: BDQ: 55% Q203: 57% Combination: 41% for BDQ, 50% for Q203 | In vitro | Drug release in simulated lung fluid: BDQ: 85% in 8 h Q203: 90% in 8 h Combination: 85% in 8 h for BDQ, 98% in 8 h for Q203 Abrupt drug release in 8 h, complete drug release in 24 h Effect against Mtb: BDQ: MIC50 of 120 nM Q203: MIC50 of 3 nM No toxicity towards A549 cells (below 500 µg/mL) |
| In vivo | - | |
| Liposomes in lactose–leucine microcapsules [141] | ||
| Preparation: thin-film hydration and extrusion Size: 90 nm–100 nm PdI: <0.1 ζ potential: −14 mV Drug encapsulation: 98% Drug loading: 8% | In vitro | Drug release: No release in lung surfactant, <10% in milli-Q water |
| In vivo | - | |
| Linezolid (LZN) | ||
| Non-structured lipid carriers in mannitol–maltodextrin–leucine microparticles [146] | ||
| Preparation: hydration Size: 809 nm–820 nm PdI: 0.21–0.25 ζ potential: −58 mV–−37 mV Drug encapsulation: 96% Drug loading: 19% Microparticles size: 1.4 µm–2.5 μm | In vitro | Drug release (in PBS, for a pH of 7.4; in citrate buffer, for a pH of 4.5): LZN-loaded NPs: 32%–35% in 1 h, 85%–90% in 24 h No toxicity towards A549 cells |
| In vivo | Animal model: mouse No toxicity 24 h after orotracheal administration compared to free LZN | |
| Gelatin NPs [142] | ||
| Preparation: desolvation Mannose-conjugated NPs: Size: 298 nm PdI: <0.148 ζ potential: 12 mV–27 mV Drug encapsulation: 51%–57% | In vitro | Drug release in PBS, for a pH of 7.4: LZN-loaded NPs: 95% in 96 h No toxicity towards J774 cells |
| In vivo | Animal model: rat Bioavailability: Free LZN: detection in the plasma for up to 10 h–12 h LZN-loaded NPs: detection in the plasma for up to 3 days–5 days; 19-fold higher half-life compared to free LZN No toxicity after 28 days of repeated administrations | |
| PLGA NPs in microparticles [147] | ||
| Preparation: emulsion-solvent evaporation Size: 45 nm–178 nm Drug encapsulation: 57%–85% Microparticles size: 3.8 μm | In vitro | Drug release in simulated lung fluid: LZN-loaded NPs: 75%–90% in 120 h Effect against Mtb: Free LZN: MIC of 1 µg/mL LZN-loaded NPs: MIC of 0.6 µg/mL |
| In vivo | - | |
| Chitosan NPs in microparticles [148] | ||
| Preparation: ionotropic gelation Size: 89 nm–223 nm Encapsulation efficiency: 37%–49% Microparticles size: 3.2 μm | In vitro | Drug release in simulated lung fluid: LZN-loaded NPs: 78%–90% in 24 h Effect against Mtb: Free LZN: MIC of 1 µg/mL LZN-loaded NPs: MIC of 0.8 µg/mL |
| In vivo | - | |
| Ethionamide (ETH) | ||
| Chitosan NPs [149] | ||
| Preparation: carrageenan-stabilized ionotropic gelation Size: 317 nm–324 nm PdI: 0.22–0.42 ζ potential: −13 mV–−24 mV | In vitro | Drug release: 0% of stabilizer: 95% in 24 h 42% of stabilizer: 95% in 24 h 59% of stabilizer: 80% in 24 h Effect against Mtb: Free ETH: MIC of 0.43 µg/mL ETH-loaded NPs: MIC of 0.61 µg/mL |
| In vivo | - | |
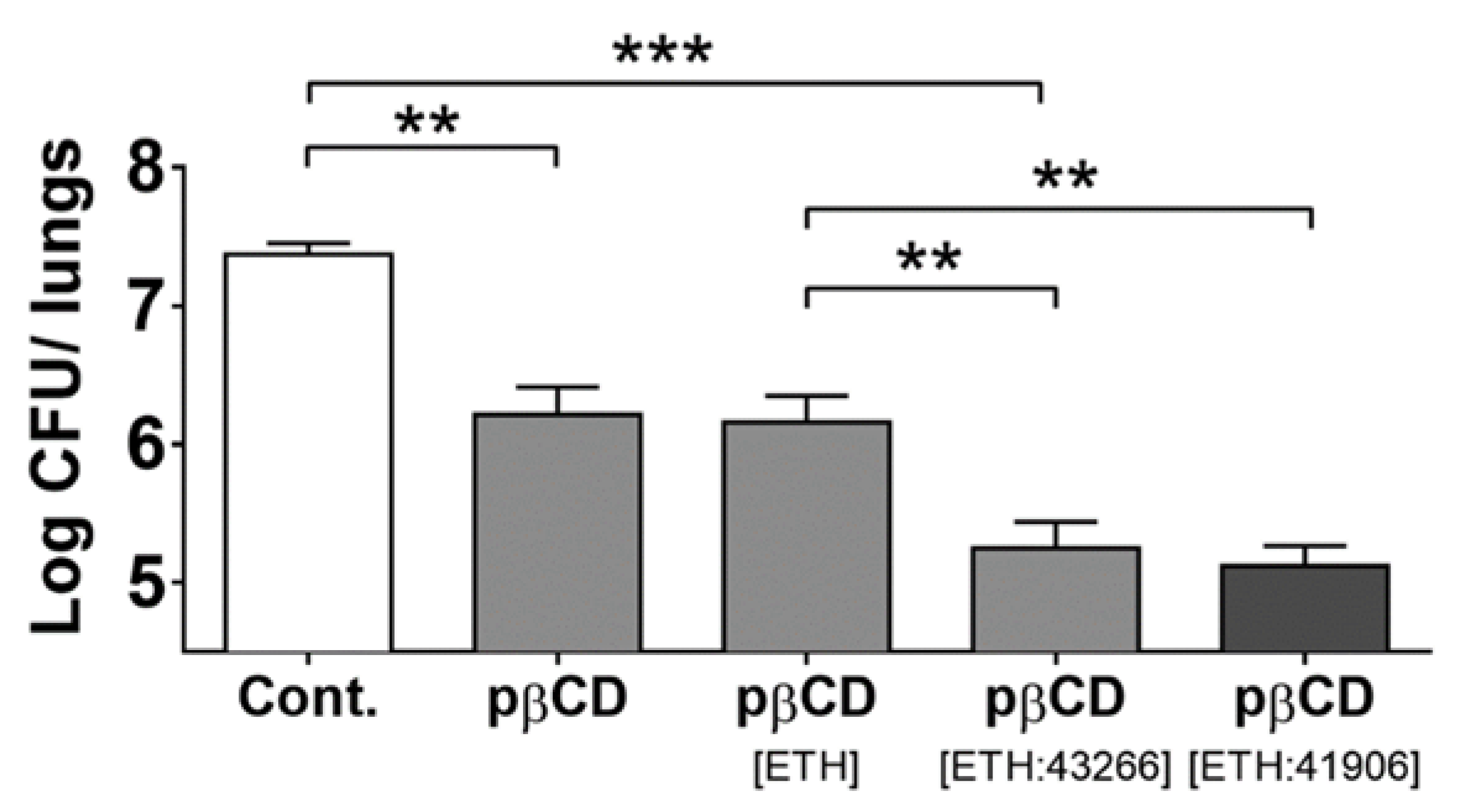
| PLA NPs, PLGA NPs and CD-based NPs: ETH + BDM41906 (booster) [127] | ||
| PLA NPs: Preparation: nanoemulsion Size: 254 nm–277 nm PdI: <0.09 ζ potential: −5 mV Drug encapsulation: ETH: 76%–77% BDM41906: 46%–51% Drug loading: ETH: 36%–38% BDM41906: 23%–26% PLGA NPs: Preparation: nanoprecipitation Size: 170 nm Drug loading: <11% CD-based NPs: Preparation: nanoprecipitation Size: 10 nm Drug loading: ETH: 25 μg for 1 mg of NPs BDM41906: 25 µg for 1 mg of NPs | In vitro | Effect against Mtb: Free ETH and free BDM41906: IC50 of 0.11 µg/mL ETH- and BDM41906-loaded PLA NPs: IC50 of 0.06 µg/mL ETH- and BDM41906-loaded CD-based NPs: IC50 of 0.06 µg/mL |
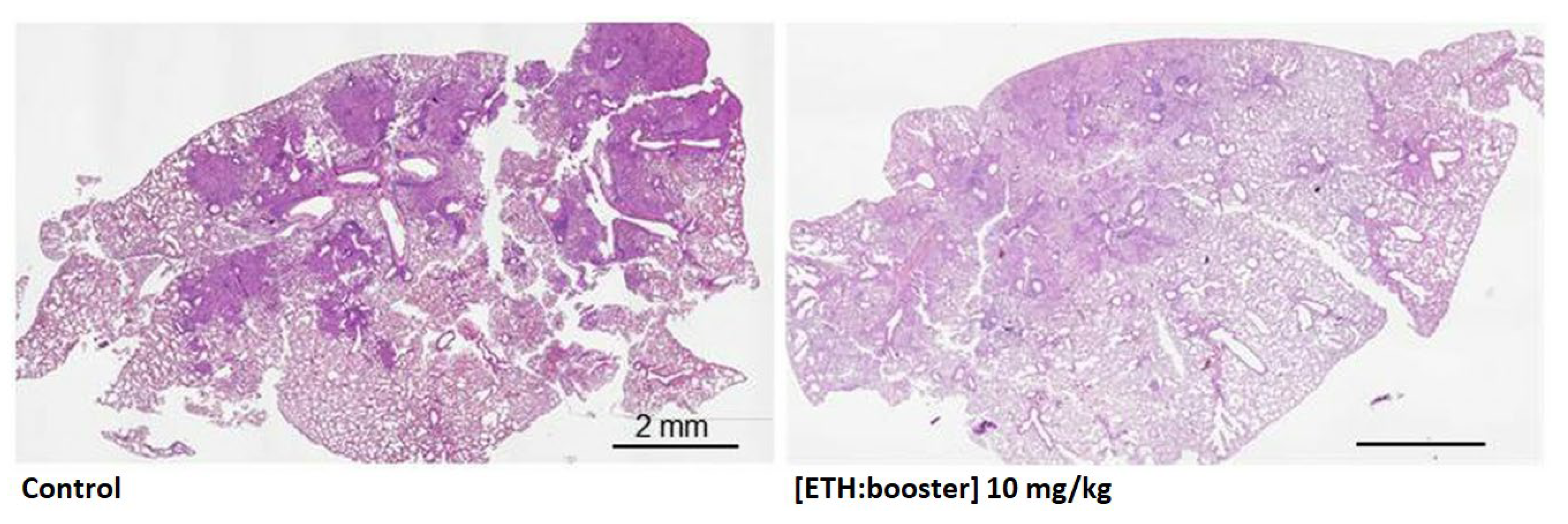
| In vivo | Animal model: mouse Effect against Mtb: ETH- and BDM41906-loaded CD-based NPs: 3-log reduction in CFUs in the lungs | |
| Codrug NPs: ETH + BDM43266 (booster) [128] | ||
| Preparation: nanoprecipitation of a codrug composed of ETH and of BDM43266 Size: 195 nm–208 nm Drug loading: 80% | In vitro | - |
| In vivo | Animal model: mouse Effect against Mtb: ETH- and BDM43266-loaded NPs: 6-fold reduction in CFUs in the lungs | |
4.8. Host-Directed Therapy Using Nanoparticles
4.9. Combined Therapies to Treat Tuberculosis
4.10. Summary of the Output of Nanoparticles to Treat Tuberculosis
5. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AuNP | gold nanoparticle |
| BCG | bacillus Calmette–Guérin |
| BDM | family of molecules boosting the activity of ethionamide |
| BDQ | bedaquiline |
| CD | cyclodextrin |
| CFU | colony forming unit |
| CFZ | clofazimine |
| DDS | drug delivery system |
| DLM | delamanid |
| EMB | ethambutol |
| ETH | ethionamide |
| INH | isoniazid |
| INP | inorganic nanoparticle |
| LVX | levofloxacin |
| LZN | linezolid |
| MDR-TB | multidrug-resistant tuberculosis |
| MOF | metal–organic framework |
| MOX | moxifloxacin |
| Mtb | Mycobacterium tuberculosis |
| NP | nanoparticle |
| PCL | poly(ε-caprolactone) |
| PEG | polyethylene glycol |
| PLA | poly(lactic acid) |
| PLGA | poly(lactic-co-glycolic acid) |
| PMD | pretomanid |
| PNP | polymeric nanoparticle |
| pre-XDR-TB | pre-extensively drug-resistant tuberculosis |
| PZA | pyrazinamide |
| RFP | rifampicin |
| RR-TB | rifampicin-resistant tuberculosis |
| SiNP | silica nanoparticle |
| SLNP | solid lipid nanoparticle |
| TB | tuberculosis |
| XDR-TB | extensively drug-resistant tuberculosis |
References
- Grotz, E.; Tateosian, N.; Amiano, N.; Cagel, M.; Bernabeu, E.; Chiappetta, D.A.; Moretton, M.A. Nanotechnology in Tuberculosis: State of the Art and the Challenges Ahead. Pharm. Res. 2018, 35, 213. [Google Scholar] [CrossRef] [PubMed]
- Muthukrishnan, L. Multidrug Resistant Tuberculosis—Diagnostic Challenges and Its Conquering by Nanotechnology Approach—An Overview. Chem. Biol. Interact. 2021, 337, 109397. [Google Scholar] [CrossRef]
- Pai, M.; Kasaeva, T.; Swaminathan, S. COVID-19’s Devastating Effect on Tuberculosis Care—A Path to Recovery. N. Engl. J. Med. 2022, 386, 1490–1493. [Google Scholar] [CrossRef]
- Daniel, T.M. The History of Tuberculosis. Respir. Med. 2006, 100, 1862–1870. [Google Scholar] [CrossRef] [PubMed]
- Barberis, I.; Bragazzi, N.L.; Galluzzo, L.; Martini, M. The History of Tuberculosis: From the First Historical Records to the Isolation of Koch’s Bacillus. J. Prev. Med. Hyg. 2017, 58, E9–E12. [Google Scholar]
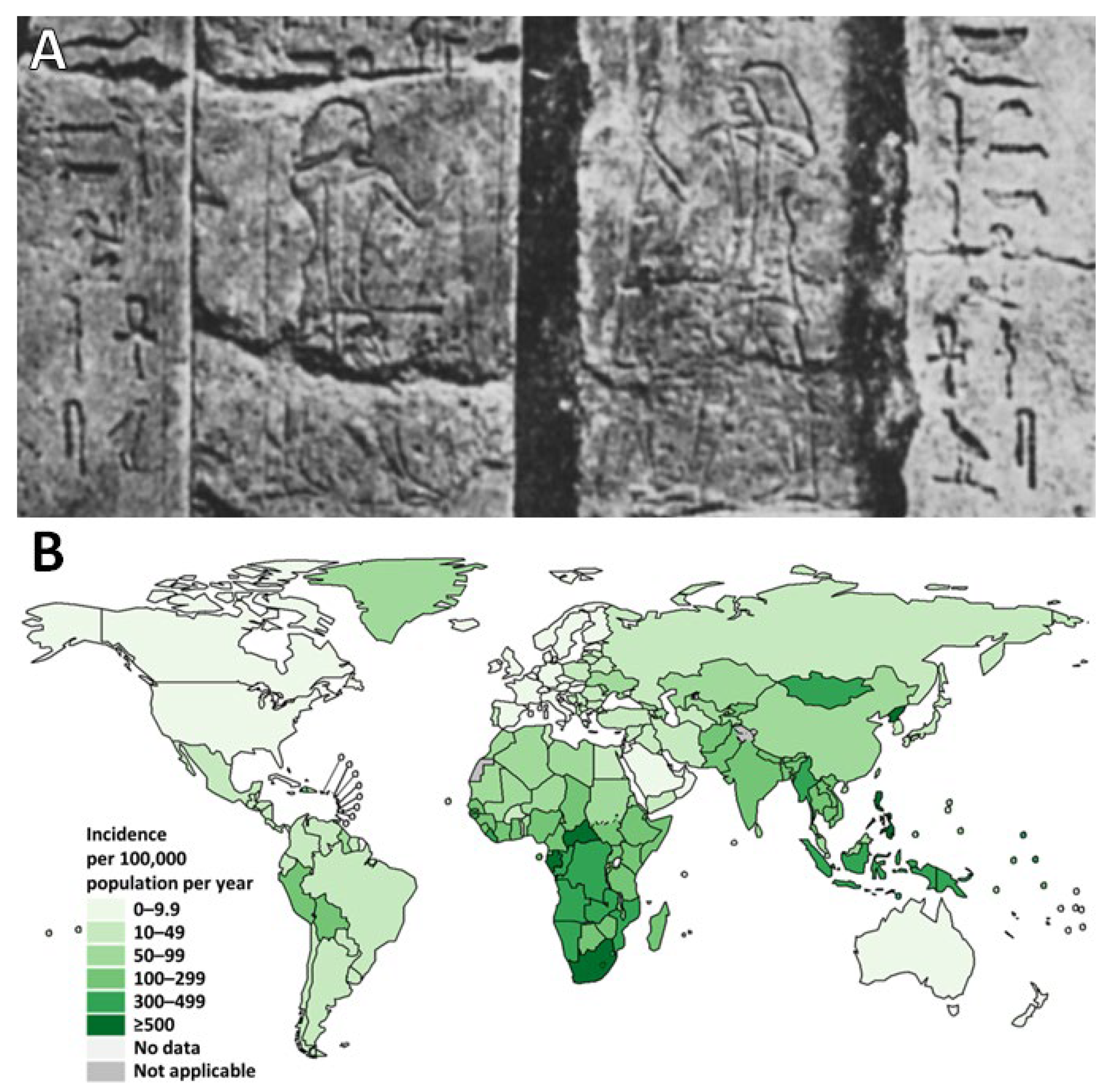
- Cave, A.J.E.; Demonstrator, A. The Evidence for the Incidence of Tuberculosis in Ancient Egypt. Br. J. Tuberc. 1939, 33, 142–152. [Google Scholar] [CrossRef]
- Morse, D.; Brothwell, D.R.; Ucko, P.J. Tuberculosis in Ancient Egypt. Am. Rev. Respir. Dis. 1964, 90, 524–541. [Google Scholar] [CrossRef] [PubMed]
- Crubézy, E.; Ludes, B.; Poveda, J.-D.; Clayton, J.; Crouau-Roy, B.; Montagnon, D. Identification of Mycobacterium DNA in an Egyptian Pott’s Disease of 5400 Years Old. Comptes Rendus Académie Sci.-Ser. III-Sci. Vie 1998, 321, 941–951. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Tuberculosis Report 2022; World Health Organization: Geneva, Switzerland, 2022. [Google Scholar]
- McCarthy, O.R. The Key to the Sanatoria. J. R. Soc. Med. 2001, 94, 413–417. [Google Scholar] [CrossRef]
- Daniel, T.M. Jean-Antoine Villemin and the Infectious Nature of Tuberculosis. Int. J. Tuberc. Lung Dis. 2015, 19, 267–268. [Google Scholar] [CrossRef]
- Sakula, A. Robert Koch: Centenary of the Discovery of the Tubercle Bacillus, 1882. Thorax 1982, 37, 246–251. [Google Scholar] [CrossRef]
- Luca, S.; Mihafscu, T. History of BCG Vaccine. Mædica 2013, 8, 53–58. [Google Scholar]
- Murray, J.F.; Schraufnagel, D.E.; Hopewell, P.C. Treatment of Tuberculosis. A Historical Perspective. Ann. Am. Thorac. Soc. 2015, 12, 1749–1759. [Google Scholar] [CrossRef] [PubMed]
- Kirby, T. Global Tuberculosis Progress Reversed by COVID-19 Pandemic. Lancet Respir. Med. 2021, 9, e118–e119. [Google Scholar] [CrossRef] [PubMed]
- McQuaid, C.F.; Vassall, A.; Cohen, T.; Fiekert, K.; COVID/TB Modelling Working Group; White, R.G. The Impact of COVID-19 on TB: A Review of the Data. Int. J. Tuberc. Lung Dis. 2021, 25, 436–446. [Google Scholar] [CrossRef]
- Dolgin, E. The Tangled History of MRNA Vaccines. Nature 2021, 597, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Berlin, J.M.; Tour, J.M. Development of Novel Drug Delivery Vehicles. Nanomed. 2010, 5, 1487–1489. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Mageed, H.M.; AbuelEzz, N.Z.; Radwan, R.A.; Mohamed, S.A. Nanoparticles in Nanomedicine: A Comprehensive Updated Review on Current Status, Challenges and Emerging Opportunities. J. Microencapsul. 2021, 38, 414–436. [Google Scholar] [CrossRef]
- Ryndak, M.B.; Laal, S. Mycobacterium Tuberculosis Primary Infection and Dissemination: A Critical Role for Alveolar Epithelial Cells. Front. Cell. Infect. Microbiol. 2019, 9, 299. [Google Scholar] [CrossRef]
- Philips, J.A.; Ernst, J.D. Tuberculosis Pathogenesis and Immunity. Annu. Rev. Pathol. Mech. Dis. 2012, 7, 353–384. [Google Scholar] [CrossRef]
- Lin, P.L.; Flynn, J.L. Understanding Latent Tuberculosis: A Moving Target. J. Immunol. 2010, 185, 15–22. [Google Scholar] [CrossRef]
- Guirado, E.; Schlesinger, L.S.; Kaplan, G. Macrophages in Tuberculosis: Friend or Foe. Semin. Immunopathol. 2013, 35, 563–583. [Google Scholar] [CrossRef]
- Kinsella, R.L.; Zhu, D.X.; Harrison, G.A.; Mayer Bridwell, A.E.; Prusa, J.; Chavez, S.M.; Stallings, C.L. Perspectives and Advances in the Understanding of Tuberculosis. Annu. Rev. Pathol. Mech. Dis. 2021, 16, 377–408. [Google Scholar] [CrossRef] [PubMed]
- Hunter, R.L. Pathology of Post Primary Tuberculosis of the Lung: An Illustrated Critical Review. Tuberculosis 2011, 91, 497–509. [Google Scholar] [CrossRef]
- Hunter, R.L. The Pathogenesis of Tuberculosis: The Early Infiltrate of Post-Primary (Adult Pulmonary) Tuberculosis: A Distinct Disease Entity. Front. Immunol. 2018, 9, 2108. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.K.; Mohan, A. Miliary Tuberculosis. In Tuberculosis and Nontuberculous Mycobacterial Infections; Schlossberg, D., Ed.; ASM Press: Washington, DC, USA, 2017; pp. 491–513. [Google Scholar]
- Blischak, J.D.; Tailleux, L.; Mitrano, A.; Barreiro, L.B.; Gilad, Y. Mycobacterial Infection Induces a Specific Human Innate Immune Response. Sci. Rep. 2015, 5, 16882. [Google Scholar] [CrossRef] [PubMed]
- Simeone, R.; Sayes, F.; Song, O.; Gröschel, M.I.; Brodin, P.; Brosch, R.; Majlessi, L. Cytosolic Access of Mycobacterium Tuberculosis: Critical Impact of Phagosomal Acidification Control and Demonstration of Occurrence In Vivo. PLoS Pathog. 2015, 11, e1004650. [Google Scholar] [CrossRef]
- Queval, C.J.; Brosch, R.; Simeone, R. The Macrophage: A Disputed Fortress in the Battle against Mycobacterium Tuberculosis. Front. Microbiol. 2017, 8, 2284. [Google Scholar] [CrossRef]
- Gould, K. Antibiotics: From Prehistory to the Present Day. J. Antimicrob. Chemother. 2016, 71, 572–575. [Google Scholar] [CrossRef]
- Lei, B.; Wei, C.-J.; Tu, S.-C. Action Mechanism of Antitubercular Isoniazid. J. Biol. Chem. 2000, 275, 2520–2526. [Google Scholar] [CrossRef]
- Campbell, E.A.; Korzheva, N.; Mustaev, A.; Murakami, K.; Nair, S.; Goldfarb, A.; Darst, S.A. Structural Mechanism for Rifampicin Inhibition of Bacterial RNA Polymerase. Cell 2001, 104, 901–912. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Shi, W.; Zhang, W.; Mitchison, D. Mechanisms of Pyrazinamide Action and Resistance. Microbiol. Spectr. 2014, 2, 2. [Google Scholar] [CrossRef]
- Zhu, C.; Liu, Y.; Hu, L.; Yang, M.; He, Z.-G. Molecular Mechanism of the Synergistic Activity of Ethambutol and Isoniazid against Mycobacterium Tuberculosis. J. Biol. Chem. 2018, 293, 16741–16750. [Google Scholar] [CrossRef] [PubMed]
- Horsburgh, C.R.; Barry, C.E.; Lange, C. Treatment of Tuberculosis. N. Engl. J. Med. 2015, 373, 2149–2160. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO Consolidated Guidelines on Tuberculosis. Module 4: Treatment—Drug-Susceptible Tuberculosis Treatment; World Health Organization: Geneva, Switzerland, 2022. [Google Scholar]
- Bahuguna, A.; Rawat, D.S. An Overview of New Antitubercular Drugs, Drug Candidates, and Their Targets. Med. Res. Rev. 2020, 40, 263–292. [Google Scholar] [CrossRef]
- Singh, V.; Chibale, K. Strategies to Combat Multi-Drug Resistance in Tuberculosis. Acc. Chem. Res. 2021, 54, 2361–2376. [Google Scholar] [CrossRef]
- Olaru, I.D.; von Groote-Bidlingmaier, F.; Heyckendorf, J.; Yew, W.W.; Lange, C.; Chang, K.C. Novel Drugs against Tuberculosis: A Clinician’s Perspective. Eur. Respir. J. 2015, 45, 1119–1131. [Google Scholar] [CrossRef]
- Provisional CDC Guidance for the Use of Pretomanid as Part of a Regimen [Bedaquiline, Pretomanid, and Linezolid (BPaL)] to Treat Drug-Resistant Tuberculosis Disease. Available online: https://www.cdc.gov/tb/topic/drtb/bpal/default.htm (accessed on 18 November 2022).
- Vale, N.; Gomes, P.; Santos, H.A. Metabolism of the Antituberculosis Drug Ethionamide. Curr. Drug Metab. 2013, 14, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Oehadian, A.; Santoso, P.; Menzies, D.; Ruslami, R. Concise Clinical Review of Hematologic Toxicity of Linezolid in Multidrug-Resistant and Extensively Drug-Resistant Tuberculosis: Role of Mitochondria. Tuberc. Respir. Dis. 2022, 85, 111–121. [Google Scholar] [CrossRef]
- Khoshnood, S.; Goudarzi, M.; Taki, E.; Darbandi, A.; Kouhsari, E.; Heidary, M.; Motahar, M.; Moradi, M.; Bazyar, H. Bedaquiline: Current Status and Future Perspectives. J. Glob. Antimicrob. Resist. 2021, 25, 48–59. [Google Scholar] [CrossRef]
- Khoshnood, S.; Taki, E.; Sadeghifard, N.; Kaviar, V.H.; Haddadi, M.H.; Farshadzadeh, Z.; Kouhsari, E.; Goudarzi, M.; Heidary, M. Mechanism of Action, Resistance, Synergism, and Clinical Implications of Delamanid Against Multidrug-Resistant Mycobacterium Tuberculosis. Front. Microbiol. 2021, 12, 717045. [Google Scholar] [CrossRef] [PubMed]
- Occhineri, S.; Matucci, T.; Rindi, L.; Tiseo, G.; Falcone, M.; Riccardi, N.; Besozzi, G. Pretomanid for Tuberculosis Treatment: An Update for Clinical Purposes. Curr. Res. Pharmacol. Drug Discov. 2022, 3, 100128. [Google Scholar] [CrossRef]
- World Health Organization. WHO Consolidated Guidelines on Tuberculosis. Module 4: Treatment—Drug-Resistant Tuberculosis Treatment, 2022 Update; World Health Organization: Geneva, Switzerland, 2022. [Google Scholar]
- Dean, A.S.; Cox, H.; Zignol, M. Epidemiology of Drug-Resistant Tuberculosis. In Strain Variation in the Mycobacterium tuberculosis Complex: Its Role in Biology, Epidemiology and Control; Gagneux, S., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2017; Volume 1019, pp. 209–220. [Google Scholar]
- Sarkar, K.; Kumar, M.; Jha, A.; Bharti, K.; Das, M.; Mishra, B. Nanocarriers for Tuberculosis Therapy: Design of Safe and Effective Drug Delivery Strategies to Overcome the Therapeutic Challenges. J. Drug Deliv. Sci. Technol. 2022, 67, 102850. [Google Scholar] [CrossRef]
- Tanner, L.; Denti, P.; Wiesner, L.; Warner, D.F. Drug Permeation and Metabolism in Mycobacterium Tuberculosis: Prioritising Local Exposure as Essential Criterion in New TB Drug Development. IUBMB Life 2018, 70, 926–937. [Google Scholar] [CrossRef]
- Prasad, R.; Singh, A.; Gupta, N. Adverse Drug Reactions in Tuberculosis and Management. Indian J. Tuberc. 2019, 66, 520–532. [Google Scholar] [CrossRef]
- Silver, L.L. Challenges of Antibacterial Discovery. Clin. Microbiol. Rev. 2011, 24, 71–109. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, B.; Mehta, P.K. Utility of Nanoparticle-Based Assays in the Diagnosis of Tuberculosis. Nanomedicine 2021, 16, 1263–1268. [Google Scholar] [CrossRef]
- Chandrasekaran, A.; Karunaratne, G.H.R.E. Use of Nanoparticles in Multidrug Resistant Tuberculosis Diagnosis. In Nanotechnology for Infectious Diseases; Hameed, S., Rehman, S., Eds.; Springer: Singapore, 2022; pp. 371–386. [Google Scholar]
- Lu, H.; Wang, J.; Wang, T.; Zhong, J.; Bao, Y.; Hao, H. Recent Progress on Nanostructures for Drug Delivery Applications. J. Nanomater. 2016, 2016, 5762431. [Google Scholar] [CrossRef]
- Beltrán-Gracia, E.; López-Camacho, A.; Higuera-Ciapara, I.; Velázquez-Fernández, J.B.; Vallejo-Cardona, A.A. Nanomedicine Review: Clinical Developments in Liposomal Applications. Cancer Nanotechnol. 2019, 10, 11. [Google Scholar] [CrossRef]
- Yasamineh, S.; Yasamineh, P.; Ghafouri Kalajahi, H.; Gholizadeh, O.; Yekanipour, Z.; Afkhami, H.; Eslami, M.; Hossein Kheirkhah, A.; Taghizadeh, M.; Yazdani, Y.; et al. A State-of-the-Art Review on the Recent Advances of Niosomes as a Targeted Drug Delivery System. Int. J. Pharm. 2022, 624, 121878. [Google Scholar] [CrossRef] [PubMed]
- Khanzode, M.; Kajale, A.; Chandewar, A.; Gawande, S. Review on Phytosomes: A Novel Drug Delivery System. GSC Biol. Pharm. Sci. 2020, 13, 203–211. [Google Scholar] [CrossRef]
- Opatha, S.A.T.; Titapiwatanakun, V.; Chutoprapat, R. Transfersomes: A Promising Nanoencapsulation Technique for Transdermal Drug Delivery. Pharmaceutics 2020, 12, 855. [Google Scholar] [CrossRef]
- Hoar, T.P.; Schulman, J.H. Transparent Water-in-Oil Dispersions: The Oleopathic Hydro-Micelle. Nature 1943, 152, 102–103. [Google Scholar] [CrossRef]
- Talegaonkar, S.; Azeem, A.; Ahmad, F.; Khar, R.; Pathan, S.; Khan, Z. Microemulsions: A Novel Approach to Enhanced Drug Delivery. Recent Pat. Drug Deliv. Formul. 2008, 2, 238–257. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.J.; Rees, G.D. Microemulsion-Based Media as Novel Drug Delivery Systems. Adv. Drug Deliv. Rev. 2000, 45, 89–121. [Google Scholar] [CrossRef]
- Danielsson, I.; Lindman, B. The Definition of Microemulsion. Colloids Surf. 1981, 3, 391–392. [Google Scholar] [CrossRef]
- Tomalia, D.A.; Fréchet, J.M.J. Discovery of Dendrimers and Dendritic Polymers: A Brief Historical Perspective. J. Polym. Sci. Part Polym. Chem. 2002, 40, 2719–2728. [Google Scholar] [CrossRef]
- Wang, J.; Li, B.; Qiu, L.; Qiao, X.; Yang, H. Dendrimer-Based Drug Delivery Systems: History, Challenges, and Latest Developments. J. Biol. Eng. 2022, 16, 18. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, E.; Aval, S.F.; Akbarzadeh, A.; Milani, M.; Nasrabadi, H.T.; Joo, S.W.; Hanifehpour, Y.; Nejati-Koshki, K.; Pashaei-Asl, R. Dendrimers: Synthesis, Applications, and Properties. Nanoscale Res. Lett. 2014, 9, 247. [Google Scholar] [CrossRef]
- Mignani, S.; Tripathi, R.P.; Chen, L.; Caminade, A.-M.; Shi, X.; Majoral, J.-P. New Ways to Treat Tuberculosis Using Dendrimers as Nanocarriers. Pharmaceutics 2018, 10, 105. [Google Scholar] [CrossRef]
- Luther, D.C.; Huang, R.; Jeon, T.; Zhang, X.; Lee, Y.-W.; Nagaraj, H.; Rotello, V.M. Delivery of Drugs, Proteins, and Nucleic Acids Using Inorganic Nanoparticles. Adv. Drug Deliv. Rev. 2020, 156, 188–213. [Google Scholar] [CrossRef]
- Siddique, S.; Chow, J.C.L. Gold Nanoparticles for Drug Delivery and Cancer Therapy. Appl. Sci. 2020, 10, 3824. [Google Scholar] [CrossRef]
- Janjua, T.I.; Cao, Y.; Yu, C.; Popat, A. Clinical Translation of Silica Nanoparticles. Nat. Rev. Mater. 2021, 6, 1072–1074. [Google Scholar] [CrossRef] [PubMed]
- Dadfar, S.M.; Roemhild, K.; Drude, N.I.; von Stillfried, S.; Knüchel, R.; Kiessling, F.; Lammers, T. Iron Oxide Nanoparticles: Diagnostic, Therapeutic and Theranostic Applications. Adv. Drug Deliv. Rev. 2019, 138, 302–325. [Google Scholar] [CrossRef] [PubMed]
- Vangijzegem, T.; Stanicki, D.; Laurent, S. Magnetic Iron Oxide Nanoparticles for Drug Delivery: Applications and Characteristics. Expert Opin. Drug Deliv. 2019, 16, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Horcajada, P.; Chalati, T.; Serre, C.; Gillet, B.; Sebrie, C.; Baati, T.; Eubank, J.F.; Heurtaux, D.; Clayette, P.; Kreuz, C.; et al. Porous Metal–Organic-Framework Nanoscale Carriers as a Potential Platform for Drug Delivery and Imaging. Nat. Mater. 2010, 9, 172–178. [Google Scholar] [CrossRef]
- Al Sharabati, M.; Sabouni, R.; Husseini, G.A. Biomedical Applications of Metal-Organic Frameworks for Disease Diagnosis and Drug Delivery: A Review. Nanomaterials 2022, 12, 277. [Google Scholar] [CrossRef]
- Muller, R.H.; Shegokar, R.; Keck, C.M. 20 Years of Lipid Nanoparticles (SLN & NLC): Present State of Development & Industrial Applications. Curr. Drug Discov. Technol. 2011, 8, 207–227. [Google Scholar] [CrossRef]
- Duan, Y.; Dhar, A.; Patel, C.; Khimani, M.; Neogi, S.; Sharma, P.; Siva Kumar, N.; Vekariya, R.L. A Brief Review on Solid Lipid Nanoparticles: Part and Parcel of Contemporary Drug Delivery Systems. RSC Adv. 2020, 10, 26777–26791. [Google Scholar] [CrossRef]
- Begines, B.; Ortiz, T.; Pérez-Aranda, M.; Martínez, G.; Merinero, M.; Argüelles-Arias, F.; Alcudia, A. Polymeric Nanoparticles for Drug Delivery: Recent Developments and Future Prospects. Nanomaterials 2020, 10, 1403. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Zhou, Y.; Liu, L.; Xu, Y.; Chen, Q.; Wang, Y.; Wu, S.; Deng, Y.; Zhang, J.; Shao, A. Nanoparticle-Based Drug Delivery in Cancer Therapy and Its Role in Overcoming Drug Resistance. Front. Mol. Biosci. 2020, 7, 193. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, D.A.; Bickerton, S.; La Cava, A. Strategies to Use Nanoparticles to Generate CD4 and CD8 Regulatory T Cells for the Treatment of SLE and Other Autoimmune Diseases. Front. Immunol. 2021, 12, 681062. [Google Scholar] [CrossRef] [PubMed]
- Gelperina, S.; Kisich, K.; Iseman, M.D.; Heifets, L. The Potential Advantages of Nanoparticle Drug Delivery Systems in Chemotherapy of Tuberculosis. Am. J. Respir. Crit. Care Med. 2005, 172, 1487–1490. [Google Scholar] [CrossRef]
- Griffiths, G.; Nyström, B.; Sable, S.B.; Khuller, G.K. Nanobead-Based Interventions for the Treatment and Prevention of Tuberculosis. Nat. Rev. Microbiol. 2010, 8, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Mazlan, M.K.N.; Mohd Tazizi, M.H.D.; Ahmad, R.; Noh, M.A.A.; Bakhtiar, A.; Wahab, H.A.; Mohd Gazzali, A. Antituberculosis Targeted Drug Delivery as a Potential Future Treatment Approach. Antibiotics 2021, 10, 908. [Google Scholar] [CrossRef] [PubMed]
- Foroozandeh, P.; Aziz, A.A. Insight into Cellular Uptake and Intracellular Trafficking of Nanoparticles. Nanoscale Res. Lett. 2018, 13, 339. [Google Scholar] [CrossRef] [PubMed]
- Panariti, A.; Miserocchi, G.; Rivolta, I. The Effect of Nanoparticle Uptake on Cellular Behavior: Disrupting or Enabling Functions? Nanotechnol. Sci. Appl. 2012, 5, 87–100. [Google Scholar] [CrossRef]
- Kladko, D.V.; Falchevskaya, A.S.; Serov, N.S.; Prilepskii, A.Y. Nanomaterial Shape Influence on Cell Behavior. Int. J. Mol. Sci. 2021, 22, 5266. [Google Scholar] [CrossRef] [PubMed]
- Chithrani, B.D.; Ghazani, A.A.; Chan, W.C.W. Determining the Size and Shape Dependence of Gold Nanoparticle Uptake into Mammalian Cells. Nano Lett. 2006, 6, 662–668. [Google Scholar] [CrossRef]
- Behzadi, S.; Serpooshan, V.; Tao, W.; Hamaly, M.A.; Alkawareek, M.Y.; Dreaden, E.C.; Brown, D.; Alkilany, A.M.; Farokhzad, O.C.; Mahmoudi, M. Cellular Uptake of Nanoparticles: Journey inside the Cell. Chem. Soc. Rev. 2017, 46, 4218–4244. [Google Scholar] [CrossRef]
- Wang, W.; Gaus, K.; Tilley, R.D.; Gooding, J.J. The Impact of Nanoparticle Shape on Cellular Internalisation and Transport: What Do the Different Analysis Methods Tell Us? Mater. Horiz. 2019, 6, 1538–1547. [Google Scholar] [CrossRef]
- Fam, S.Y.; Chee, C.F.; Yong, C.Y.; Ho, K.L.; Mariatulqabtiah, A.R.; Tan, W.S. Stealth Coating of Nanoparticles in Drug-Delivery Systems. Nanomaterials 2020, 10, 787. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Gui, J.; Xiong, K.; Chen, M.; Gao, H.; Fu, Y. A Roadmap to Pulmonary Delivery Strategies for the Treatment of Infectious Lung Diseases. J. Nanobiotechnol. 2022, 20, 101. [Google Scholar] [CrossRef]
- Chakraborty, P.; Bajeli, S.; Kaushal, D.; Radotra, B.D.; Kumar, A. Biofilm Formation in the Lung Contributes to Virulence and Drug Tolerance of Mycobacterium Tuberculosis. Nat. Commun. 2021, 12, 1606. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Kłodzińska, S.N.; Wan, F.; Nielsen, H.M. Nanoparticle-Mediated Pulmonary Drug Delivery: State of the Art towards Efficient Treatment of Recalcitrant Respiratory Tract Bacterial Infections. Drug Deliv. Transl. Res. 2021, 11, 1634–1654. [Google Scholar] [CrossRef] [PubMed]
- Praphawatvet, T.; Peters, J.I.; Williams, R.O. Inhaled Nanoparticles—An Updated Review. Int. J. Pharm. 2020, 587, 119671. [Google Scholar] [CrossRef]
- Pourshahab, P.S.; Gilani, K.; Moazeni, E.; Eslahi, H.; Fazeli, M.R.; Jamalifar, H. Preparation and Characterization of Spray Dried Inhalable Powders Containing Chitosan Nanoparticles for Pulmonary Delivery of Isoniazid. J. Microencapsul. 2011, 28, 605–613. [Google Scholar] [CrossRef]
- Fernández-Paz, C.; Fernández-Paz, E.; Salcedo-Abraira, P.; Rojas, S.; Barrios-Esteban, S.; Csaba, N.; Horcajada, P.; Remuñán-López, C. Microencapsulated Isoniazid-Loaded Metal–Organic Frameworks for Pulmonary Administration of Antituberculosis Drugs. Molecules 2021, 26, 6408. [Google Scholar] [CrossRef]
- Ma, L.-J.; Niu, R.; Wu, X.; Wu, J.; Zhou, E.; Xiao, X.-P.; Chen, J. Quantitative Evaluation of Cellular Internalization of Polymeric Nanoparticles within Laryngeal Cancer Cells and Immune Cells for Enhanced Drug Delivery. Nanoscale Res. Lett. 2021, 16, 40. [Google Scholar] [CrossRef]
- Sung, J.C.; Padilla, D.J.; Garcia-Contreras, L.; VerBerkmoes, J.L.; Durbin, D.; Peloquin, C.A.; Elbert, K.J.; Hickey, A.J.; Edwards, D.A. Formulation and Pharmacokinetics of Self-Assembled Rifampicin Nanoparticle Systems for Pulmonary Delivery. Pharm. Res. 2009, 26, 1847–1855. [Google Scholar] [CrossRef]
- Chuan, J.; Li, Y.; Yang, L.; Sun, X.; Zhang, Q.; Gong, T.; Zhang, Z. Enhanced Rifampicin Delivery to Alveolar Macrophages by Solid Lipid Nanoparticles. J. Nanoparticle Res. 2013, 15, 1634. [Google Scholar] [CrossRef]
- Scolari, I.R.; Páez, P.L.; Musri, M.M.; Petiti, J.P.; Torres, A.; Granero, G.E. Rifampicin Loaded in Alginate/Chitosan Nanoparticles as a Promising Pulmonary Carrier against Staphylococcus Aureus. Drug Deliv. Transl. Res. 2020, 10, 1403–1417. [Google Scholar] [CrossRef] [PubMed]
- Pham, D.-D.; Fattal, E.; Tsapis, N. Pyrazinamide-Loaded Poly(Lactide-Co-Glycolide) Nanoparticles: Optimization by Experimental Design. J. Drug Deliv. Sci. Technol. 2015, 30, 384–390. [Google Scholar] [CrossRef]
- Ravi Varma, J.; Kumar, T.; Prasanthi, B.; Ratna, J. Formulation and Characterization of Pyrazinamide Polymeric Nanoparticles for Pulmonary Tuberculosis: Efficiency for Alveolar Macrophage Targeting. Indian J. Pharm. Sci. 2015, 77, 258. [Google Scholar] [CrossRef] [PubMed]
- Nemati, E.; Mokhtarzadeh, A.; Panahi-Azar, V.; Mohammadi, A.; Hamishehkar, H.; Mesgari-Abbasi, M.; Ezzati Nazhad Dolatabadi, J.; de la Guardia, M. Ethambutol-Loaded Solid Lipid Nanoparticles as Dry Powder Inhalable Formulation for Tuberculosis Therapy. AAPS PharmSciTech 2019, 20, 120. [Google Scholar] [CrossRef] [PubMed]
- Shete, H.K.; Vyas, S.S.; Patravale, V.B.; Disouza, J.I. Pulmonary Multifunctional Nano-Oncological Modules for Lung Cancer Treatment and Prevention. J. Biomed. Nanotechnol. 2014, 10, 1863–1893. [Google Scholar] [CrossRef]
- Prabhu, P.; Fernandes, T.; Chaubey, P.; Kaur, P.; Narayanan, S.; Vk, R.; Sawarkar, S.P. Mannose-Conjugated Chitosan Nanoparticles for Delivery of Rifampicin to Osteoarticular Tuberculosis. Drug Deliv. Transl. Res. 2021, 11, 1509–1519. [Google Scholar] [CrossRef]
- Singh, M.; Guzman-Aranguez, A.; Hussain, A.; Srinivas, C.S.; Kaur, I.P. Solid Lipid Nanoparticles for Ocular Delivery of Isoniazid: Evaluation, Proof of Concept and in Vivo Safety & Kinetics. Nanomedicine 2019, 14, 465–491. [Google Scholar] [CrossRef]
- Bazán Henostroza, M.A.; Curo Melo, K.J.; Nishitani Yukuyama, M.; Löbenberg, R.; Araci Bou-Chacra, N. Cationic Rifampicin Nanoemulsion for the Treatment of Ocular Tuberculosis. Colloids Surf. Physicochem. Eng. Asp. 2020, 597, 124755. [Google Scholar] [CrossRef]
- Bhandari, R.; Kaur, I.P. Pharmacokinetics, Tissue Distribution and Relative Bioavailability of Isoniazid-Solid Lipid Nanoparticles. Int. J. Pharm. 2013, 441, 202–212. [Google Scholar] [CrossRef]
- Baijnath, S.; Moodley, C.; Ngcobo, B.; Singh, S.D.; Kruger, H.G.; Arvidsson, P.I.; Naicker, T.; Pym, A.; Govender, T. Clofazimine Protects against Mycobacterium Tuberculosis Dissemination in the Central Nervous System Following Aerosol Challenge in a Murine Model. Int. J. Antimicrob. Agents 2018, 51, 77–81. [Google Scholar] [CrossRef] [PubMed]
- de Castro, R.R.; do Carmo, F.A.; Martins, C.; Simon, A.; de Sousa, V.P.; Rodrigues, C.R.; Cabral, L.M.; Sarmento, B. Clofazimine Functionalized Polymeric Nanoparticles for Brain Delivery in the Tuberculosis Treatment. Int. J. Pharm. 2021, 602, 120655. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, A.; Jain, P.; Mohapatra, S.; Mustafa, G.; Ansari, M.J.; Aldawsari, M.F.; Alalaiwe, A.S.; Mirza; Mohd, A.; Iqbal, Z. A Novel Approach of Targeting Linezolid Nanoemulsion for the Management of Lymph Node Tuberculosis. ACS Omega 2022, 7, 15688–15694. [Google Scholar] [CrossRef]
- Hussain, A.; Altamimi, M.A.; Alshehri, S.; Imam, S.S.; Shakeel, F.; Singh, S.K. Novel Approach for Transdermal Delivery of Rifampicin to Induce Synergistic Antimycobacterial Effects Against Cutaneous and Systemic Tuberculosis Using a Cationic Nanoemulsion Gel. Int. J. Nanomed. 2020, 15, 1073–1094. [Google Scholar] [CrossRef] [PubMed]
- Pi, J.; Shen, L.; Yang, E.; Shen, H.; Huang, D.; Wang, R.; Hu, C.; Jin, H.; Cai, H.; Cai, J.; et al. Macrophage-Targeted Isoniazid–Selenium Nanoparticles Promote Antimicrobial Immunity and Synergize Bactericidal Destruction of Tuberculosis Bacilli. Angew. Chem. 2020, 132, 3252–3260. [Google Scholar] [CrossRef]
- Saraogi, G.K.; Sharma, B.; Joshi, B.; Gupta, P.; Gupta, U.D.; Jain, N.K.; Agrawal, G.P. Mannosylated Gelatin Nanoparticles Bearing Isoniazid for Effective Management of Tuberculosis. J. Drug Target. 2011, 19, 219–227. [Google Scholar] [CrossRef]
- Rauf, A.; Tabish, T.A.; Ibrahim, I.M.; Rauf ul Hassan, M.; Tahseen, S.; Abdullah Sandhu, M.; Shahnaz, G.; Rahdar, A.; Cucchiarini, M.; Pandey, S. Design of Mannose-Coated Rifampicin Nanoparticles Modulating the Immune Response and Rifampicin Induced Hepatotoxicity with Improved Oral Drug Delivery. Arab. J. Chem. 2021, 14, 103321. [Google Scholar] [CrossRef]
- Marcianes, P.; Negro, S.; Barcia, E.; Montejo, C.; Fernández-Carballido, A. Potential Active Targeting of Gatifloxacin to Macrophages by Means of Surface-Modified PLGA Microparticles Destined to Treat Tuberculosis. AAPS PharmSciTech 2019, 21, 15. [Google Scholar] [CrossRef] [PubMed]
- Denkbas, E.B.; Kaitian, X.; Tuncel, A.; Piskin, E. Rifampicin-Carrying Poly(D,L-Iactide) Microspheres: Loading and Release. J. Biomater. Sci. Polym. Ed. 1995, 6, 815–825. [Google Scholar] [CrossRef]
- Patel, B.K.; Parikh, R.H.; Aboti, P.S. Development of Oral Sustained Release Rifampicin Loaded Chitosan Nanoparticles by Design of Experiment. J. Drug Deliv. 2013, 2013, 370938. [Google Scholar] [CrossRef] [PubMed]
- Hwang, A.A.; Lee, B.-Y.; Clemens, D.L.; Dillon, B.J.; Zink, J.I.; Horwitz, M.A. PH-Responsive Isoniazid-Loaded Nanoparticles Markedly Improve Tuberculosis Treatment in Mice. Small 2015, 11, 5066–5078. [Google Scholar] [CrossRef]
- Vemuri, N.; Khuller, G.K.; Prabhakar, T.; Pal, N.; Gupta, P.; Gupta, U. Nanoformulations of Moxifloxacin, Econozole and Ethionamide as Novel Treatment Regimens Against MDR TB—An Experimental Study. Curr. Nanosci. 2016, 12, 110–117. [Google Scholar] [CrossRef]
- Zargarnezhad, S.; Gholami, A.; Khoshneviszadeh, M.; Abootalebi, S.N.; Ghasemi, Y. Antimicrobial Activity of Isoniazid in Conjugation with Surface-Modified Magnetic Nanoparticles against Mycobacterium Tuberculosis and Nonmycobacterial Microorganisms. J. Nanomater. 2020, 2020, 7372531. [Google Scholar] [CrossRef]
- Khatak, S.; Mehta, M.; Awasthi, R.; Paudel, K.R.; Singh, S.K.; Gulati, M.; Hansbro, N.G.; Hansbro, P.M.; Dua, K.; Dureja, H. Solid Lipid Nanoparticles Containing Anti-Tubercular Drugs Attenuate the Mycobacterium Marinum Infection. Tuberculosis 2020, 125, 102008. [Google Scholar] [CrossRef] [PubMed]
- Changsan, N.; Sinsuebpol, C. Dry Powder Inhalation Formulation of Chitosan Nanoparticles for Co-Administration of Isoniazid and Pyrazinamide. Pharm. Dev. Technol. 2021, 26, 181–192. [Google Scholar] [CrossRef]
- Zahoor, A.; Sharma, S.; Khuller, G.K. Inhalable Alginate Nanoparticles as Antitubercular Drug Carriers against Experimental Tuberculosis. Int. J. Antimicrob. Agents 2005, 26, 298–303. [Google Scholar] [CrossRef]
- Pandey, R.; Sharma, S.; Khuller, G.K. Oral Solid Lipid Nanoparticle-Based Antitubercular Chemotherapy. Tuberculosis 2005, 85, 415–420. [Google Scholar] [CrossRef]
- Pandey, R.; Khuller, G.K. Oral Nanoparticle-Based Antituberculosis Drug Delivery to the Brain in an Experimental Model. J. Antimicrob. Chemother. 2006, 57, 1146–1152. [Google Scholar] [CrossRef] [PubMed]
- Abdelghany, S.; Parumasivam, T.; Pang, A.; Roediger, B.; Tang, P.; Jahn, K.; Britton, W.J.; Chan, H.-K. Alginate Modified-PLGA Nanoparticles Entrapping Amikacin and Moxifloxacin as a Novel Host-Directed Therapy for Multidrug-Resistant Tuberculosis. J. Drug Deliv. Sci. Technol. 2019, 52, 642–651. [Google Scholar] [CrossRef]
- Costa-Gouveia, J.; Pancani, E.; Jouny, S.; Machelart, A.; Delorme, V.; Salzano, G.; Iantomasi, R.; Piveteau, C.; Queval, C.J.; Song, O.-R.; et al. Combination Therapy for Tuberculosis Treatment: Pulmonary Administration of Ethionamide and Booster Co-Loaded Nanoparticles. Sci. Rep. 2017, 7, 5390. [Google Scholar] [CrossRef] [PubMed]
- Pastor, A.; Machelart, A.; Li, X.; Willand, N.; Baulard, A.; Brodin, P.; Gref, R.; Desmaële, D. A Novel Codrug Made of the Combination of Ethionamide and Its Potentiating Booster: Synthesis, Self-Assembly into Nanoparticles and Antimycobacterial Evaluation. Org. Biomol. Chem. 2019, 17, 5129–5137. [Google Scholar] [CrossRef]
- Salzano, G.; Wankar, J.; Ottani, S.; Villemagne, B.; Baulard, A.R.; Willand, N.; Brodin, P.; Manet, I.; Gref, R. Cyclodextrin-Based Nanocarriers Containing a Synergic Drug Combination: A Potential Formulation for Pulmonary Administration of Antitubercular Drugs. Int. J. Pharm. 2017, 531, 577–587. [Google Scholar] [CrossRef] [PubMed]
- Wankar, J.; Salzano, G.; Pancani, E.; Benkovics, G.; Malanga, M.; Manoli, F.; Gref, R.; Fenyvesi, E.; Manet, I. Efficient Loading of Ethionamide in Cyclodextrin-Based Carriers Offers Enhanced Solubility and Inhibition of Drug Crystallization. Int. J. Pharm. 2017, 531, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Machelart, A.; Salzano, G.; Li, X.; Demars, A.; Debrie, A.-S.; Menendez-Miranda, M.; Pancani, E.; Jouny, S.; Hoffmann, E.; Deboosere, N.; et al. Intrinsic Antibacterial Activity of Nanoparticles Made of β-Cyclodextrins Potentiates Their Effect as Drug Nanocarriers against Tuberculosis. ACS Nano 2019, 13, 3992–4007. [Google Scholar] [CrossRef]
- Mistry, N.; Bandyopadhyaya, R.; Mehra, S. ZnO Nanoparticles and Rifampicin Synergistically Damage the Membrane of Mycobacteria. ACS Appl. Nano Mater. 2020, 3, 3174–3184. [Google Scholar] [CrossRef]
- Gajendiran, M.; Gopi, V.; Elangovan, V.; Murali, R.V.; Balasubramanian, S. Isoniazid Loaded Core Shell Nanoparticles Derived from PLGA–PEG–PLGA Tri-Block Copolymers: In Vitro and in Vivo Drug Release. Colloids Surf. B Biointerfaces 2013, 104, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Wu, M.; Ye, W.; Huang, Z.; Ma, X.; Wang, W.; Wang, W.; Huang, Y.; Pan, X.; Wu, C. Inhalable Solid Lipid Nanoparticles for Intracellular Tuberculosis Infection Therapy: Macrophage-Targeting and PH-Sensitive Properties. Drug Deliv. Transl. Res. 2021, 11, 1218–1235. [Google Scholar] [CrossRef]
- Maghrebi, S.; Joyce, P.; Jambhrunkar, M.; Thomas, N.; Prestidge, C.A. Poly(Lactic-Co-Glycolic) Acid–Lipid Hybrid Microparticles Enhance the Intracellular Uptake and Antibacterial Activity of Rifampicin. ACS Appl. Mater. Interfaces 2020, 12, 8030–8039. [Google Scholar] [CrossRef] [PubMed]
- Vieira, A.C.C.; Chaves, L.L.; Pinheiro, M.; Lima, S.C.; Neto, P.J.R.; Ferreira, D.; Sarmento, B.; Reis, S. Lipid Nanoparticles Coated with Chitosan Using a One-Step Association Method to Target Rifampicin to Alveolar Macrophages. Carbohydr. Polym. 2021, 252, 116978. [Google Scholar] [CrossRef]
- Saifullah, B.; Maitra, A.; Chrzastek, A.; Naeemullah, B.; Fakurazi, S.; Bhakta, S.; Hussein, M. Nano-Formulation of Ethambutol with Multifunctional Graphene Oxide and Magnetic Nanoparticles Retains Its Anti-Tubercular Activity with Prospects of Improving Chemotherapeutic Efficacy. Molecules 2017, 22, 1697. [Google Scholar] [CrossRef]
- Helal-Neto, E.; Rocha Pinto, S.; Portilho, F.L.; da Costa, M.D.; Pereira, J.X.; Nigro, F.; Ricci-Junior, E.; Candéa, A.L.P.; das Graças Muller de Oliveira Henri, M.; Santos-Oliveira, R. Development and Biological Evaluation of a New Nanotheranostic for Tuberculosis. Drug Deliv. Transl. Res. 2019, 9, 97–105. [Google Scholar] [CrossRef]
- Jary, D.; Hibbitts, A.; Lozano-Fernandez, T.; Pérez, D.; Lucia, A.; Ainsa, J.A.; Codony, D.; Freire, C.; Redinger, N.; Schaible, U.; et al. Bedaquiline Loaded Lipid Nanoparticles: A Promising Candidate for TB Treatment. TechConnect Briefs 2018, 3, 59–62. [Google Scholar]
- De Matteis, L.; Jary, D.; Lucía, A.; García-Embid, S.; Serrano-Sevilla, I.; Pérez, D.; Ainsa, J.A.; Navarro, F.P.; de la Fuente, J.M. New Active Formulations against M. Tuberculosis: Bedaquiline Encapsulation in Lipid Nanoparticles and Chitosan Nanocapsules. Chem. Eng. J. 2018, 340, 181–191. [Google Scholar] [CrossRef]
- Huck, B.C.; Thiyagarajan, D.; Bali, A.; Boese, A.; Besecke, K.F.W.; Hozsa, C.; Gieseler, R.K.; Furch, M.; Carvalho-Wodarz, C.; Waldow, F.; et al. Nano-in-Microparticles for Aerosol Delivery of Antibiotic-Loaded, Fucose-Derivatized, and Macrophage-Targeted Liposomes to Combat Mycobacterial Infections: In Vitro Deposition, Pulmonary Barrier Interactions, and Targeted Delivery. Adv. Healthc. Mater. 2022, 11, 2102117. [Google Scholar] [CrossRef]
- Patil, K.D.; Bagade, S.B.; Bonde, S.C. Biodistribution, Pharmacokinetics and Toxicity Evaluation of Mannosylated Gelatin Nanoparticles of Linezolid for Anti-Tubercular Therapy. Mater. Technol. 2022, 37, 95–103. [Google Scholar] [CrossRef]
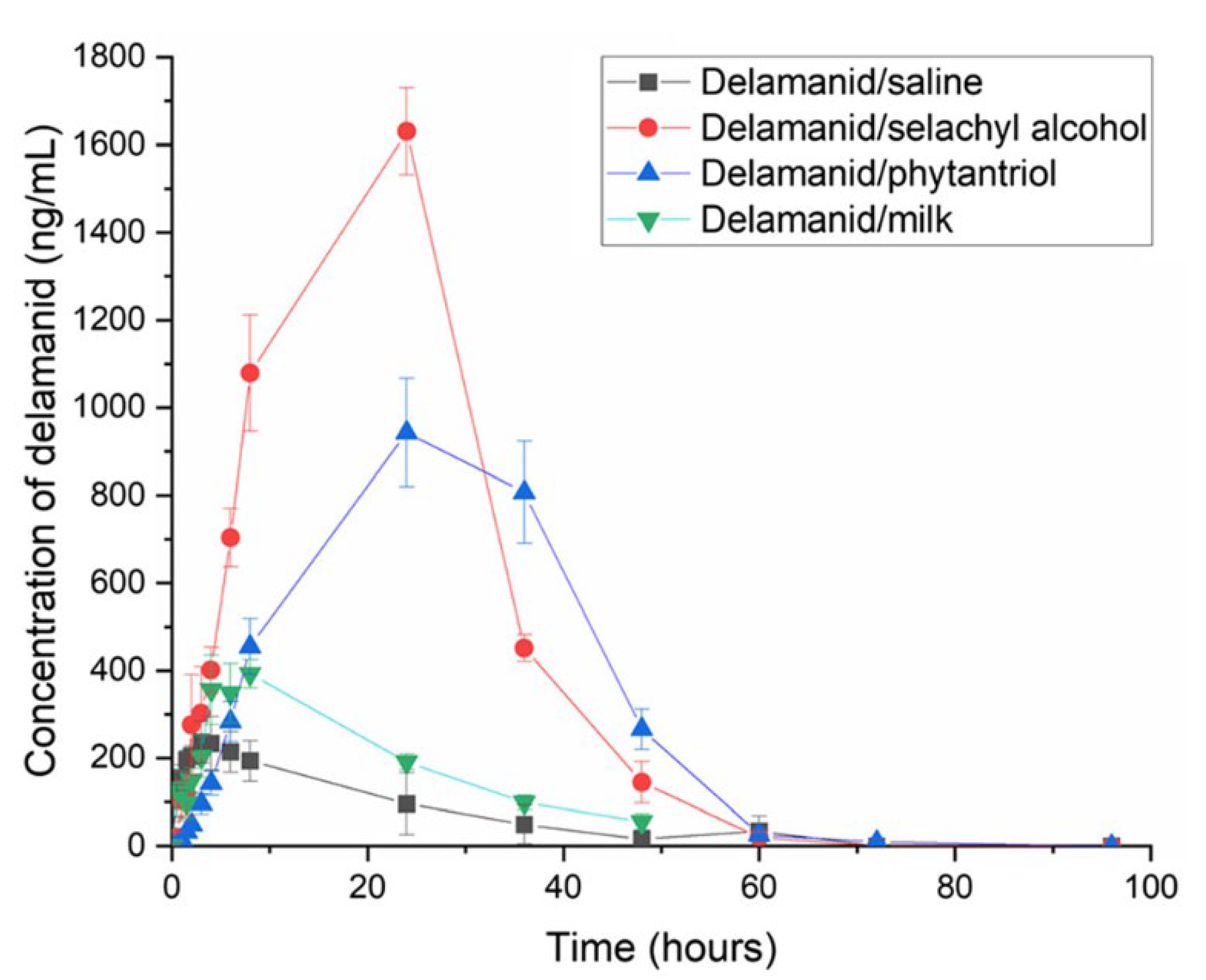
- Ramirez, G.; Pham, A.C.; Clulow, A.J.; Salim, M.; Hawley, A.; Boyd, B.J. Sustained Absorption of Delamanid from Lipid-Based Formulations as a Path to Reduced Frequency of Administration. Drug Deliv. Transl. Res. 2021, 11, 1236–1244. [Google Scholar] [CrossRef]
- Ang, C.W.; Tan, L.; Qu, Z.; West, N.P.; Cooper, M.A.; Popat, A.; Blaskovich, M.A.T. Mesoporous Silica Nanoparticles Improve Oral Delivery of Antitubercular Bicyclic Nitroimidazoles. ACS Biomater. Sci. Eng. 2022, 8, 4196–4206. [Google Scholar] [CrossRef]
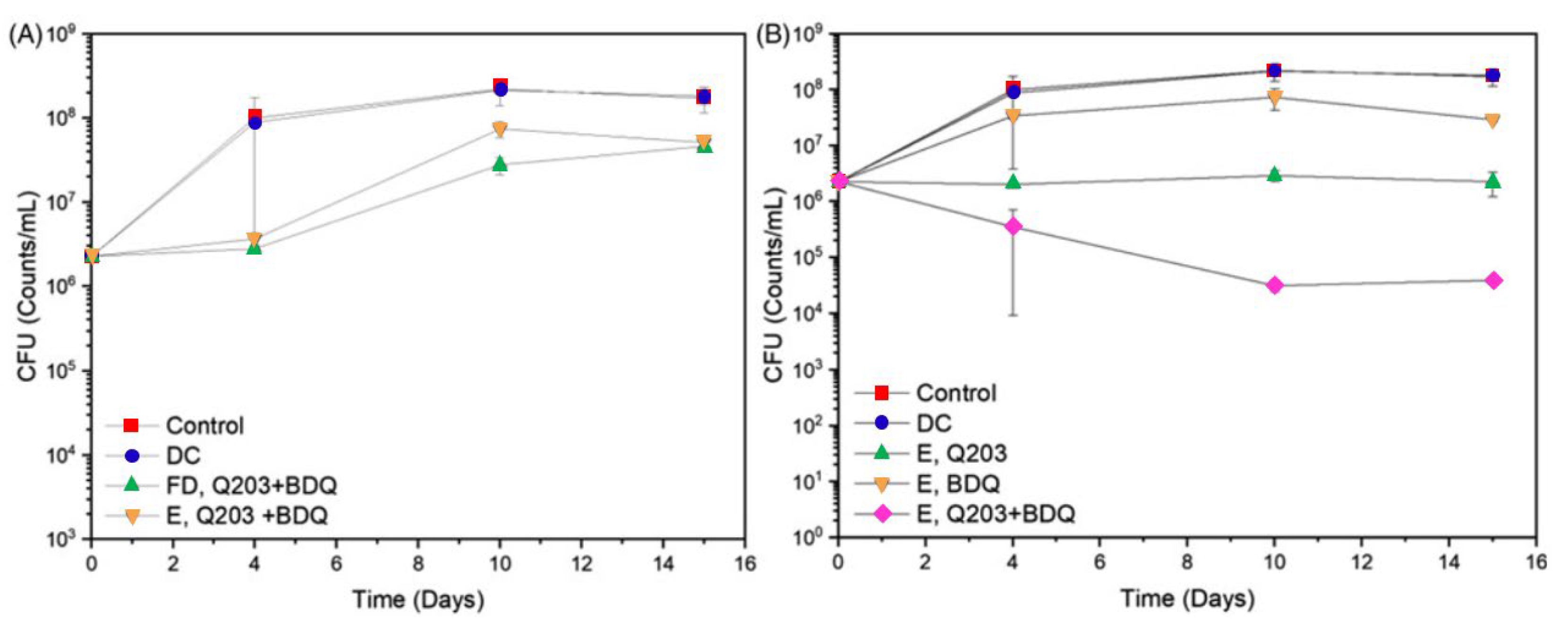
- Poh, W.; Ab Rahman, N.; Ostrovski, Y.; Sznitman, J.; Pethe, K.; Loo, S.C.J. Active Pulmonary Targeting against Tuberculosis (TB) via Triple-Encapsulation of Q203, Bedaquiline and Superparamagnetic Iron Oxides (SPIOs) in Nanoparticle Aggregates. Drug Deliv. 2019, 26, 1039–1048. [Google Scholar] [CrossRef]
- Makled, S.; Boraie, N.; Nafee, N. Nanoparticle-Mediated Macrophage Targeting—A New Inhalation Therapy Tackling Tuberculosis. Drug Deliv. Transl. Res. 2021, 11, 1037–1055. [Google Scholar] [CrossRef]
- Shah, S.; Cristopher, D.; Sharma, S.; Soniwala, M.; Chavda, J. Inhalable Linezolid Loaded PLGA Nanoparticles for Treatment of Tuberculosis: Design, Development and in Vitro Evaluation. J. Drug Deliv. Sci. Technol. 2020, 60, 102013. [Google Scholar] [CrossRef]
- Shah, S.; Maheshwari, H.; Soniwala, M.; Chavda, J. Pulmonary Delivery of Linezolid Nanoparticles for Treatment of Tuberculosis: Design, Development, and Optimization. J. Pharm. Innov. 2022, 17, 46–59. [Google Scholar] [CrossRef]
- Abdelghany, S.; Alkhawaldeh, M.; AlKhatib, H.S. Carrageenan-Stabilized Chitosan Alginate Nanoparticles Loaded with Ethionamide for the Treatment of Tuberculosis. J. Drug Deliv. Sci. Technol. 2017, 39, 442–449. [Google Scholar] [CrossRef]
- Krug, S.; Parveen, S.; Bishai, W.R. Host-Directed Therapies: Modulating Inflammation to Treat Tuberculosis. Front. Immunol. 2021, 12, 660916. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, S.H.E.; Dorhoi, A.; Hotchkiss, R.S.; Bartenschlager, R. Host-Directed Therapies for Bacterial and Viral Infections. Nat. Rev. Drug Discov. 2018, 17, 35–56. [Google Scholar] [CrossRef] [PubMed]
- Goletti, D.; Petruccioli, E.; Romagnoli, A.; Piacentini, M.; Fimia, G.M. Autophagy in Mycobacterium Tuberculosis Infection: A Passepartout to Flush the Intruder Out? Cytokine Growth Factor Rev. 2013, 24, 335–343. [Google Scholar] [CrossRef]
- Khoza, L.J.; Kumar, P.; Dube, A.; Demana, P.H.; Choonara, Y.E. Insights into Innovative Therapeutics for Drug-Resistant Tuberculosis: Host-Directed Therapy and Autophagy Inducing Modified Nanoparticles. Int. J. Pharm. 2022, 622, 121893. [Google Scholar] [CrossRef]
- Dube, A.; Reynolds, J.L.; Law, W.-C.; Maponga, C.C.; Prasad, P.N.; Morse, G.D. Multimodal Nanoparticles That Provide Immunomodulation and Intracellular Drug Delivery for Infectious Diseases. Nanomed. Nanotechnol. Biol. Med. 2014, 10, 831–838. [Google Scholar] [CrossRef]
- Tukulula, M.; Hayeshi, R.; Fonteh, P.; Meyer, D.; Ndamase, A.; Madziva, M.T.; Khumalo, V.; Lubuschagne, P.; Naicker, B.; Swai, H.; et al. Curdlan-Conjugated PLGA Nanoparticles Possess Macrophage Stimulant Activity and Drug Delivery Capabilities. Pharm. Res. 2015, 32, 2713–2726. [Google Scholar] [CrossRef]
- D’Souza, S.; Du Plessis, S.; Egieyeh, S.; Bekale, R.; Maphasa, R.; Irabin, A.; Sampson, S.; Dube, A. Physicochemical and Biological Evaluation of Curdlan-Poly(Lactic-Co-Glycolic Acid) Nanoparticles as a Host-Directed Therapy Against Mycobacterium Tuberculosis. J. Pharm. Sci. 2022, 111, 469–478. [Google Scholar] [CrossRef]
- Bahlool, A.Z.; Fattah, S.; O’Sullivan, A.; Cavanagh, B.; MacLoughlin, R.; Keane, J.; O’Sullivan, M.P.; Cryan, S.-A. Development of Inhalable ATRA-Loaded PLGA Nanoparticles as Host-Directed Immunotherapy against Tuberculosis. Pharmaceutics 2022, 14, 1745. [Google Scholar] [CrossRef]
- Tousif, S.; Singh, D.K.; Mukherjee, S.; Ahmad, S.; Arya, R.; Nanda, R.; Ranganathan, A.; Bhattacharyya, M.; Van Kaer, L.; Kar, S.K.; et al. Nanoparticle-Formulated Curcumin Prevents Posttherapeutic Disease Reactivation and Reinfection with Mycobacterium Tuberculosis Following Isoniazid Therapy. Front. Immunol. 2017, 8, 739. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Vaghasiya, K.; Gupta, P.; Gupta, U.D.; Verma, R.K. Reclaiming Hijacked Phagosomes: Hybrid Nano-in-Micro Encapsulated MIAP Peptide Ensures Host Directed Therapy by Specifically Augmenting Phagosome-Maturation and Apoptosis in TB Infected Macrophage Cells. Int. J. Pharm. 2018, 536, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, T.K.; Fatima, N.; Sharma, A.; Sharma, D.; Sharma, R. Nano-Rifabutin Entrapment within Glucan Microparticles Enhances Protection against Intracellular Mycobacterium Tuberculosis. Artif. Cells Nanomedicine Biotechnol. 2019, 47, 427–435. [Google Scholar] [CrossRef]
- Puri, V.; Chaudhary, K.R.; Singh, A.; Singh, C. Inhalation Potential of N-Acetylcysteine Loaded PLGA Nanoparticles for the Management of Tuberculosis: In Vitro Lung Deposition and Efficacy Studies. Curr. Res. Pharmacol. Drug Discov. 2022, 3, 100084. [Google Scholar] [CrossRef]
- Amaral, E.P.; Conceição, E.L.; Costa, D.L.; Rocha, M.S.; Marinho, J.M.; Cordeiro-Santos, M.; D’Império-Lima, M.R.; Barbosa, T.; Sher, A.; Andrade, B.B. N-Acetyl-Cysteine Exhibits Potent Anti-Mycobacterial Activity in Addition to Its Known Anti-Oxidative Functions. BMC Microbiol. 2016, 16, 251. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, P.; van der Meulen, S.A.; Simões Caetano, J.M.; Goojani, H.G.; Botman, D.; van Spanning, R.; Lill, H.; Bald, D. Response of Mycobacterium Smegmatis to the Cytochrome Bcc Inhibitor Q203. Int. J. Mol. Sci. 2022, 23, 10331. [Google Scholar] [CrossRef]
- Li, G.; Li, J.; Hou, Y.; Xie, S.; Xu, J.; Yang, M.; Li, D.; Du, Y. Levofloxacin-Loaded Nanosonosensitizer as a Highly Efficient Therapy for Bacillus Calmette-Guérin Infections Based on Bacteria-Specific Labeling and Sonotheranostic Strategy. Int. J. Nanomed. 2021, 16, 6553–6573. [Google Scholar] [CrossRef]
- Xie, S.; Li, G.; Hou, Y.; Yang, M.; Li, F.; Li, J.; Li, D.; Du, Y. A Synergistic Bactericidal Effect of Low-Frequency and Low-Intensity Ultrasound Combined with Levofloxacin-Loaded PLGA Nanoparticles on M. Smegmatis in Macrophages. J. Nanobiotechnol. 2020, 18, 107. [Google Scholar] [CrossRef]
- Tian, N.; Duan, H.; Cao, T.; Dai, G.; Sheng, G.; Chu, H.; Sun, Z. Macrophage-Targeted Nanoparticles Mediate Synergistic Photodynamic Therapy and Immunotherapy of Tuberculosis. RSC Adv. 2023, 13, 1727–1737. [Google Scholar] [CrossRef]
- Sharma, A.; Vaghasiya, K.; Gupta, P.; Singh, A.K.; Gupta, U.D.; Verma, R.K. Dynamic Mucus Penetrating Microspheres for Efficient Pulmonary Delivery and Enhanced Efficacy of Host Defence Peptide (HDP) in Experimental Tuberculosis. J. Control. Release 2020, 324, 17–33. [Google Scholar] [CrossRef]
- Anselmo, A.C.; Mitragotri, S. Nanoparticles in the Clinic: An Update. Bioeng. Transl. Med. 2019, 4, e10143. [Google Scholar] [CrossRef] [PubMed]
- Laghari, M.; Darwis, Y.; Memon, A.H.; Khan, A.A.; Abdulbaqi, I.M.T.; Assi, R.A. Nanoformulations and Clinical Trial Candidates as Probably Effective and Safe Therapy for Tuberculosis. Trop. J. Pharm. Res. 2016, 15, 201–211. [Google Scholar] [CrossRef]
- Saramago, S.; Magalhães, J.; Pinheiro, M. Tuberculosis Vaccines: An Update of Recent and Ongoing Clinical Trials. Appl. Sci. 2021, 11, 9250. [Google Scholar] [CrossRef]
- Cobelens, F.; Suri, R.K.; Helinski, M.; Makanga, M.; Weinberg, A.L.; Schaffmeister, B.; Deege, F.; Hatherill, M. Accelerating Research and Development of New Vaccines against Tuberculosis: A Global Roadmap. Lancet Infect. Dis. 2022, 22, e108–e120. [Google Scholar] [CrossRef]
- Ma, R.; Zhang, J.; Chen, Z.; Ma, H.; Liu, X.; Liang, S.; Wu, P.; Ge, Z. Treatment of Spinal Tuberculosis in Rabbits Using Bovine Serum Albumin Nanoparticles Loaded with Isoniazid and Rifampicin. Neurol. Res. 2022, 44, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Knudsen Dal, N.-J.; Speth, M.; Johann, K.; Barz, M.; Beauvineau, C.; Wohlmann, J.; Fenaroli, F.; Gicquel, B.; Griffiths, G.; Alonso-Rodriguez, N. The Zebrafish Embryo as an in Vivo Model for Screening Nanoparticle-Formulated Lipophilic Anti-Tuberculosis Compounds. Dis. Model. Mech. 2022, 15, dmm049147. [Google Scholar] [CrossRef]
- Elkington, P.; Lerm, M.; Kapoor, N.; Mahon, R.; Pienaar, E.; Huh, D.; Kaushal, D.; Schlesinger, L.S. In Vitro Granuloma Models of Tuberculosis: Potential and Challenges. J. Infect. Dis. 2019, 219, 1858–1866. [Google Scholar] [CrossRef] [PubMed]
- Ladavière, C.; Gref, R. Toward an Optimized Treatment of Intracellular Bacterial Infections: Input of Nanoparticulate Drug Delivery Systems. Nanomedicine 2015, 10, 3033–3055. [Google Scholar] [CrossRef]
- Bourguignon, T.; Torrano, A.A.; Houel-Renault, L.; Machelart, A.; Brodin, P.; Gref, R. An Original Methodology to Study Polymeric Nanoparticle-Macrophage Interactions: Nanoparticle Tracking Analysis in Cell Culture Media and Quantification of the Internalized Objects. Int. J. Pharm. 2021, 610, 121202. [Google Scholar] [CrossRef]
- Shao, L.; Shen, S.; Liu, H. Recent Advances in PLGA Micro/Nanoparticle Delivery Systems as Novel Therapeutic Approach for Drug-Resistant Tuberculosis. Front. Bioeng. Biotechnol. 2022, 10, 941077. [Google Scholar] [CrossRef]
- Guan, X.; Zhang, W. Applications of Chitosan in Pulmonary Drug Delivery. In Role of Novel Drug Delivery Vehicles in Nanobiomedicine; IntechOpen: London, UK, 2019. [Google Scholar]
- Maphasa, R.E.; Meyer, M.; Dube, A. The Macrophage Response to Mycobacterium Tuberculosis and Opportunities for Autophagy Inducing Nanomedicines for Tuberculosis Therapy. Front. Cell. Infect. Microbiol. 2021, 10, 618414. [Google Scholar] [CrossRef] [PubMed]
















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bourguignon, T.; Godinez-Leon, J.A.; Gref, R. Nanosized Drug Delivery Systems to Fight Tuberculosis. Pharmaceutics 2023, 15, 393. https://doi.org/10.3390/pharmaceutics15020393
Bourguignon T, Godinez-Leon JA, Gref R. Nanosized Drug Delivery Systems to Fight Tuberculosis. Pharmaceutics. 2023; 15(2):393. https://doi.org/10.3390/pharmaceutics15020393
Chicago/Turabian StyleBourguignon, Tom, Jesus Alfredo Godinez-Leon, and Ruxandra Gref. 2023. "Nanosized Drug Delivery Systems to Fight Tuberculosis" Pharmaceutics 15, no. 2: 393. https://doi.org/10.3390/pharmaceutics15020393
APA StyleBourguignon, T., Godinez-Leon, J. A., & Gref, R. (2023). Nanosized Drug Delivery Systems to Fight Tuberculosis. Pharmaceutics, 15(2), 393. https://doi.org/10.3390/pharmaceutics15020393