

Synthesis and Anti-Angiogenic Activity of Novel c(RGDyK) Peptide-Based JH-VII-139-1 Conjugates
 , , ,
, , ,  ,
,  , and
, and
Abstract
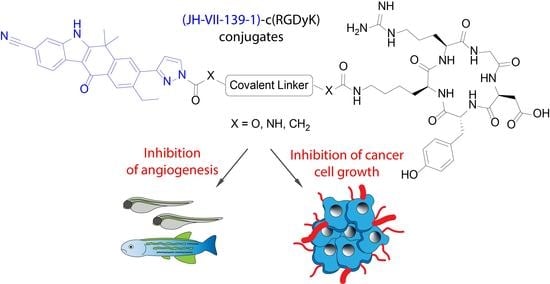
1. Introduction
2. Materials and Methods
2.1. Chemistry
2.1.1. General Experimental Details
2.1.2. Synthetic Procedures
2.2. Chemostability Assays
2.2.1. Stability in Buffer Solutions
2.2.2. Stability in Dulbecco’s Modified Eagle Medium
2.2.3. Stability in Human Plasma
2.3. In Vitro Kinase Assays
2.4. Cell Toxicity
2.4.1. Cell Culture
2.4.2. MTT Assays
2.5. Fluorescence Microscopy
2.6. Zebrafish Angiogenesis Assay
2.6.1. Zebrafish Screening Assays
2.6.2. Zebrafish Imaging Analysis
3. Results
3.1. Synthesis of Compounds
3.1.1. Optimized Synthesis of SRPK1 Inhibitor, JH-VII-139-1
3.1.2. Synthesis of (JH-VII-139-1)-c(RGDyK) Peptide–Drug Conjugates
3.1.3. Synthesis of Fluorescein-Tagged (JH-VII-139-1)-c(RGDyK) Conjugate
3.2. In Vitro Stability Studies
3.2.1. Chemostability Profile of Conjugate geo75
3.2.2. Chemostability Profile of Conjugate geo77
3.2.3. Chemostability Profile of Conjugate geo85
3.2.4. Chemostability Profile of geo107
3.3. Biological Activity of the Synthesized Compounds
3.3.1. Inhibition of Kinase Activity by JH-VII-139-1-c(RGDyK) Conjugates
3.3.2. Cytotoxicity of JH-VII-139-1-c(RGDyK) Conjugates
3.3.3. Cellular Uptake of c(RGDyK) Conjugate geo106
3.3.4. In Vivo Zebrafish Angiogenesis Studies
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Yoo, S.Y.; Kwon, S.M. Angiogenesis and its therapeutic opportunities. Mediators Inflamm. 2013, 2013, 127170. [Google Scholar] [CrossRef] [PubMed]
- Potente, M.; Gerhardt, H.; Carmeliet, P. Basic and therapeutic aspects of angiogenesis. Cell 2011, 146, 873–887. [Google Scholar] [CrossRef] [PubMed]
- Garcea, G.; Lloyd, T.D.; Gescher, A.; Dennison, A.R.; Steward, W.P.; Berry, D.P. Angiogenesis of gastrointestinal tumours and their metastases--a target for intervention? Eur. J. Cancer 2004, 40, 1302–1313. [Google Scholar] [CrossRef]
- Folkman, J. Role of angiogenesis in tumor growth and metastasis. Semin. Oncol. 2002, 29, 15–18. [Google Scholar] [CrossRef]
- Bielenberg, D.R.; Zetter, B.R. The Contribution of Angiogenesis to the Process of Metastasis. Cancer J. 2015, 21, 267–273. [Google Scholar] [CrossRef]
- Fallah, A.; Sadeghinia, A.; Kahroba, H.; Samadi, A.; Heidari, H.R.; Bradaran, B.; Zeinali, S.; Molavi, O. Therapeutic targeting of angiogenesis molecular pathways in angiogenesis-dependent diseases. Biomed. Pharmacother. 2019, 110, 775–785. [Google Scholar] [CrossRef]
- Adamis, A.P.; Aiello, L.P.; D’Amato, R.A. Angiogenesis and ophthalmic disease. Angiogenesis 1999, 3, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Girolamo, F.; Coppola, C.; Ribatti, D.; Trojano, M. Angiogenesis in multiple sclerosis and experimental autoimmune encephalomyelitis. Acta Neuropathol. Commun. 2014, 2, 84. [Google Scholar] [CrossRef] [PubMed]
- Elshabrawy, H.A.; Chen, Z.; Volin, M.V.; Ravella, S.; Virupannavar, S.; Shahrara, S. The pathogenic role of angiogenesis in rheumatoid arthritis. Angiogenesis 2015, 18, 433–448. [Google Scholar] [CrossRef] [PubMed]
- Camaré, C.; Pucelle, M.; Nègre-Salvayre, A.; Salvayre, R. Angiogenesis in the atherosclerotic plaque. Redox Biol. 2017, 12, 18–34. [Google Scholar] [CrossRef]
- Jaipersad, A.S.; Lip, G.Y.; Silverman, S.; Shantsila, E. The role of monocytes in angiogenesis and atherosclerosis. J. Am. Coll. Cardiol. 2014, 63, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Serini, G.; Valdembri, D.; Bussolino, F. Integrins and angiogenesis: A sticky business. Exp. Cell Res. 2006, 312, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, Z.; Bicknell, R. Angiogenic signalling pathways. Methods Mol. Biol. 2009, 467, 3–24. [Google Scholar] [PubMed]
- Thurston, G.; Kitajewski, J. VEGF and Delta-Notch: Interacting signalling pathways in tumour angiogenesis. Br. J. Cancer 2008, 99, 1204–1209. [Google Scholar] [CrossRef]
- Uemura, A.; Fruttiger, M.; D’Amore, P.A.; De Falco, S.; Joussen, A.M.; Sennlaub, F.; Brunck, L.R.; Johnson, K.T.; Lambrou, G.N.; Rittenhouse, K.D.; et al. VEGFR1 signaling in retinal angiogenesis and microinflammation. Prog. Retin. Eye Res. 2021, 84, 100954. [Google Scholar] [CrossRef]
- Shibuya, M. Role of VEGF-flt receptor system in normal and tumor angiogenesis. Adv. Cancer Res. 1995, 67, 281–316. [Google Scholar]
- Ferrara, N.; Kerbel, R.S. Angiogenesis as a therapeutic target. Nature 2005, 438, 967–974. [Google Scholar] [CrossRef]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef]
- Gammons, M.V.; Dick, A.D.; Harper, S.J.; Bates, D.O. SRPK1 inhibition modulates VEGF splicing to reduce pathological neovascularization in a rat model of retinopathy of prematurity. Investig. Ophthalmol. Vis. Sci. 2013, 54, 5797–5806. [Google Scholar] [CrossRef]
- Gammons, M.V.; Lucas, R.; Dean, R.; Coupland, S.E.; Oltean, S.; Bates, D.O. Targeting SRPK1 to control VEGF-mediated tumour angiogenesis in metastatic melanoma. Br. J. Cancer 2014, 111, 477–485. [Google Scholar] [CrossRef]
- Mavrou, A.; Brakspear, K.; Hamdollah-Zadeh, M.; Damodaran, G.; Babaei-Jadidi, R.; Oxley, J.; Gillatt, D.A.; Ladomery, M.R.; Harper, S.J.; Bates, D.O.; et al. Serine-arginine protein kinase 1 (SRPK1) inhibition as a potential novel targeted therapeutic strategy in prostate cancer. Oncogene 2015, 34, 4311–4319. [Google Scholar] [CrossRef] [PubMed]
- Chatzisideri, T.; Leonidis, G.; Karampelas, T.; Skavatsou, E.; Velentza-Almpani, A.; Bianchini, F.; Tamvakopoulos, C.; Sarli, V. Integrin-Mediated Targeted Cancer Therapy Using c(RGDyK)-Based Conjugates of Gemcitabine. J. Med. Chem. 2022, 65, 271–284. [Google Scholar] [CrossRef]
- Chatzisideri, T.; Dalezis, P.; Leonidis, G.; Bousis, S.; Trafalis, D.; Bianchini, F.; Sarli, V. Synthesis and biological studies of c(RGDyK) conjugates of cucurbitacins. Future Med. Chem. 2021, 13, 877–895. [Google Scholar] [CrossRef]
- Chatzisideri, T.; Thysiadis, S.; Katsamakas, S.; Dalezis, P.; Sigala, I.; Lazarides, T.; Nikolakaki, E.; Trafalis, D.; Gederaas, O.A.; Lindgren, M.; et al. Synthesis and biological evaluation of a Platinum(II)-c(RGDyK) conjugate for integrin-targeted photodynamic therapy. Eur. J. Med. Chem. 2017, 141, 221–231. [Google Scholar] [CrossRef]
- Kapp, T.G.; Rechenmacher, F.; Neubauer, S.; Maltsev, O.V.; Cavalcanti-Adam, E.A.; Zarka, R.; Reuning, U.; Notni, J.; Wester, H.J.; Mas-Moruno, C.; et al. A Comprehensive Evaluation of the Activity and Selectivity Profile of Ligands for RGD-binding Integrins. Sci. Rep. 2017, 7, 39805. [Google Scholar] [CrossRef]
- Leonidis, G.; Dalezis, P.; Trafalis, D.; Beis, D.; Giardoglou, P.; Koukiali, A.; Sigala, I.; Nikolakaki, E.; Sarli, V. Synthesis and Biological Evaluation of a c(RGDyK) Peptide Conjugate of SRPIN803. ACS Omega 2021, 6, 28379–28393. [Google Scholar] [CrossRef] [PubMed]
- Hatcher, J.M.; Wu, G.; Zeng, C.; Zhu, J.; Meng, F.; Patel, S.; Wang, W.; Ficarro, S.B.; Leggett, A.L.; Powell, C.E.; et al. SRPKIN-1: A Covalent SRPK1/2 Inhibitor that Potently Converts VEGF from Pro-angiogenic to Anti-angiogenic Isoform. Cell Chem. Biol. 2018, 25, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Larkins, E.; Blumenthal, G.M.; Chen, H.; He, K.; Agarwal, R.; Gieser, G.; Stephens, O.; Zahalka, E.; Ringgold, K.; Helms, W.; et al. FDA Approval: Alectinib for the Treatment of Metastatic, ALK-Positive Non-Small Cell Lung Cancer Following Crizotinib. Clin. Cancer Res. 2016, 22, 5171–5176. [Google Scholar] [CrossRef] [PubMed]
- Song, D.; Lee, M.; Park, C.H.; Ahn, S.; Yun, C.S.; Lee, C.O.; Kim, H.R.; Hwang, J.Y. Novel 2,4-diaminopyrimidines bearing tetrahydronaphthalenyl moiety against anaplastic lymphoma kinase (ALK): Synthesis, in vitro, ex vivo, and in vivo efficacy studies. Bioorg. Med. Chem. Lett. 2016, 26, 1720–1725. [Google Scholar] [CrossRef]
- Kinoshita, K.; Kobayashi, T.; Asoh, K.; Furuichi, N.; Ito, T.; Kawada, H.; Hara, S.; Ohwada, J.; Hattori, K.; Miyagi, T.; et al. 9-substituted 6,6-dimethyl-11-oxo-6,11-dihydro-5H-benzo[b]carbazoles as highly selective and potent anaplastic lymphoma kinase inhibitors. J. Med. Chem. 2011, 54, 6286–6294. [Google Scholar] [CrossRef]
- John, A.A.; Ramil, C.P.; Tian, Y.; Cheng, G.; Lin, Q. Synthesis and Site-Specific Incorporation of Red-Shifted Azobenzene Amino Acids into Proteins. Org. Lett. 2015, 17, 6258–6261. [Google Scholar] [CrossRef] [PubMed]
- Wan, T.; Capaldo, L.; Laudadio, G.; Nyuchev, A.V.; Rincón, J.A.; García-Losada, P.; Mateos, C.; Frederick, M.O.; Nuño, M.; Noël, T. Decatungstate-Mediated C(sp3)–H Heteroarylation via Radical-Polar Crossover in Batch and Flow. Angew. Chem. Int. Ed. 2021, 60, 17893–17897. [Google Scholar] [CrossRef] [PubMed]
- Young, M.B.; Barrow, J.C.; Glass, K.L.; Lundell, G.F.; Newton, C.L.; Pellicore, J.M.; Rittle, K.E.; Selnick, H.G.; Stauffer, K.J.; Vacca, J.P.; et al. Discovery and evaluation of potent P1 aryl heterocycle-based thrombin inhibitors. J. Med. Chem. 2004, 47, 2995–3008. [Google Scholar] [CrossRef] [PubMed]
- Yeager, A.R.; Finney, N.S. The first direct evaluation of the two-active site mechanism for chitin synthase. J. Org. Chem. 2004, 69, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Grissom, C.B. Design, Synthesis, and Characterization of Fluorescent Cobalamin Analogues with High Quantum Efficiencies. Org. Lett. 2009, 11, 2499–2502. [Google Scholar] [CrossRef] [PubMed]
- Voukkalis, N.; Koutroumani, M.; Zarkadas, C.; Nikolakaki, E.; Vlassi, M.; Giannakouros, T. SRPK1 and Akt Protein Kinases Phosphorylate the RS Domain of Lamin B Receptor with Distinct Specificity: A Combined Biochemical and In Silico Approach. PLoS ONE 2016, 11, e0154198. [Google Scholar] [CrossRef]
- Sigala, I.; Tsamis, K.I.; Gousia, A.; Alexiou, G.; Voulgaris, S.; Giannakouros, T.; Kyritsis, A.P.; Nikolakaki, E. Expression of SRPK1 in gliomas and its role in glioma cell lines viability. Tumour Biol. 2016, 37, 8699–8707. [Google Scholar] [CrossRef]
- Jin, S.W.; Beis, D.; Mitchell, T.; Chen, J.N.; Stainier, D.Y.R. Cellular and molecular analyses of vascular tube and lumen formation in zebrafish. Development 2005, 132, 5199–5209. [Google Scholar] [CrossRef]
- Aleström, P.; D’Angelo, L.; Midtlyng, P.J.; Schorderet, D.F.; Schulte-Merker, S.; Sohm, F.; Warner, S. Zebrafish: Housing and husbandry recommendations. Lab. Anim. 2020, 54, 213–224. [Google Scholar] [CrossRef]
- Davis, R.A.; Lau, K.; Hausner, S.H.; Sutcliffe, J.L. Solid-phase synthesis and fluorine-18 radiolabeling of cycloRGDyK. Org. Biomol. Chem. 2016, 14, 8659–8663. [Google Scholar] [CrossRef]
- Hammershøj, P.; Kumar, E.K.P.; Harris, P.; Andresen, T.L.; Clausen, M.H. Facile Large-Scale Synthesis of 5- and 6-Carboxyfluoresceins: Application for the Preparation of New Fluorescent Dyes. Eur. J. Org. Chem. 2015, 2015, 7301–7309. [Google Scholar] [CrossRef]
- Adamczyk, M.; Fishpaugh, J.R.; Heuser, K.J. Preparation of succinimidyl and pentafluorophenyl active esters of 5- and 6-carboxyfluorescein. Bioconjug. Chem. 1997, 8, 253–255. [Google Scholar] [CrossRef]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Czarnomysy, R.; Surażyński, A.; Popławska, B.; Rysiak, E.; Pawłowska, N.; Czajkowska, A.; Bielawski, K.; Bielawska, A. Synergistic action of cisplatin and echistatin in MDA-MB-231 breast cancer cells. Mol. Cell. Biochem. 2017, 27, 13–22. [Google Scholar] [CrossRef]
- Kazmierczak, P.M.; Todica, A.; Gildehaus, F.J.; Hirner-Eppeneder, H.; Brendel, M.; Eschbach, R.S.; Hellmann, M.; Nikolaou, K.; Reiser, M.F.; Wester, H.J.; et al. 68Ga-TRAP-(RGD)3 Hybrid Imaging for the In Vivo Monitoring of αvß3-Integrin Expression as Biomarker of Anti-Angiogenic Therapy Effects in Experimental Breast Cancer. PLoS ONE 2016, 11, e0168248. [Google Scholar] [CrossRef] [PubMed]
- Braun, K.; Wiessler, M.; Pipkorn, R.; Ehemann, V.; Bäuerle, T.; Fleischhacker, H.; Müller, G.; Lorenz, P.; Waldeck, W. A cyclic-RGD-BioShuttle functionalized with TMZ by DARinv “Click Chemistry” targeted to αvβ3 integrin for therapy. Int. J. Med. Sci. 2010, 7, 326–339. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.A.; Nam, K.; Kim, S.W. Tumor targeting RGD conjugated bio-reducible polymer for VEGF siRNA expressing plasmid delivery. Biomaterials 2014, 35, 7543–7552. [Google Scholar] [CrossRef]
- Xiong, L.; Yu, M.; Cheng, M.; Zhang, M.; Zhang, X.; Xu, C.; Li, F. A photostable fluorescent probe for targeted imaging of tumour cells possessing integrin alpha(v)beta(3). Mol. Biosyst. 2009, 5, 241–243. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Kim, J.Y.; Han, J.H.; Bhuniya, S.; Sessler, J.L.; Kang, C.; Kim, J.S. Direct fluorescence monitoring of the delivery and cellular uptake of a cancer-targeted RGD peptide-appended naphthalimide theragnostic prodrug. J. Am. Chem. Soc. 2012, 134, 12668–12674. [Google Scholar] [CrossRef]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Temp. (°C) | Solvent | Base | Time (h) | Ratio (%) * 10 | Ratio (%) * 14: 9: JH-VII-139-1 | Isolated Yield for JH-VII-139-1 (%) |
|---|---|---|---|---|---|---|
| 85 | DME/H2O | K2CO3 | 24 | 0 | 40: 57: 3 | ND |
| 120 | DME/H2O | Cs2CO3 | 24 | 0 | 12: 80: 8 | ND |
| 120 (MW) | dioxane | K2CO3 | 2 | 96 | 0: 4: 0 | 0 |
| 140 (MW) | dioxane | K2CO3 | 1 | 90 | 0: 10: 0 | 0 |
| 85 | DME/H2O | K2CO3 | 24 | 0 | 22: 64: 14 | 8 |
| 85 (MW) | dioxane/H2O | K2CO3 | 2 | 0 | 0: 45: 55 | 50 |
| 100 (MW) | dioxane/H2O | K2CO3 | 2 | 0 | 0: 82: 18 | 17 |
| 60 (MW) | dioxane/H2O | K2CO3 | 6 | 30 | 0: 4: 66 | 59 |
| 70 (MW) | dioxane/H2O | K2CO3 | 2 | 0 | 0: 12: 88 | 80 |
| Compound | IC50 (nΜ) for SRPK1 |
|---|---|
| JH-VII-139-1 | 32 ± 0.5 |
| geo75 | 35 ± 1.1 |
| geo77 | 30 ± 1.7 |
| geo85 | 42 ± 1.9 |
| Compound (μΜ) | HeLa | K562 | MDA-MB-231 | MCF7 | |
|---|---|---|---|---|---|
| JH-VII-139-1 | GI50 | 8 ± 0.2 | 8 ± 0.5 | 11 ± 0.3 | 9 ± 0.2 |
| TGI | 19 ± 0.2 | 15 ± 0.5 | 21 ± 0.4 | 19 ± 0.3 | |
| IC50 | 14 ± 0.8 | 13 ± 0.3 | 18 ± 0.5 | 8 ± 0.6 | |
| geo75 | GI50 TGI IC50 | 5 ± 0.4 11 ± 0.3 11 ± 0.3 | 5 ± 0.3 10 ± 0.3 12 ± 0.2 | 8 ± 0.2 16 ± 0.3 11 ± 0.4 | 5 ± 0.5 10 ± 0.5 4 ± 0.5 |
| geo77 | GI50 | 5 ± 0.2 | 12 ± 0.9 | 8 ± 0.5 | 4 ± 0.4 |
| TGI | 11 ± 0.3 | 23 ± 0.5 | 15 ± 0.7 | 9 ± 0.4 | |
| IC50 | 9 ± 0.2 | 17 ± 0.5 | 10 ± 0.3 | 3 ± 0.4 | |
| geo85 | GI50 TGI IC50 | >100 >100 >100 | >100 >100 >100 | >100 >100 >100 | 33 ± 0.9 64 ± 0.9 45 ± 1.0 |
| c(RGDyK) | GI50 | 79 ± 2.2 | >100 | 80 ± 1.2 | >100 |
| TGI | >100 | >100 | >100 | >100 | |
| IC50 | >100 | >100 | >100 | >100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leonidis, G.; Koukiali, A.; Sigala, I.; Tsimaratou, K.; Beis, D.; Giannakouros, T.; Nikolakaki, E.; Sarli, V. Synthesis and Anti-Angiogenic Activity of Novel c(RGDyK) Peptide-Based JH-VII-139-1 Conjugates. Pharmaceutics 2023, 15, 381. https://doi.org/10.3390/pharmaceutics15020381
Leonidis G, Koukiali A, Sigala I, Tsimaratou K, Beis D, Giannakouros T, Nikolakaki E, Sarli V. Synthesis and Anti-Angiogenic Activity of Novel c(RGDyK) Peptide-Based JH-VII-139-1 Conjugates. Pharmaceutics. 2023; 15(2):381. https://doi.org/10.3390/pharmaceutics15020381
Chicago/Turabian StyleLeonidis, George, Anastasia Koukiali, Ioanna Sigala, Katerina Tsimaratou, Dimitris Beis, Thomas Giannakouros, Eleni Nikolakaki, and Vasiliki Sarli. 2023. "Synthesis and Anti-Angiogenic Activity of Novel c(RGDyK) Peptide-Based JH-VII-139-1 Conjugates" Pharmaceutics 15, no. 2: 381. https://doi.org/10.3390/pharmaceutics15020381
APA StyleLeonidis, G., Koukiali, A., Sigala, I., Tsimaratou, K., Beis, D., Giannakouros, T., Nikolakaki, E., & Sarli, V. (2023). Synthesis and Anti-Angiogenic Activity of Novel c(RGDyK) Peptide-Based JH-VII-139-1 Conjugates. Pharmaceutics, 15(2), 381. https://doi.org/10.3390/pharmaceutics15020381