
Optimisation of AAV-NDI1 Significantly Enhances Its Therapeutic Value for Correcting Retinal Mitochondrial Dysfunction

 ,
,  ,
,  , ,
, ,
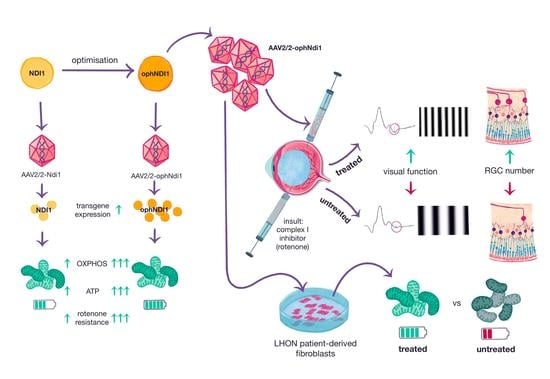
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Codon Optimisation
2.2. Predictions of Immunogenic Codons
2.3. Vector Construction
2.4. Cell Culture
2.5. Culture of Primary Human Dermal Fibroblasts
2.6. NADH Oxidation Assay
2.7. ROS Assay
2.8. Seahorse Mitochondrial Metabolism Assays
2.9. Adeno-Associated Virus (AAV)
2.10. Intravitreal Injection
2.11. RT-qPCR Analysis
2.12. Optokinetic (OKR) Analysis
2.13. Photopic Negative Response
2.14. Histological Analysis
2.15. Statistical Analysis
3. Results
3.1. Optimisation of NDI1
3.2. ophNdi1 Functions More Efficiently Than Ndi1 In Vitro
3.2.1. Mitochondrial Localisation and Expression of ophNdi1
3.2.2. OCR
3.2.3. ATP
3.2.4. Rotenone Resistance
3.2.5. NADH Oxidation and ROS
3.3. ophNdi1 Provides Functional Benefit and Preserves RGCs In Vivo
3.3.1. mRNA Expression In Vivo
3.3.2. Optokinetic Response
3.3.3. Photopic Negative Response
3.3.4. RGC Preservation
3.4. AAV-ophNdi1 Provides Benefit in Complex I Deficient LHON Patient Cells
4. Discussion
5. Patent
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Meyer, J.G.; Garcia, T.Y.; Schilling, B.; Gibson, B.W.; Lamba, D.A. Proteome and Secretome Dynamics of Human Retinal Pigment Epithelium in Response to Reactive Oxygen Species. Sci. Rep. 2019, 9, 15440. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-L.; Lin, T.-Y.; Chen, P.-L.; Guo, T.-N.; Huang, S.-Y.; Chen, C.-H.; Lin, C.-H.; Chan, C.-C. Mitochondrial Function and Parkinson’s Disease: From the Perspective of the Electron Transport Chain. Front. Mol. Neurosci. 2021, 14, 797833. [Google Scholar] [CrossRef]
- Damiano, M.; Galvan, L.; Déglon, N.; Brouillet, E. Mitochondria in Huntington’s disease. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2010, 1802, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Maloney, D.M.; Chadderton, N.; Millington-Ward, S.; Palfi, A.; Shortall, C.; O’Byrne, J.J.; Cassidy, L.; Keegan, D.; Humphries, P.; Kenna, P.; et al. Optimized OPA1 Isoforms 1 and 7 Provide Therapeutic Benefit in Models of Mitochondrial Dysfunction. Front. Neurosci. 2020, 14, 571479. [Google Scholar] [CrossRef]
- Strachan, E.L.; Mac White-Begg, D.; Crean, J.; Reynolds, A.L.; Kennedy, B.N.; O’Sullivan, N.C. The Role of Mitochondria in Optic Atrophy With Autosomal Inheritance. Front. Neurosci. 2021, 15, 784987. [Google Scholar] [CrossRef] [PubMed]
- Man, P.; Griffiths, P.; Brown, D.; Howell, N.; Turnbull, D.; Chinnery, P. The Epidemiology of Leber Hereditary Optic Neuropathy in the North East of England. Am. J. Hum. Genet. 2003, 72, 333–339. [Google Scholar] [CrossRef]
- Abu-Amero, K.K.; Morales, J.; Bosley, T.M. Mitochondrial Abnormalities in Patients with Primary Open-Angle Glaucoma. Investig. Opthalmol. Vis. Sci. 2006, 47, 2533–2541. [Google Scholar] [CrossRef]
- Dammak, A.; Huete-Toral, F.; Carpena-Torres, C.; Martin-Gil, A.; Pastrana, C.; Carracedo, G. From Oxidative Stress to Inflammation in the Posterior Ocular Diseases: Diagnosis and Treatment. Pharmaceutics 2021, 13, 1376. [Google Scholar] [CrossRef]
- Sasaki, M.; Ozawa, Y.; Kurihara, T.; Kubota, S.; Yuki, K.; Noda, K.; Kobayashi, S.; Ishida, S.; Tsubota, K. Neurodegenerative influence of oxidative stress in the retina of a murine model of diabetes. Diabetologia 2010, 53, 971–979. [Google Scholar] [CrossRef]
- Kang, Q.; Yang, C. Oxidative stress and diabetic retinopathy: Molecular mechanisms, pathogenetic role and therapeutic implications. Redox Biol. 2020, 37, 101799. [Google Scholar] [CrossRef]
- Ferrington, D.A.; Ebeling, M.C.; Kapphahn, R.J.; Terluk, M.R.; Fisher, C.R.; Polanco, J.R.; Roehrich, H.; Leary, M.M.; Geng, Z.; Dutton, J.R.; et al. Altered bioenergetics and enhanced resistance to oxidative stress in human retinal pigment epithelial cells from donors with age-related macular degeneration. Redox Biol. 2017, 13, 255–265. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Uusitalo, H.; Blasiak, J.; Felszeghy, S.; Kannan, R.; Kauppinen, A.; Salminen, A.; Sinha, D.; Ferrington, D. Mechanisms of mitochondrial dysfunction and their impact on age-related macular degeneration. Prog. Retin. Eye Res. 2020, 79, 100858. [Google Scholar] [CrossRef]
- Morgan, J.E.; Uchida, H.; Caprioli, J. Retinal ganglion cell death in experimental glaucoma. Br. J. Ophthalmol. 2000, 84, 303–310. [Google Scholar] [CrossRef]
- Lenaers, G.; Neutzner, A.; Le Dantec, Y.; Jüschke, C.; Xiao, T.; Decembrini, S.; Swirski, S.; Kieninger, S.; Agca, C.; Kim, U.S.; et al. Dominant optic atrophy: Culprit mitochondria in the optic nerve. Prog. Retin. Eye Res. 2021, 83, 100935. [Google Scholar] [CrossRef]
- Kim, U.S.; Mahroo, O.A.; Mollon, J.D.; Yu-Wai-Man, P. Retinal Ganglion Cells-Diversity of Cell Types and Clinical Relevance. Front. Neurol. 2021, 12, 661938. [Google Scholar] [CrossRef]
- Andrews, R.M.; Griffiths, P.G.; Johnson, M.A.; Turnbull, D.M. Histochemical localisation of mitochondrial enzyme activity in human optic nerve and retina. Br. J. Ophthalmol. 1999, 83, 231–235. [Google Scholar] [CrossRef]
- Bristow, E.A.; Griffiths, P.G.; Andrews, R.M.; Johnson, M.A.; Turnbull, D.M. The Distribution of Mitochondrial Activity in Relation to Optic Nerve Structure. Arch. Ophthalmol. 2002, 120, 791–796. [Google Scholar] [CrossRef]
- Fraser, J.A.; Biousse, V.; Newman, N.J. The Neuro-ophthalmology of Mitochondrial Disease. J. Surv. Ophthalmol. 2010, 55, 299–334. [Google Scholar] [CrossRef]
- Catalani, E.; Cervia, D. Diabetic retinopathy: A matter of retinal ganglion cell homeostasis. Neural Regen. Res. 2020, 15, 1253–1254. [Google Scholar] [CrossRef]
- Yang, T.-C.; Yarmishyn, A.A.; Yang, Y.-P.; Lu, P.-C.; Chou, S.-J.; Wang, M.-L.; Lin, T.-C.; Hwang, D.-K.; Chou, Y.-B.; Chen, S.-J.; et al. Mitochondrial transport mediates survival of retinal ganglion cells in affected LHON patients. Hum. Mol. Genet. 2020, 29, 1454–1464. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Chen, D.; Tan, Q.; Xia, X.; Jiang, H.; Jiang, J. Outcome of Selective Laser Trabeculoplasty in Young Patients with Primary Open-Angle Glaucoma and Ocular Hypertension. J. Ophthalmol. 2020, 2020, 5742832. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, T.; Nørby, S.; Schwartz, M.; Saillard, J.; Magalhães, P.J.; Leroy, D.; Kann, E.C.; Duno, M. Prevalence and Genetics of Leber Hereditary Optic Neuropathy in the Danish Population. Investig. Opthalmol. Vis. Sci. 2016, 57, 1370–1375. [Google Scholar] [CrossRef] [PubMed]
- Puomila, A.; Hämäläinen, P.; Kivioja, S.; Savontaus, M.-L.; Koivumäki, S.; Huoponen, K.; Nikoskelainen, E. Epidemiology and penetrance of Leber hereditary optic neuropathy in Finland. Eur. J. Hum. Genet. 2007, 15, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C.; Lott, M.T. Leber Hereditary Optic Neuropathy: Exemplar of an mtDNA Disease. Handb. Exp. Pharmacol. 2017, 240, 339–376. [Google Scholar] [CrossRef]
- Theodorou-Kanakari, A.; Karampitianis, S.; Karageorgou, V.; Kampourelli, E.; Kapasakis, E.; Theodossiadis, P.; Chatziralli, I. Current and Emerging Treatment Modalities for Leber’s Hereditary Optic Neuropathy: A Review of the Literature. Adv. Ther. 2018, 35, 1510–1518. [Google Scholar] [CrossRef]
- Manickam, A.H.; Michael, M.J.; Ramasamy, S. Mitochondrial genetics and therapeutic overview of Leber’s hereditary optic neuropathy. Indian J. Ophthalmol. 2017, 65, 1087–1092. [Google Scholar]
- Toomes, C.; Marchbank, N.J.; Mackey, D.A.; Craig, J.E.; Newbury-Ecob, R.A.; Bennett, C.P.; Vize, C.J.; Desai, S.P.; Black, G.C.; Patel, N.; et al. Spectrum, frequency and penetrance of OPA1 mutations in dominant optic atrophy. Hum. Mol. Genet. 2001, 10, 1369–1378. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P.; Newman, N.J.; Carelli, V.; Moster, M.L.; Biousse, V.; Sadun, A.A.; Klopstock, T.; Vignal-Clermont, C.; Sergott, R.C.; Rudolph, G.; et al. Bilateral visual improvement with unilateral gene therapy injection for Leber hereditary optic neuropathy. Sci. Transl. Med. 2020, 12, eaaz7423. [Google Scholar] [CrossRef]
- Almind, G.J.; Ek, J.; Rosenberg, T.; Eiberg, H.; Larsen, M.; LuCamp, L.; Brøndum-Nielsen, K.; Grønskov, K. Dominant optic atrophy in Denmark—Report of 15 novel mutations in OPA1, using a strategy with a detection rate of 90%. BMC Med. Genet. 2012, 13, 65. [Google Scholar] [CrossRef]
- Altanbyek, V.; Cha, S.-J.; Kang, G.-U.; Im, D.S.; Lee, S.; Kim, H.-J.; Kim, K. Imbalance of mitochondrial dynamics in Drosophila models of amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2016, 481, 259–264. [Google Scholar] [CrossRef]
- Ge, Y.; Shi, X.; Boopathy, S.; McDonald, J.; Smith, A.W.; Chao, L.H. Two forms of Opa1 cooperate to complete fusion of the mitochondrial inner-membrane. eLife 2020, 9, e50973. [Google Scholar] [CrossRef] [PubMed]
- Gilkerson, R.; De La Torre, P.; Vallier, S.S. Mitochondrial OMA1 and OPA1 as Gatekeepers of Organellar Structure/Function and Cellular Stress Response. Front. Cell Dev. Biol. 2021, 9, 626117. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Wang, J.; Li, Y.; Jiang, B. Prevalence of primary open angle glaucoma in the last 20 years: A meta-analysis and systematic review. Sci. Rep. 2021, 11, 13762. [Google Scholar] [CrossRef] [PubMed]
- Allison, K.; Patel, D.; Alabi, O. Epidemiology of Glaucoma: The Past, Present, and Predictions for the Future. Cureus 2020, 12, e11686. [Google Scholar] [CrossRef] [PubMed]
- Osborne, N.N.; Núñez-Álvarez, C.; Joglar, B.; del Olmo-Aguado, S. Glaucoma: Focus on mitochondria in relation to pathogenesis and neuroprotection. Eur. J. Pharmacol. 2016, 787, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Van Bergen, N.J.; Crowston, J.G.; Craig, J.E.; Burdon, K.P.; Kearns, L.S.; Sharma, S.; Hewitt, A.W.; Mackey, D.A.; Trounce, I.A. Measurement of Systemic Mitochondrial Function in Advanced Primary Open-Angle Glaucoma and Leber Hereditary Optic Neuropathy. PLoS ONE 2015, 10, e0140919. [Google Scholar] [CrossRef]
- Stenton, S.L.; Sheremet, N.L.; Catarino, C.B.; Andreeva, N.A.; Assouline, Z.; Barboni, P.; Barel, O.; Berutti, R.; Bychkov, I.; Caporali, L.; et al. Impaired complex I repair causes recessive Leber’s hereditary optic neuropathy. J. Clin. Investig. 2021, 131, e138267. [Google Scholar] [CrossRef]
- Marella, M.; Seo, B.B.; Thomas, B.B.; Matsuno-Yagi, A.; Yagi, T. Successful Amelioration of Mitochondrial Optic Neuropathy Using the Yeast NDI1 Gene in a Rat Animal Model. PLoS ONE 2010, 5, e11472. [Google Scholar] [CrossRef]
- Chadderton, N.; Palfi, A.; Millington-Ward, S.; Gobbo, O.; Overlack, N.; Carrigan, M.; O’Reilly, M.; Campbell, M.; Ehrhardt, C.; Wolfrum, U.; et al. Intravitreal delivery of AAV-NDI1 provides functional benefit in a murine model of Leber hereditary optic neuropathy. Eur. J. Hum. Genet. 2012, 21, 62–68. [Google Scholar] [CrossRef]
- Talla, V.; Koilkonda, R.; Guy, J. Gene Therapy with Single-Subunit Yeast NADH-Ubiquinone Oxidoreductase (NDI1) Improves the Visual Function in Experimental Autoimmune Encephalomyelitis (EAE) Mice Model of Multiple Sclerosis (MS). Mol. Neurobiol. 2020, 57, 1952–1965. [Google Scholar] [CrossRef]
- Barber-Singh, J.; Seo, B.B.; Matsuno-Yagi, A.; Yagi, T. Protective Role of rAAV-NDI1, Serotype 5, in an Acute MPTP Mouse Parkinson’s Model. Park. Dis. 2011, 2011, 428370. [Google Scholar] [CrossRef] [PubMed]
- Colella, P.; Ronzitti, G.; Mingozzi, F. Emerging Issues in AAV-Mediated In Vivo Gene Therapy. Mol. Ther. Methods Clin. Dev. 2017, 8, 87–104. [Google Scholar] [CrossRef] [PubMed]
- Fleri, W.; Paul, S.; Dhanda, S.K.; Mahajan, S.; Xu, X.; Peters, B.; Sette, A. The Immune Epitope Database and Analysis Resource in Epitope Discovery and Synthetic Vaccine Design. Front. Immunol. 2017, 8, 278. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Galarza, F.F.; Christmas, S.; Middleton, D.; Jones, A.R. Allele frequency net: A database and online repository for immune gene frequencies in worldwide populations. Nucleic Acids Res. 2011, 39, D913–D919. [Google Scholar] [CrossRef] [PubMed]
- Henikoff, S.; Henikoff, J.G. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA 1992, 89, 10915–10919. [Google Scholar] [CrossRef] [PubMed]
- Hauck, B.; Murphy, S.L.; Smith, P.H.; Qu, G.; Liu, X.; Zelenaia, O.; Mingozzi, F.; Sommer, J.M.; High, K.A.; Wright, J.F. Undetectable Transcription of cap in a Clinical AAV Vector: Implications for Preformed Capsid in Immune Responses. Mol. Ther. 2009, 17, 144–152. [Google Scholar] [CrossRef]
- Palfi, A.; Yesmambetov, A.; Millington-Ward, S.; Shortall, C.; Humphries, P.; Kenna, P.F.; Chadderton, N.; Farrar, G.J. AAV-Delivered Tulp1Supplementation Therapy Targeting Photoreceptors Provides Minimal Benefit in Tulp1-/- Retinas. Front. Neurosci. 2020, 14, 891. [Google Scholar] [CrossRef]
- Spinazzi, M.; Casarin, A.; Pertegato, V.; Salviati, L.; Angelini, C. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat. Protoc. 2012, 7, 1235–1246. [Google Scholar] [CrossRef]
- Finnegan, L.K.; Chadderton, N.; Kenna, P.F.; Palfi, A.; Carty, M.; Bowie, A.G.; Millington-Ward, S.; Farrar, G.J. SARM1 Ablation Is Protective and Preserves Spatial Vision in an In Vivo Mouse Model of Retinal Ganglion Cell Degeneration. Int. J. Mol. Sci. 2022, 23, 1606. [Google Scholar] [CrossRef]
- O’Reilly, M.; Palfi, A.; Chadderton, N.; Millington-Ward, S.; Ader, M.; Cronin, T.; Tuohy, T.; Auricchio, A.; Hildinger, M.; Tivnan, A.; et al. RNA Interference-Mediated Suppression and Replacement of Human Rhodopsin In Vivo. Am. J. Hum. Genet. 2007, 81, 127–135. [Google Scholar] [CrossRef]
- Bennicelli, J.; Wright, J.F.; Komaromy, A.; Jacobs, J.B.; Hauck, B.; Zelenaia, O.; Mingozzi, F.; Hui, D.; Chung, D.; Rex, T.S.; et al. Reversal of Blindness in Animal Models of Leber Congenital Amaurosis Using Optimized AAV2-mediated Gene Transfer. Mol. Ther. 2008, 16, 458–465. [Google Scholar] [CrossRef]
- Rohr, U.-P.; Wulf, M.-A.; Stahn, S.; Steidl, U.; Haas, R.; Kronenwett, R. Fast and reliable titration of recombinant adeno-associated virus type-2 using quantitative real-time PCR. J. Virol. Methods 2002, 106, 81–88. [Google Scholar] [CrossRef]
- Millington-Ward, S.; Chadderton, N.; O’Reilly, M.; Palfi, A.; Goldmann, T.; Kilty, C.; Humphries, M.; Wolfrum, U.; Bennett, J.; Humphries, P.; et al. Suppression and Replacement Gene Therapy for Autosomal Dominant Disease in a Murine Model of Dominant Retinitis Pigmentosa. Mol. Ther. 2011, 19, 642–649. [Google Scholar] [CrossRef]
- Palfi, A.; Chadderton, N.; O’Reilly, M.; Nagel-Wolfrum, K.; Wolfrum, U.; Bennett, J.; Humphries, P.; Kenna, P.F.; Millington-Ward, S.; Farrar, G.J. Efficient gene delivery to photoreceptors using AAV2/rh10 and rescue of the Rho(-/-) mouse. Mol. Ther. Methods Clin. Dev. 2015, 2, 15016. [Google Scholar] [CrossRef]
- Chadderton, N.; Millington-Ward, S.; Palfi, A.; O’Reilly, M.; Tuohy, G.; Humphries, M.M.; Li, T.; Humphries, P.; Kenna, P.F.; Farrar, G.J. Improved Retinal Function in a Mouse Model of Dominant Retinitis Pigmentosa Following AAV-delivered Gene Therapy. Mol. Ther. 2009, 17, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Prusky, G.T.; Alam, N.M.; Beekman, S.; Douglas, R.M. Rapid Quantification of Adult and Developing Mouse Spatial Vision Using a Virtual Optomotor System. Investig. Opthalmol. Vis. Sci. 2004, 45, 4611–4616. [Google Scholar] [CrossRef]
- Douglas, R.; ALAM, N.; Silver, B.; Mcgill, T.; Tschetter, W.; Prusky, G. Independent visual threshold measurements in the two eyes of freely moving rats and mice using a virtual-reality optokinetic system. Vis. Neurosci. 2005, 22, 677–684. [Google Scholar] [CrossRef]
- Palfi, A.; Hokamp, K.; Hauck, S.M.; Vencken, S.; Millington-Ward, S.; Chadderton, N.; Carrigan, M.; Kortvely, E.; Greene, C.M.; Kenna, P.F.; et al. microRNA regulatory circuits in a mouse model of inherited retinal degeneration. Sci. Rep. 2016, 6, 31431. [Google Scholar] [CrossRef]
- Nadal-Nicolás, F.M.; Jiménez-López, M.; Sobrado-Calvo, P.; Nieto-López, L.; Cánovas-Martínez, I.; Salinas-Navarro, M.; Vidal-Sanz, M.; Agudo, M. Brn3a as a Marker of Retinal Ganglion Cells: Qualitative and Quantitative Time Course Studies in Naive and Optic Nerve-Injured Retinas. Investig. Opthalmol. Vis. Sci. 2009, 50, 3860–3868. [Google Scholar] [CrossRef]
- Trost, A.; Motloch, K.; Bruckner, D.; Schroedl, F.; Bogner, B.; Kaser-Eichberger, A.; Runge, C.; Strohmaier, C.; Klein, B.; Aigner, L.; et al. Time-dependent retinal ganglion cell loss, microglial activation and blood-retina-barrier tightness in an acute model of ocular hypertension. Exp. Eye Res. 2015, 136, 59–71. [Google Scholar] [CrossRef]
- Williams, R.W.; Strom, R.C.; Rice, D.S.; Goldowitz, D. Genetic and Environmental Control of Variation in Retinal Ganglion Cell Number in Mice. J. Neurosci. 1996, 16, 7193–7205. [Google Scholar] [CrossRef] [PubMed]
- Millington-Ward, S.; Chadderton, N.; Berkeley, M.; Finnegan, L.K.; Hanlon, K.S.; Carrigan, M.; Humphries, P.; Kenna, P.F.; Palfi, A.; Farrar, G.J. Novel 199 base pair NEFH promoter drives expression in retinal ganglion cells. Sci. Rep. 2020, 10, 16515. [Google Scholar] [CrossRef] [PubMed]
- Farrar, G.J.; Millington-Ward, S.; Chadderton, N.; Carrigan, M.A.; Kenna, P. Variants of Yeast NDI1 Gene, and Uses Thereof in the Treatment of Disease Associated with Mitochondrial Dysfunction. US Patent US20150099798A1, 5 March 2019. [Google Scholar]
- Seo, B.B.; Kitajima-Ihara, T.; Chan, E.K.L.; Scheffler, I.E.; Matsuno-Yagi, A.; Yagi, T. Molecular remedy of complex I defects: Rotenone-insensitive internal NADH-quinone oxidoreductase of Saccharomyces cerevisiae mitochondria restores the NADH oxidase activity of complex I-deficient mammalian cells. Proc. Natl. Acad. Sci. USA 1998, 95, 9167–9171. [Google Scholar] [CrossRef]
- Seo, B.B.; Wang, J.; Flotte, T.R.; Yagi, T.; Matsuno-Yagi, A. Use of the NADH-Quinone Oxidoreductase (NDI1) Gene ofSaccharomyces cerevisiae as a Possible Cure for Complex I Defects in Human Cells. J. Biol. Chem. 2000, 275, 37774–37778. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Hájek, P.; Chomyn, A.; Chan, E.; Seo, B.B.; Matsuno-Yagi, A.; Yagi, T.; Attardi, G. Lack of complex I activity in human cells carrying a mutation in MtDNA-encoded ND4 subunit is corrected by the Saccharomyces cerevisiae NADH-quinone oxidoreductase (NDI1) gene. J. Biol. Chem. 2001, 276, 38808–38813. [Google Scholar] [CrossRef]
- Cronin-Furman, E.N.; Barber-Singh, J.; Bergquist, K.E.; Yagi, T.; Trimmer, P.A. Differential Effects of Yeast NADH Dehydrogenase (Ndi1) Expression on Mitochondrial Function and Inclusion Formation in a Cell Culture Model of Sporadic Parkinson’s Disease. Biomolecules 2019, 9, 119. [Google Scholar] [CrossRef]
- Fato, R.; Bergamini, C.; Bortolus, M.; Maniero, A.L.; Leoni, S.; Ohnishi, T.; Lenaz, G. Differential effects of mitochondrial Complex I inhibitors on production of reactive oxygen species. Biochim. Biophys. Acta 2009, 1787, 384–392. [Google Scholar] [CrossRef]
- Heinz, S.; Freyberger, A.; Lawrenz, B.; Schladt, L.; Schmuck, G.; Ellinger-Ziegelbauer, H. Mechanistic Investigations of the Mitochondrial Complex I Inhibitor Rotenone in the Context of Pharmacological and Safety Evaluation. Sci. Rep. 2017, 7, 45465. [Google Scholar] [CrossRef]
- Zhang, X.; Jones, D.; Gonzalez-Lima, F. Mouse model of optic neuropathy caused by mitochondrial complex I dysfunction. Neurosci. Lett. 2002, 326, 97–100. [Google Scholar] [CrossRef]
- Zhang, X.; Jones, D.; Gonzalez-Lima, F. Neurodegeneration Produced by Rotenone in the Mouse Retina: A Potential Model to Investigate Environmental Pesticide Contributions to Neurodegenerative Diseases. J. Toxicol. Environ. Health A 2006, 69, 1681–1697. [Google Scholar] [CrossRef]
- Hayworth, C.R.; Rojas, J.C.; Gonzalez-Lima, F. Transgenic Mice Expressing Cyan Fluorescent Protein as a Reporter Strain to Detect the Effects of Rotenone Toxicity on Retinal Ganglion Cells. J. Toxicol. Environ. Health A 2008, 71, 1582–1592. [Google Scholar] [CrossRef] [PubMed]
- Yu-Wai-Man, P.; Soiferman, D.; Moore, D.G.; Burté, F.; Saada, A. Evaluating the therapeutic potential of idebenone and related quinone analogues in Leber hereditary optic neuropathy. Mitochondrion 2017, 36, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Antony, P.M.A.; Kondratyeva, O.; Mommaerts, K.; Ostaszewski, M.; Sokolowska, K.; Baumuratov, A.S.; Longhino, L.; Poulain, J.F.; Grossmann, D.; Balling, R.; et al. Fibroblast mitochondria in idiopathic Parkinson’s disease display morphological changes and enhanced resistance to depolarization. Sci. Rep. 2020, 10, 1569. [Google Scholar] [CrossRef] [PubMed]
- Millington-Ward, S.; Chadderton, N.; Finnegan, L.K.; Post, I.J.; Carrigan, M.; Gardiner, T.; Peixoto, E.; Maloney, D.; Humphries, M.M.; Stitt, A.; et al. AAV-mediated gene therapy improving mitochondrial function provides benefit in age-related macular degeneration models. Clin. Transl. Med. 2022, 12, e952. [Google Scholar] [CrossRef]
- Marella, M.; Seo, B.B.; Nakamaru-Ogiso, E.; Greenamyre, J.T.; Matsuno-Yagi, A.; Yagi, T. Protection by the NDI1 gene against neurodegeneration in a rotenone rat model of Parkinson’s disease. PLoS ONE 2008, 3, e1433. [Google Scholar] [CrossRef]
- Shirley, J.L.; de Jong, Y.P.; Terhorst, C.; Herzog, R.W. Immune Responses to Viral Gene Therapy Vectors. Mol. Ther. 2020, 28, 709–722. [Google Scholar] [CrossRef]
- MacLachlan, T.K.; Lukason, M.; Collins, M.; Munger, R.; Isenberger, E.; Rogers, C.; Malatos, S.; DuFresne, E.; Morris, J.; Calcedo, R.; et al. Preclinical Safety Evaluation of AAV2-sFLT01—A Gene Therapy for Age-related Macular Degeneration. Mol. Ther. 2011, 19, 326–334. [Google Scholar] [CrossRef]
- Marangoni, D.; Bush, R.A.; Zeng, Y.; Wei, L.L.; Ziccardi, L.; Vijayasarathy, C.; Bartoe, J.T.; Palyada, K.; Santos, M.; Hiriyanna, S.; et al. Ocular and systemic safety of a recombinant AAV8 vector for X-linked retinoschisis gene therapy: GLP studies in rabbits and Rs1-KO mice. Mol. Ther. Methods Clin. Dev. 2014, 5, 16011. [Google Scholar] [CrossRef]
- Ye, G.J.; Budzynski, E.; Sonnentag, P.; Nork, T.M.; Miller, P.E.; Sharma, A.K.; Ver Hoeve, J.N.; Smith, L.M.; Arndt, T.; Calcedo, R.; et al. Safety and Biodistribution Evaluation in Cynomolgus Macaques of rAAV2tYF-PR1.7-hCNGB3, a Recombinant AAV Vector for Treatment of Achromatopsia. Hum. Gene Ther. Clin. Dev. 2016, 27, 37–48. [Google Scholar] [CrossRef]
- Morales, L.; Gambhir, Y.; Bennett, J.; Stedman, H.H. Broader Implications of Progressive Liver Dysfunction and Lethal Sepsis in Two Boys following Systemic High-Dose AAV. Mol. Ther. 2020, 28, 1753–1755. [Google Scholar] [CrossRef]
- Adverum Biotechnologies, Inc. Update on ADVM-022 and INFINITY Trial 07/22/201. Available online: https://investors.adverum.com/news/news-details/2021/Adverum-Provides-Update-on-ADVM-022-and-the-INFINITY-Trial-in-Patients-with-Diabetic-Macular-Edema/default.aspx (accessed on 20 June 2022).
- Shieh, P.; Kuntz, N.; Dowling, J.; Müller-Felber, W.; Bönneman, C.; Foley, D.; Seferian, A.; Servais, L.; Lawlor, M.; Noursalehi, M.; et al. OP018: ASPIRO gene therapy trial in X-Linked Myotubular Myopathy (XLMTM): Update on preliminary efficacy and safety findings. Genet. Med. 2022, 24, S350. [Google Scholar] [CrossRef]
- Molnar, M.J.; Kovacs, G.G. Mitochondrial diseases. Handb. Clin. Neurol. 2017, 145, 147–155. [Google Scholar] [PubMed]
- De Barcelos, I.P.; Troxell, R.M.; Graves, J.S. Mitochondrial Dysfunction and Multiple Sclerosis. Biology 2019, 8, 37. [Google Scholar] [CrossRef] [PubMed]
- Bostick, B.; Ghosh, A.; Yue, Y.; Long, C.; Duan, D. Systemic AAV-9 transduction in mice is influenced by animal age but not by the route of administration. Gene Ther. 2007, 14, 1605–1609. [Google Scholar] [CrossRef] [PubMed]
- Foust, K.D.; Nurre, E.; Montgomery, C.L.; Hernandez, A.; Chan, C.M.; Kaspar, B.K. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat. Biotechnol. 2009, 27, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Al-Zaidy, S.; Shell, R.; Arnold, W.D.; Rodino-Klapac, L.R.; Prior, T.W.; Lowes, L.; Alfano, L.; Berry, K.; Church, K.; et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1713–1722. [Google Scholar] [CrossRef]
- Dalkara, D.; Byrne, L.C.; Lee, T.; Hoffmann, N.; Schaffer, D.V.; Flannery, J. Enhanced gene delivery to the neonatal retina through systemic administration of tyrosine-mutated AAV9. Gene Ther. 2012, 19, 176–181. [Google Scholar] [CrossRef]
- Hanlon, K.S.; Meltzer, J.C.; Buzhdygan, T.; Cheng, M.J.; Sena-Esteves, M.; Bennett, R.E.; Sullivan, T.P.; Razmpour, R.; Gong, Y.; Ng, C.; et al. Selection of an Efficient AAV Vector for Robust CNS Transgene Expression. Mol. Ther. Methods Clin. Dev. 2019, 15, 320–332. [Google Scholar] [CrossRef]
- Chan, K.Y.; Jang, M.J.; Yoo, B.B.; Greenbaum, A.; Ravi, N.; Wu, W.-L.; Sanchez-Guardado, L.; Lois, C.; Mazmanian, S.K.; Deverman, B.E.; et al. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat. Neurosci. 2017, 20, 1172–1179. [Google Scholar] [CrossRef]
- Dayton, R.D.; Grames, M.S.; Klein, R.L. More expansive gene transfer to the rat CNS: AAV PHP.EB vector dose–response and comparison to AAV PHP.B. Gene Ther. 2018, 25, 392–400. [Google Scholar] [CrossRef]
- Reynaud-Dulaurier, R.; Benegiamo, G.; Marrocco, E.; Al-Tannir, R.; Surace, E.M.; Auwerx, J.; Decressac, M. Gene replacement therapy provides benefit in an adult mouse model of Leigh syndrome. Brain 2020, 143, 1686–1696. [Google Scholar] [CrossRef] [PubMed]
- Palfi, A.; Chadderton, N.; Millington-Ward, S.; Post, I.; Humphries, P.; Kenna, P.F.; Farrar, G.J. AAV-PHP.eB transduces both the inner and outer retina with high efficacy in mice. Mol. Ther. Methods Clin. Dev. 2022, 25, 236–249. [Google Scholar] [CrossRef] [PubMed]
- Goertsen, D.; Flytzanis, N.C.; Goeden, N.; Chuapoco, M.R.; Cummins, A.; Chen, Y.; Fan, Y.; Zhang, Q.; Sharma, J.; Duan, Y.; et al. AAV capsid variants with brain-wide transgene expression and decreased liver targeting after intravenous delivery in mouse and marmoset. Nat. Neurosci. 2022, 25, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Amore, G.; Romagnoli, M.; Carbonelli, M.; Barboni, P.; Carelli, V.; La Morgia, C. Therapeutic Options in Hereditary Optic Neuropathies. Drugs 2021, 81, 57–86. [Google Scholar] [CrossRef]
- Zuccarelli, M.; Vella-Szijj, J.; Serracino-Inglott, A.; Borg, J.-J. Treatment of Leber’s hereditary optic neuropathy: An overview of recent developments. Eur. J. Ophthalmol. 2020, 30, 1220–1227. [Google Scholar] [CrossRef]
- Sadun, A.; Ammar, M.; Chahal, J.; Karanjia, R. Treatment of Leber’s Hereditary Optic Neuropathy. Curr. Pharm. Des. 2017, 23, 624–628. [Google Scholar] [CrossRef]
- Klopstock, T.; Yu-Wai-Man, P.; Dimitriadis, K.; Rouleau, J.; Heck, S.; Bailie, M.; Atawan, A.; Chattopadhyay, S.; Schubert, M.; Garip, A.; et al. A randomized placebo-controlled trial of idebenone in Leber’s hereditary optic neuropathy. Brain 2011, 134, 2677–2686. [Google Scholar] [CrossRef]
- Klopstock, T.; Metz, G.; Yu-Wai-Man, P.; Büchner, B.; Gallenmüller, C.; Bailie, M.; Nwali, N.; Griffiths, P.G.; von Livonius, B.; Reznicek, L.; et al. Persistence of the treatment effect of idebenone in Leber’s hereditary optic neuropathy. Brain 2013, 136, e230. [Google Scholar] [CrossRef]
- Catarino, C.B.; von Livonius, B.; Priglinger, C.; Banik, R.; Matloob, S.; Tamhankar, M.A.; Castillo, L.; Friedburg, C.; Halfpenny, C.A.; Lincoln, J.A.; et al. Real-World Clinical Experience with Idebenone in the Treatment of Leber Hereditary Optic Neuropathy. J. Neuro Ophthalmol. 2020, 40, 558–565. [Google Scholar] [CrossRef]
- Newman, N.J.; Yu-Wai-Man, P.; Carelli, V.; Moster, M.L.; Biousse, V.; Vignal-Clermont, C.; Sergott, R.C.; Klopstock, T.; Sadun, A.A.; Barboni, P.; et al. Efficacy and Safety of Intravitreal Gene Therapy for Leber Hereditary Optic Neuropathy Treated within 6 Months of Disease Onset. Ophthalmology 2021, 128, 649–660. [Google Scholar] [CrossRef]
- Chen, J.J.; Bhatti, M.T. Gene Therapy for Leber Hereditary Optic Neuropathy: Is Vision Truly RESCUED? Ophthalmology 2021, 128, P661–P662. [Google Scholar] [CrossRef] [PubMed]
- Yu-Wai-Man, P.; Newman, N.J.; Carelli, V.; La Morgia, C.; Biousse, V.; Bandello, F.M.; Clermont, C.V.; Campillo, L.C.; Leruez, S.; Moster, M.L.; et al. Natural history of patients with Leber hereditary optic neuropathy—Results from the REALITY study. Eye 2022, 36, 818–826. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chadderton, N.; Palfi, A.; Maloney, D.M.; Carrigan, M.; Finnegan, L.K.; Hanlon, K.S.; Shortall, C.; O’Reilly, M.; Humphries, P.; Cassidy, L.; et al. Optimisation of AAV-NDI1 Significantly Enhances Its Therapeutic Value for Correcting Retinal Mitochondrial Dysfunction. Pharmaceutics 2023, 15, 322. https://doi.org/10.3390/pharmaceutics15020322
Chadderton N, Palfi A, Maloney DM, Carrigan M, Finnegan LK, Hanlon KS, Shortall C, O’Reilly M, Humphries P, Cassidy L, et al. Optimisation of AAV-NDI1 Significantly Enhances Its Therapeutic Value for Correcting Retinal Mitochondrial Dysfunction. Pharmaceutics. 2023; 15(2):322. https://doi.org/10.3390/pharmaceutics15020322
Chicago/Turabian StyleChadderton, Naomi, Arpad Palfi, Daniel M. Maloney, Matthew Carrigan, Laura K. Finnegan, Killian S. Hanlon, Ciara Shortall, Mary O’Reilly, Peter Humphries, Lorraine Cassidy, and et al. 2023. "Optimisation of AAV-NDI1 Significantly Enhances Its Therapeutic Value for Correcting Retinal Mitochondrial Dysfunction" Pharmaceutics 15, no. 2: 322. https://doi.org/10.3390/pharmaceutics15020322
APA StyleChadderton, N., Palfi, A., Maloney, D. M., Carrigan, M., Finnegan, L. K., Hanlon, K. S., Shortall, C., O’Reilly, M., Humphries, P., Cassidy, L., Kenna, P. F., Millington-Ward, S., & Farrar, G. J. (2023). Optimisation of AAV-NDI1 Significantly Enhances Its Therapeutic Value for Correcting Retinal Mitochondrial Dysfunction. Pharmaceutics, 15(2), 322. https://doi.org/10.3390/pharmaceutics15020322