
Physiologically Based Biopharmaceutics Model (PBBM) of Minimally Absorbed Locally Acting Drugs in the Gastrointestinal Tract—Case Study: Tenapanor

 , ,
, ,
Abstract
:1. Introduction
2. Methods
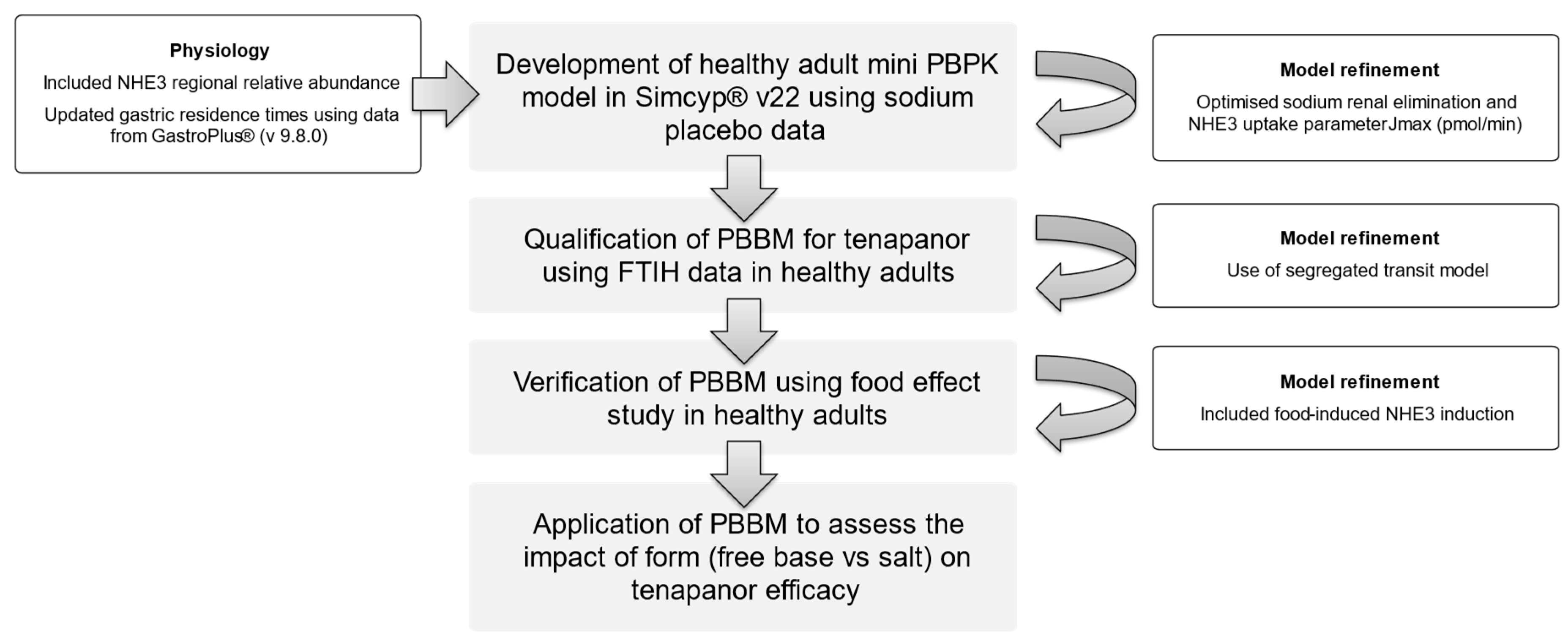
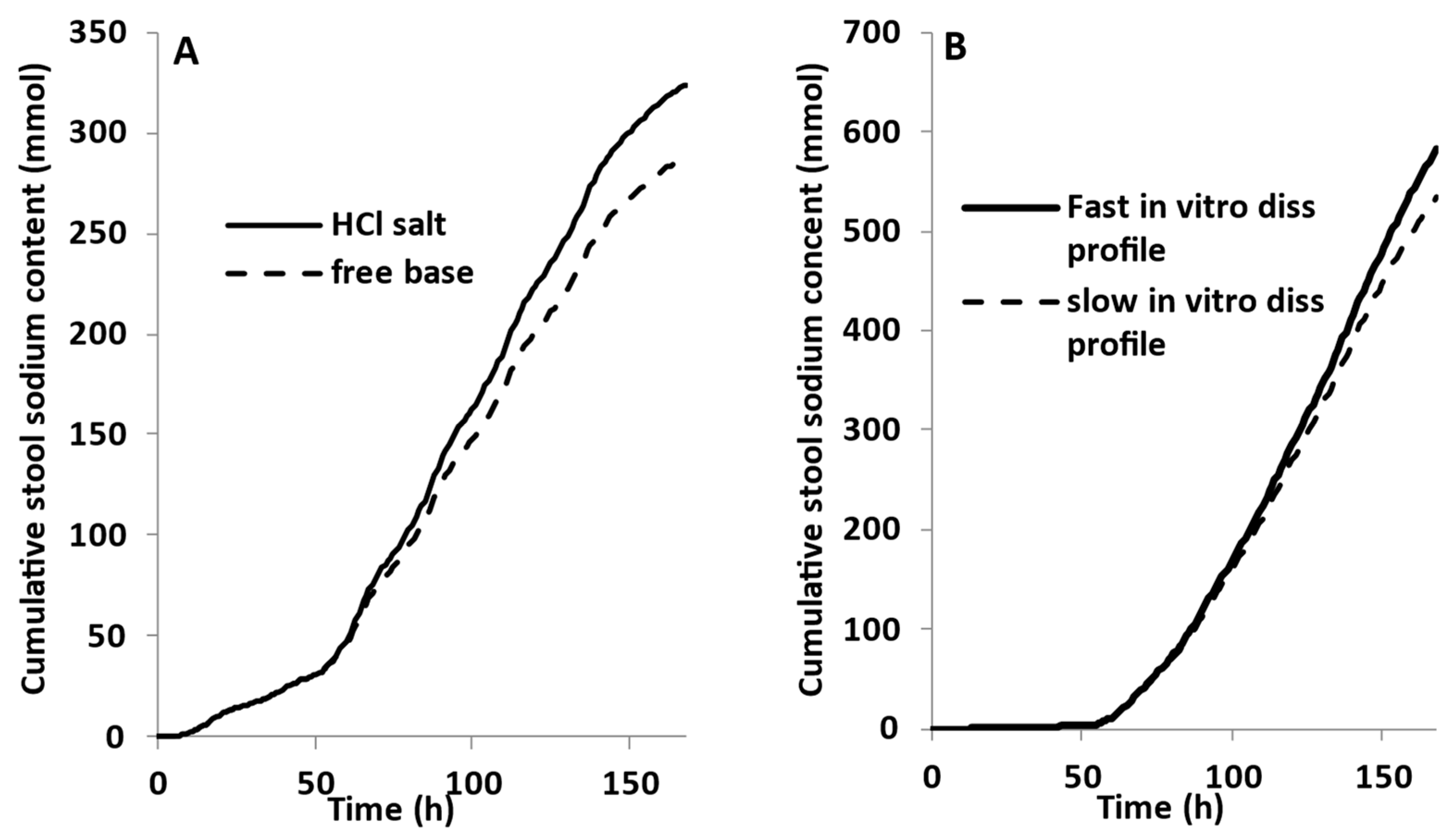
2.1. PBBM Development
2.2. Sodium, Tenapanor, and Food Effect Modelling
2.3. Clinical Studies
- Model development and qualification:
- Model verification:
2.4. Statistical Analysis
3. Results
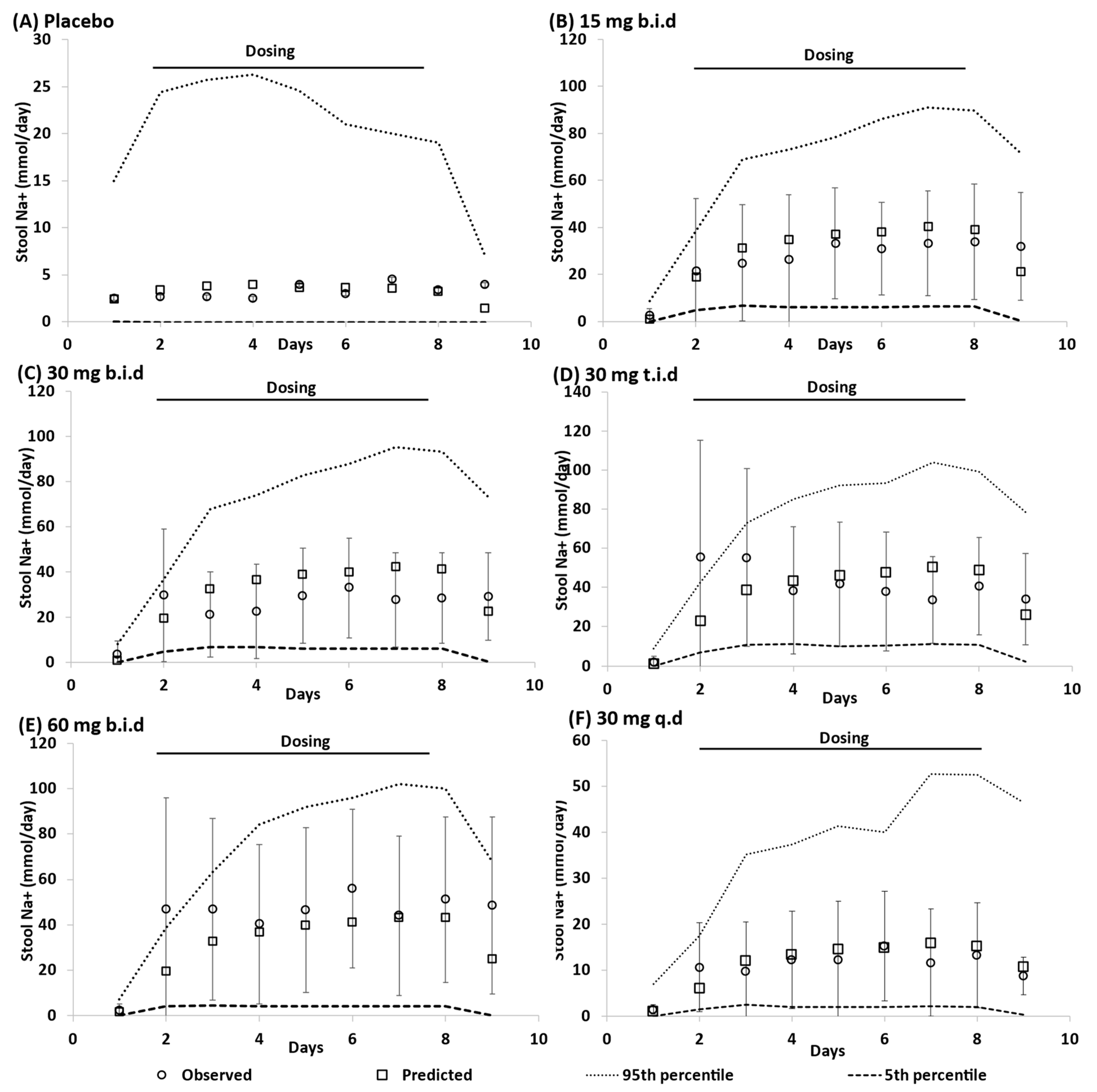
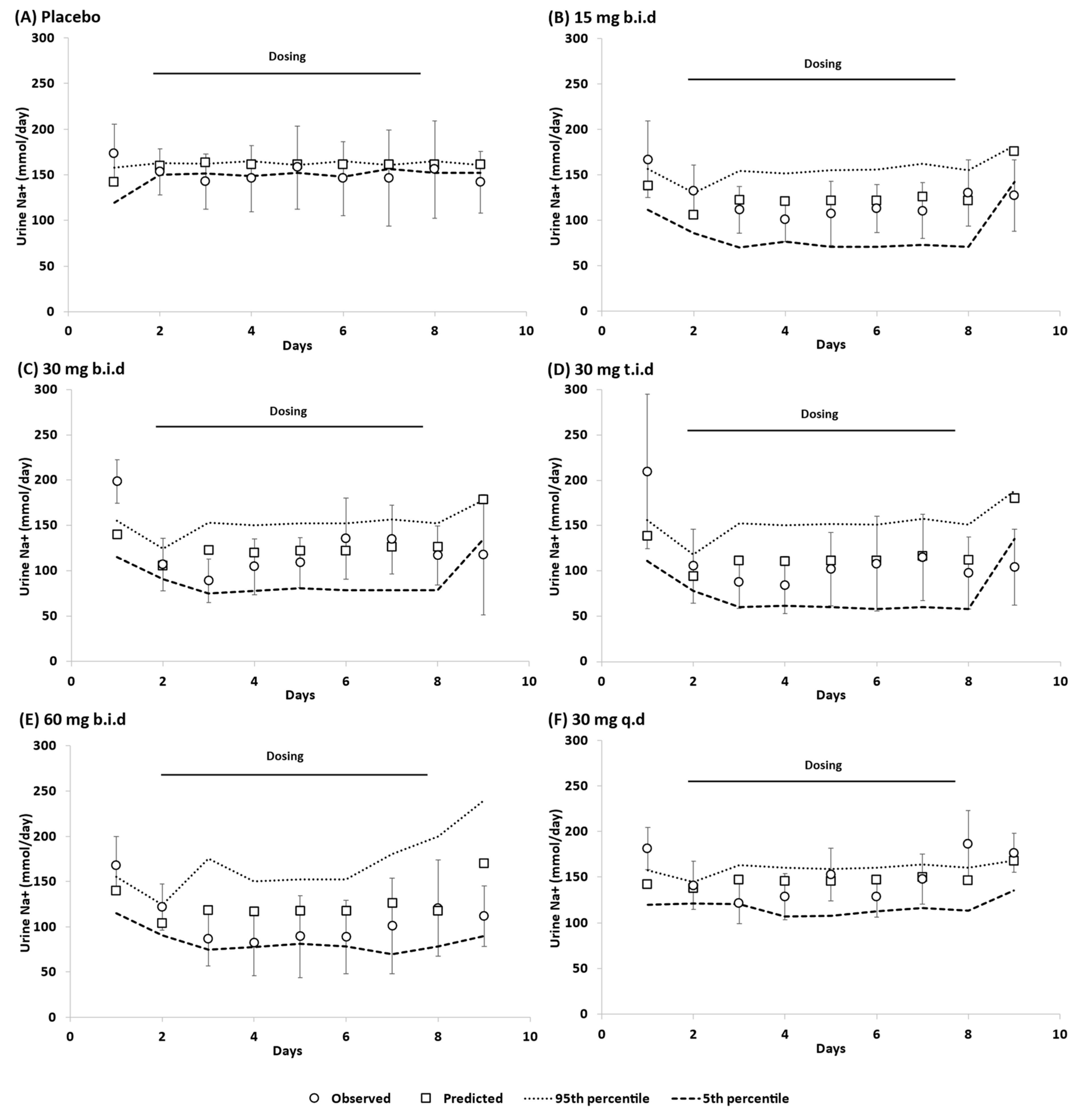
3.1. Model Qulaification
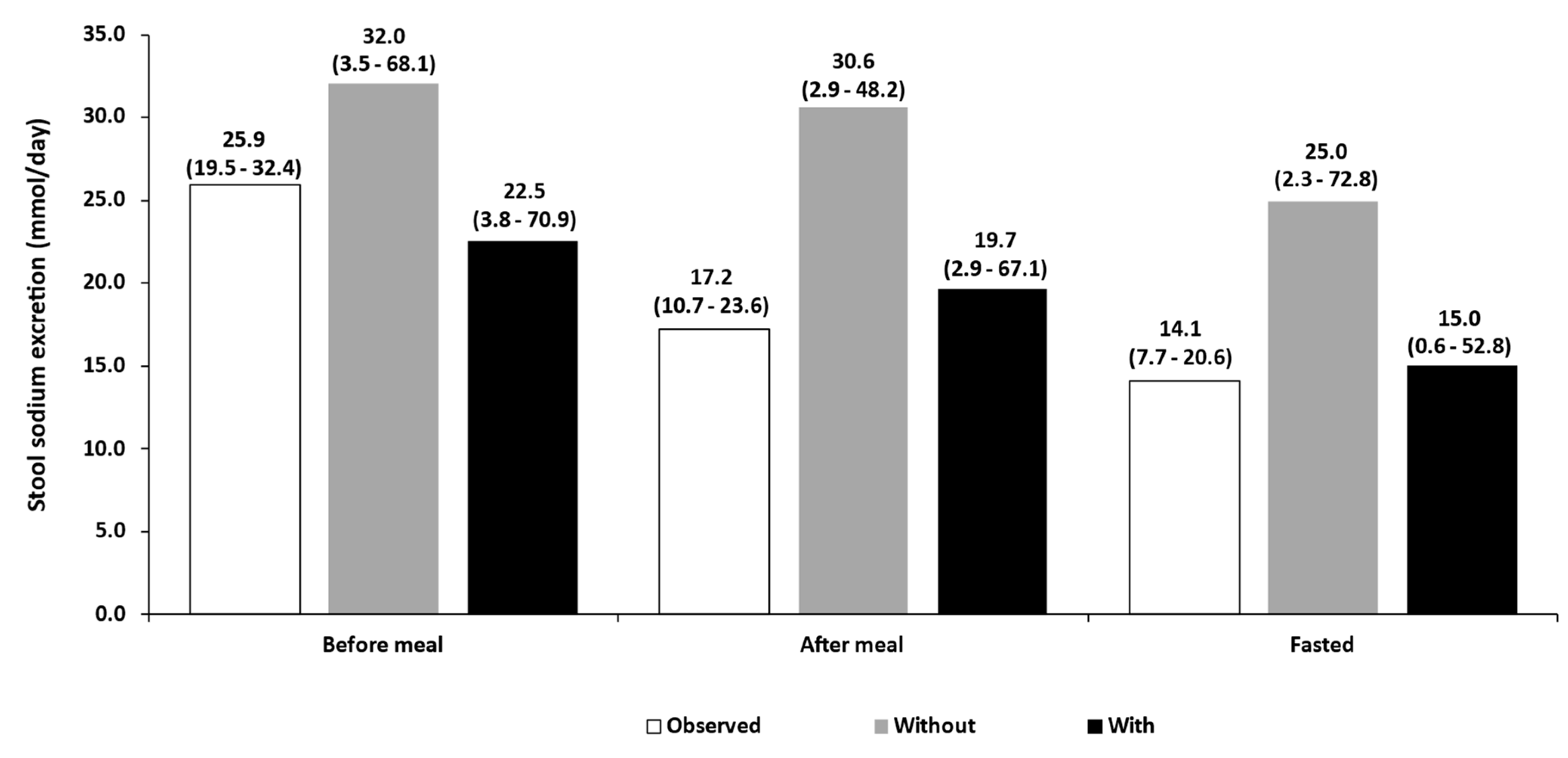
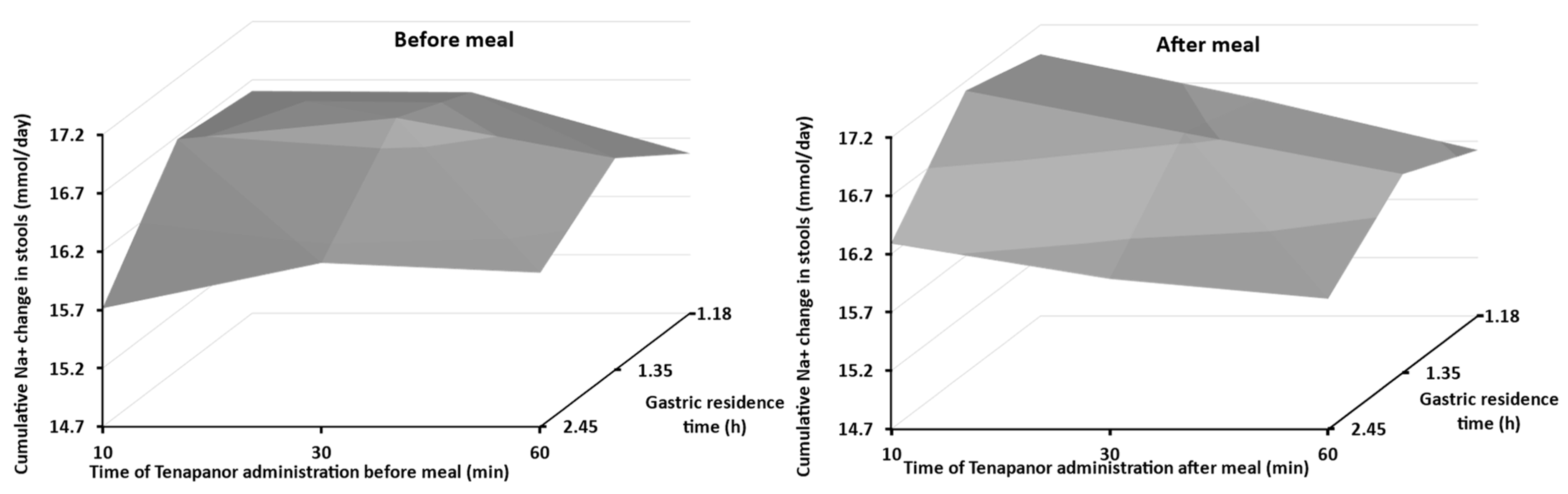
3.2. Food Effect
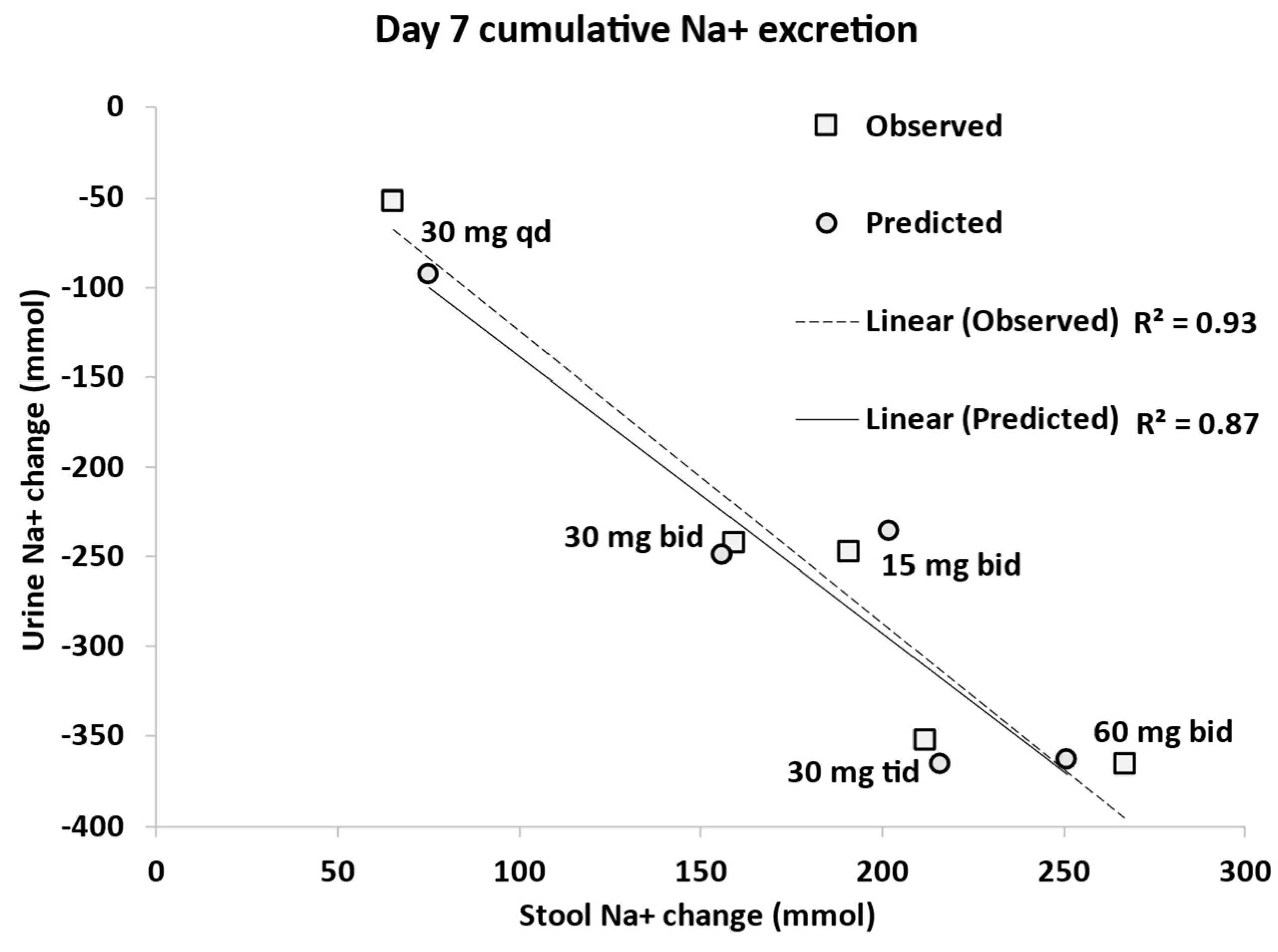
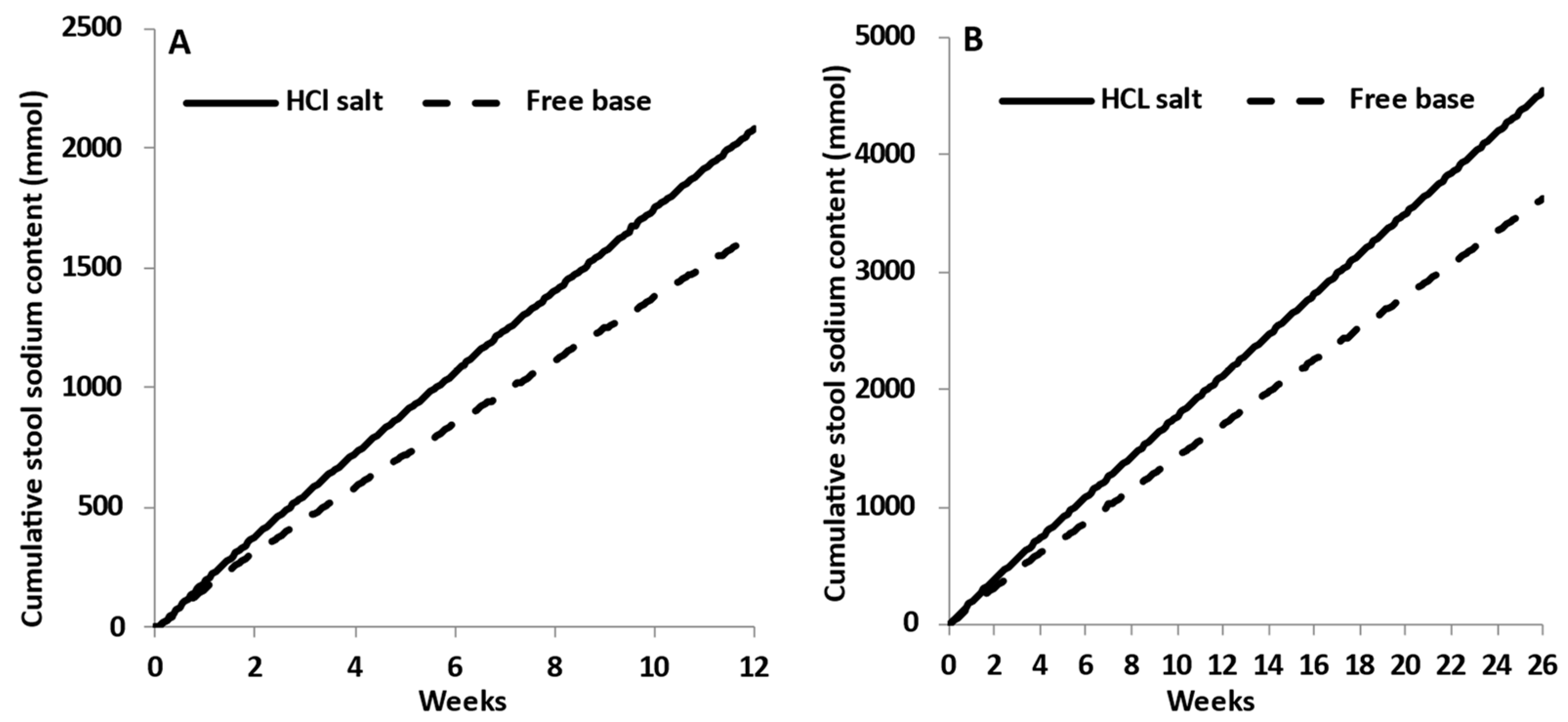
3.3. Dose Escalation Study
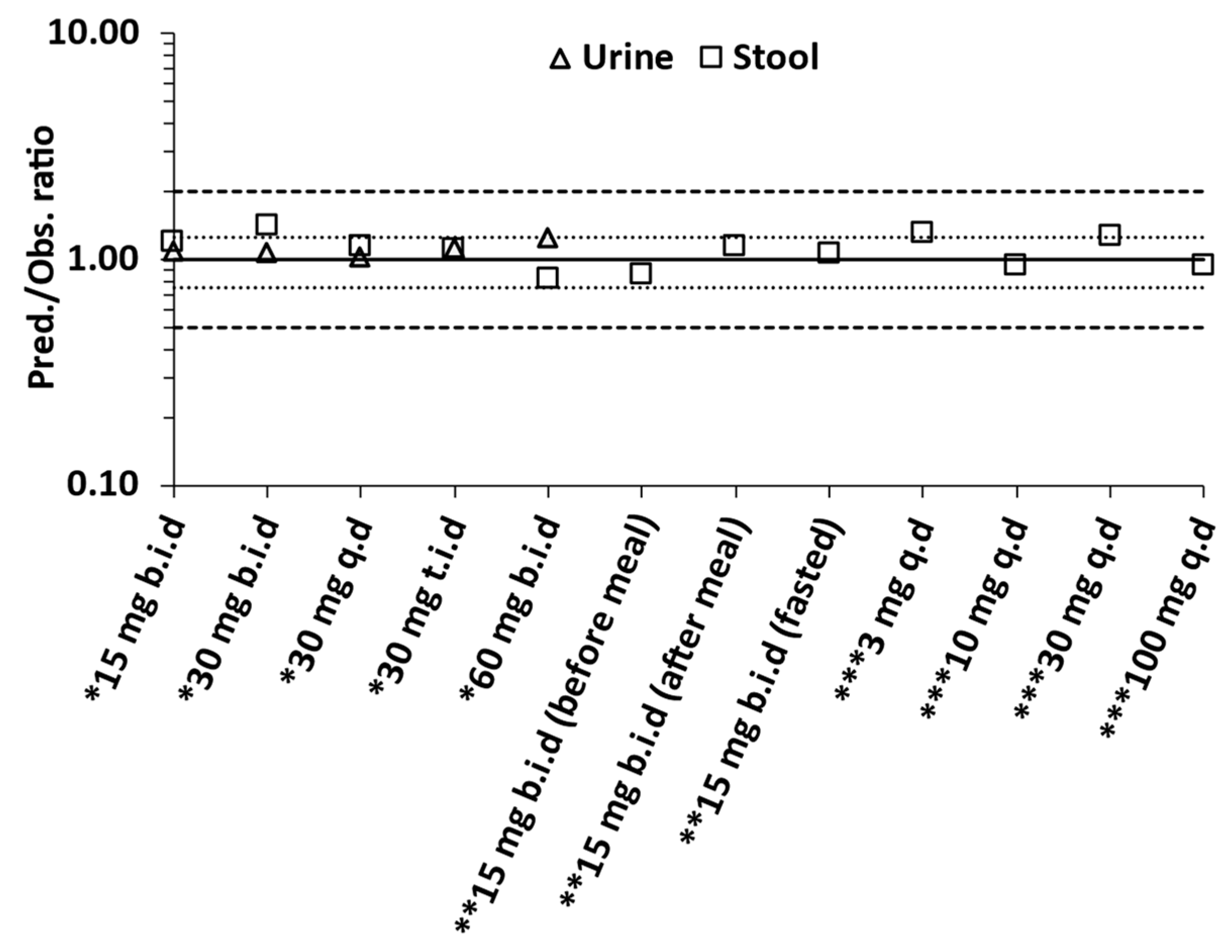
3.4. Overall Predictive Accuracy
3.5. Model Application
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhao, L.; Seo, P.; Lionberger, R. Current Scientific Considerations to Verify Physiologically-Based Pharmacokinetic Models and Their Implications for Locally Acting Products. CPT Pharmacomet. Syst. Pharmacol. 2019, 8, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, D.P.; Yan, A.; Jacobs, J.W. Pharmacodynamics, Safety, and Tolerability of the NHE3 Inhibitor Tenapanor: Two Trials in Healthy Volunteers. Clin. Drug Investig. 2018, 38, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Spencer, A.G.; Labonte, E.D.; Rosenbaum, D.P.; Plato, C.F.; Carreras, C.W.; Leadbetter, M.R.; Kozuka, K.; Kohler, J.; Koo-McCoy, S.; He, L.; et al. Intestinal inhibition of the Na+/H+ exchanger 3 prevents cardiorenal damage in rats and inhibits Na+ uptake in humans. Sci. Transl. Med. 2014, 6, 227ra236. [Google Scholar] [CrossRef] [PubMed]
- Johansson, S.A.; Knutsson, M.; Leonsson-Zachrisson, M.; Rosenbaum, D.P. Effect of Food Intake on the Pharmacodynamics of Tenapanor: A Phase 1 Study. Clin. Pharmacol. Drug Dev. 2017, 6, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.W.; Leadbetter, M.R.; Bell, N.; Koo-McCoy, S.; Carreras, C.W.; He, L.; Kohler, J.; Kozuka, K.; Labonté, E.D.; Navre, M.; et al. Discovery of Tenapanor: A First-in-Class Minimally Systemic Inhibitor of Intestinal Na+/H+ Exchanger Isoform 3. ACS Med. Chem. Lett. 2022, 13, 1043–1051. [Google Scholar] [CrossRef]
- Broere, N.; Chen, M.; Cinar, A.; Singh, A.K.; Hillesheim, J.; Riederer, B.; Lünnemann, M.; Rottinghaus, I.; Krabbenhöft, A.; Engelhardt, R.; et al. Defective jejunal and colonic salt absorption and alteredNa+/H+ exchanger 3 (NHE3) activity in NHE regulatory factor 1 (NHERF1) adaptor protein-deficient mice. Pflug. Arch. 2009, 457, 1079–1091. [Google Scholar] [CrossRef]
- He, P.; Yun, C.C. Mechanisms of the regulation of the intestinal Na+/H+ exchanger NHE3. J. Biomed. Biotechnol. 2010, 2010, 238080. [Google Scholar] [CrossRef]
- Alexander, R.T.; Grinstein, S. Tethering, recycling and activation of the epithelial sodium-proton exchanger, NHE3. J. Exp. Biol. 2009, 212 Pt 11, 1630–1637. [Google Scholar] [CrossRef]
- Kovesdy, C.P.; Adebiyi, A.; Rosenbaum, D.; Jacobs, J.W.; Quarles, L.D. Novel Treatments from Inhibition of the Intestinal Sodium-Hydrogen Exchanger 3. Int. J. Nephrol. Renov. Dis. 2021, 14, 411–420. [Google Scholar] [CrossRef]
- Laubitz, D.; Larmonier, C.B.; Bai, A.; Midura-Kiela, M.T.; Lipko, M.A.; Thurston, R.D.; Kiela, P.R.; Ghishan, F.K. Colonic gene expression profile in NHE3-deficient mice: Evidence for spontaneous distal colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G63–G77. [Google Scholar] [CrossRef]
- Maher, M.M.; Gontarek, J.D.; Jimenez, R.E.; Donowitz, M.; Yeo, C.J. Role of brush border Na+/H+ exchange in canine ileal absorption. Dig. Dis. Sci. 1996, 41, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Pasham, V.; Rotte, A.; Gu, S.; Yang, W.; Bhandaru, M.; Rexhepaj, R.; Pathare, G.; Lang, F. Upregulation of Intestinal NHE3 Following Saline Ingestion. Kidney Blood Press. Res. 2013, 37, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Musch, M.W.; Bookstein, C.; Xie, Y.; Sellin, J.H.; Chang, E.B. SCFA increase intestinal Na absorption by induction of NHE3 in rat colon and human intestinal C2/bbe cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 280, G687–G693. [Google Scholar] [CrossRef] [PubMed]
- Sugano, K. Theoretical comparison of hydrodynamic diffusion layer models used for dissolution simulation in drug discovery and development. Int. J. Pharm. 2008, 363, 73–77. [Google Scholar] [CrossRef]
- Shroff, H. Final Recommendation for NDA 211801. 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/211801Orig1s000ChemR.pdf (accessed on 1 April 2023).
- Yu, F.H.; Shull, G.E.; Orlowski, J. Functional properties of the rat Na/H exchanger NHE-2 isoform expressed in Na/H exchanger-deficient Chinese hamster ovary cells. J. Biol. Chem. 1993, 268, 25536–25541. [Google Scholar] [CrossRef]
- Dudeja, P.K.; Rao, D.D.; Syed, I.; Joshi, V.; Dahdal, R.Y.; Gardner, C.; Risk, M.C.; Schmidt, L.; Bavishi, D.; Kim, K.E.; et al. Intestinal distribution of human Na+/H+ exchanger isoforms NHE-1, NHE-2, and NHE-3 mRNA. Am. J. Physiol. 1996, 271 Pt 1, G483–G493. [Google Scholar] [CrossRef] [PubMed]
- Multi-Discipline Review. 2018. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/211801Orig1s000MultidisciplineR.pdf (accessed on 1 April 2023).
- Wright, E.M.; Loo, D.D.; Hirayama, B.A. Biology of human sodium glucose transporters. Physiol. Rev. 2011, 91, 733–794. [Google Scholar] [CrossRef]
- Fyfe, M.C.T. Chapter One—Non-systemic Intestine-Targeted Drugs. In Progress in Medicinal Chemistry; Lawton, G., Witty, D.R., Eds.; Elsevier: Amsterdam, The Netherlands, 2016; Volume 55, pp. 1–44. [Google Scholar]
- Stamatopoulos, K.; Ferrini, P.; Nguyen, D.; Zhang, Y.; Butler, J.M.; Hall, J.; Mistry, N. Integrating In Vitro Biopharmaceutics into Physiologically Based Biopharmaceutic Model (PBBM) to Predict Food Effect of BCS IV Zwitterionic Drug (GSK3640254). Pharmaceutics 2023, 15, 521. [Google Scholar] [CrossRef]
- Shimizu, H.; Yoshida, K.; Nakada, T.; Kojima, K.; Ogasawara, A.; Nakamaru, Y.; Yamazaki, H. Prediction of Human Distribution Volumes of Compounds in Various Elimination Phases Using Physiologically Based Pharmacokinetic Modeling and Experimental Pharmacokinetics in Animals. Drug Metab. Dispos. 2019, 47, 114–123. [Google Scholar] [CrossRef]
- Stamatopoulos, K.; O’Farrell, C.; Simmons, M.J.H.; Batchelor, H.K.; Mistry, N. Use of In Vitro Dynamic Colon Model (DCM) to Inform a Physiologically Based Biopharmaceutic Model (PBBM) to Predict the In Vivo Performance of a Modified-Release Formulation of Theophylline. Pharmaceutics 2023, 15, 882. [Google Scholar] [CrossRef]
- Tannergren, C.; Jadhav, H.; Eckernäs, E.; Fagerberg, J.; Augustijns, P.; Sjögren, E. Physiologically based biopharmaceutics modeling of regional and colon absorption in humans. Eur. J. Pharm. Biopharm. 2023, 186, 144–159. [Google Scholar] [CrossRef]
- Vasant, D.H.; Paine, P.A.; Black, C.J.; Houghton, L.A.; Everitt, H.A.; Corsetti, M.; Agrawal, A.; Aziz, I.; Farmer, A.D.; Eugenicos, M.P.; et al. British Society of Gastroenterology guidelines on the management of irritable bowel syndrome. Gut 2021, 70, 1214–1240. [Google Scholar] [CrossRef]
- Black, C.J.; Ford, A.C. Best management of irritable bowel syndrome. Frontline Gastroenterol. 2021, 12, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Chey, W.D.; Lembo, A.J.; Rosenbaum, D.P. Efficacy of Tenapanor in Treating Patients With Irritable Bowel Syndrome With Constipation: A 12-Week, Placebo-Controlled Phase 3 Trial (T3MPO-1). Am. J. Gastroenterol. 2020, 115, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Chey, W.D.; Lembo, A.J.; Yang, Y.; Rosenbaum, D.P. Efficacy of Tenapanor in Treating Patients With Irritable Bowel Syndrome With Constipation: A 26-Week, Placebo-Controlled Phase 3 Trial (T3MPO-2). Am. J. Gastroenterol. 2021, 116, 1294–1303. [Google Scholar] [CrossRef] [PubMed]
- Du Souich, P.; Besner, J.G.; Caillé, G. Pharmacodynamics as a tool to assess the bioequivalence of non-systemically available drugs: Size of the sample required. Biopharm. Drug Dispos. 1992, 13, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Zachos, N.C.; Tse, M.; Donowitz, M. Molecular physiology of intestinal Na+/H+ exchange. Annu. Rev. Physiol. 2005, 67, 411–443. [Google Scholar] [CrossRef] [PubMed]
- Jakab, R.L.; Collaco, A.M.; Ameen, N.A. Cell-specific effects of luminal acid, bicarbonate, cAMP, and carbachol on transporter trafficking in the intestine. Am. J. Physiol.-Gastrointest. Liver Physiol. 2012, 303, G937–G950. [Google Scholar] [CrossRef]
- Yu, A.; Baker, J.R.; Fioritto, A.F.; Wang, Y.; Luo, R.; Li, S.; Wen, B.; Bly, M.; Tsume, Y.; Koenigsknecht, M.J.; et al. Measurement of in vivo Gastrointestinal Release and Dissolution of Three Locally Acting Mesalamine Formulations in Regions of the Human Gastrointestinal Tract. Mol. Pharm. 2017, 14, 345–358. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Value (Unit) | |
|---|---|---|
| Sodium (Na+) | Tenapanor HCl | |
| Physico-chemical properties | ||
| Molecular Weight | 23 | 1217.97 [15] |
| log P | −10 a | 4.07 [15] |
| Compound Type | Neutral | Monoprotic base |
| pKa | - | 6.6 (estimated with SIVA) |
| B/P | 0.55 | 0.63 |
| Fraction unbound in plasma, fu | 1.0 | 0.001 |
| Absorption | ||
| Absorption model option | ADAM | |
| Formulation type | Solution | IR |
| Passive Permeability (10−6 cm/s) | 0 (Caco2) | 0.04 (MDCK) [3] |
| Dissolution | ||
| Intrinsic solubility (mg/mL) | - | 1.73 10−4 (estimated with SIVA) |
| CSR | - | 10 (default) |
| PRC (h−1) | - | 4 (default) |
| Salt limited solubility model | - | active |
| Drug solubility at pHmax (mg/mL) | - | 8.267@pH1.0 [15] (free base) |
| Ksp value (mM2) | - | 43.429 (back calculated from solubility at pHmax) |
| Solubility factor | - | 17,328 free base-(estimated with SIVA) |
| Counterion | Hydrochloric acid | |
| Distribution model | Mini-PBPK | |
| Distribution volume input type | Predicted | |
| Vss | 0.495 | 0.370 |
| Global tissue to plasma (Kp) Scalar | 1 | 1 |
| Elimination model | ||
| Renal Clearance (L/h) | 5.5 (%CV:30) (optimized) b | - |
| Hepatic Clearance (L/h) | 8.1 (%CV:30) c | |
| Transport model | ||
| Gut NHE3 (Apical Influx) | ||
| Jmax (pmol/min) | 4900 (optimized) b | - |
| Km (μM) | 4700 [16] d | - |
| Regional relative abundance of NHE3 [17] | ||
| Duodenum | 0 e | |
| Jejunum I–II | 1 e | |
| Ileum I–IV | 1.5 e | |
| Colon | 0.4 e | |
| Transporter interaction—NHE3 | ||
| Transporter | - | Apical influx (intestine) |
| Ki (μM) | - | 0.005 [3] |
| Food-stimulated NHE3 activity | ||
| Emax | 5.72 f | - |
| EC50 (μM) | 1500 f | - |
| γ | 1.93 f | |
| Dose Schedule | Urine (mmol/Day) | Stool (mmol/Day) | |||||
|---|---|---|---|---|---|---|---|
| Predicted | Observed | Pred/Obs | Predicted | Observed | Pred/Obs | ||
| Dose regimen study [2] | 15 mg b.i.d | 122.5 | 112.30 | 1.09 | 37.00 | 30.50 | 1.21 |
| 30 mg b.i.d | 123.00 | 114.90 | 1.07 | 38.4 | 27.00 | 1.42 | |
| 30 mg q.d | 147.1 | 143.00 | 1.03 | 14.3 | 12.40 | 1.16 | |
| 30mg t.i.d | 112.00 | 98.90 | 1.13 | 45.90 | 41.30 | 1.11 | |
| 60mg b.i.d | 118.8 | 94.80 | 1.25 | 39.30 | 47.50 | 0.83 | |
| Food effect (15 mg b.i.d) [4] | Before meal | 22.50 | 25.90 | 0.87 | |||
| After meal | 19.70 | 17.20 | 1.14 | ||||
| Fasting | 15.00 | 14.10 | 1.07 | ||||
| Dose escalation study [3] | 3 mg q.d | 6.20 | 4.70 | 1.33 | |||
| 10 mg q.d | 9.00 | 9.50 | 0.95 | ||||
| 30 mg q.d | 15.00 | 11.70 | 1.28 | ||||
| 100 mg q.d | 14.70 | 15.40 | 0.95 | ||||
| AAFE | 1.05 | 1.18 | |||||
| AFE | 1.11 | 1.10 | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stamatopoulos, K.; Mistry, N.; Fotaki, N.; Turner, D.B.; Swift, B. Physiologically Based Biopharmaceutics Model (PBBM) of Minimally Absorbed Locally Acting Drugs in the Gastrointestinal Tract—Case Study: Tenapanor. Pharmaceutics 2023, 15, 2726. https://doi.org/10.3390/pharmaceutics15122726
Stamatopoulos K, Mistry N, Fotaki N, Turner DB, Swift B. Physiologically Based Biopharmaceutics Model (PBBM) of Minimally Absorbed Locally Acting Drugs in the Gastrointestinal Tract—Case Study: Tenapanor. Pharmaceutics. 2023; 15(12):2726. https://doi.org/10.3390/pharmaceutics15122726
Chicago/Turabian StyleStamatopoulos, Konstantinos, Nena Mistry, Nikoletta Fotaki, David B. Turner, and Brandon Swift. 2023. "Physiologically Based Biopharmaceutics Model (PBBM) of Minimally Absorbed Locally Acting Drugs in the Gastrointestinal Tract—Case Study: Tenapanor" Pharmaceutics 15, no. 12: 2726. https://doi.org/10.3390/pharmaceutics15122726
APA StyleStamatopoulos, K., Mistry, N., Fotaki, N., Turner, D. B., & Swift, B. (2023). Physiologically Based Biopharmaceutics Model (PBBM) of Minimally Absorbed Locally Acting Drugs in the Gastrointestinal Tract—Case Study: Tenapanor. Pharmaceutics, 15(12), 2726. https://doi.org/10.3390/pharmaceutics15122726