1. Introduction
Pancreatic cancer is a devasting disease with a current five-year survival rate of just 12%. The American Cancer Society estimates that about 79% of the 64,050 people diagnosed with pancreatic cancer in 2023 will die (
Cancer Facts & Figures 2023, American Cancer Society, Atlanta, GA, USA, 2023). Moreover, pancreatic ductal adenocarcinoma (PDAC) accounts for over 90% of pancreatic cancer cases. The challenge with PDAC is the lack of symptoms in the early disease, thus obfuscating surgery, the only potentially curative treatment option. While resection and standard-of-care chemotherapy combinations such as gemcitabine plus nab-paclitaxel and FOLFIRINOX have managed to benefit patients with early-stage pancreatic cancer, chemotherapy has limited and only short-term activity for patients in the late stages of the disease [
1]. While immunotherapies have dramatically affected some solid tumors, such as lung and melanoma, such success has not manifested in PDAC, which is commonly considered nonimmunogenic and exhibits an immune-suppressive microenvironment [
2,
3].
The aggressive and therapeutic-resistant nature of PDAC is thought to exist both at the genetic and cellular levels, where it is challenging to target oncogenes, and where tumor cell heterogeneity predominates in the disease. While new KRAS inhibitors are exciting in this disease where mutant KRAS is a near-universal driver, resistance is likely to develop rapidly, based on initial trials [
4]. In addition, cellular plasticity and the presence of cancer stem-like cells contribute to therapeutic resistance. Current therapies do not adequately target cancer stem cells, which make up approximately 1% of pancreatic cancer cells and have been shown to contribute to tumor development and treatment resistance [
5]. In addition, the high metastatic potential of PDAC further reduces the efficacy of current treatments. These factors suggest that therapeutic interventions targeting individual pathways will eventually result in drug resistance, limiting our clinical treatment options for pancreatic disease. This scenario indicates that using natural compounds, which typically target multiple molecular pathways and manifest various mechanisms of action, may result in higher PDAC survival rates. Experiments on different cancer types have demonstrated the anticancer efficacy of natural compounds [
6,
7,
8]. In the last five years, approximately 70 natural products have been studied and reported in relation to pancreatic cancer [
9], seven of which have been evaluated in clinical trials for treating pancreatic cancer, of which five were conducted in the United States. Four of the seven clinical trials were completed in Phase 2, one was in Phase 3, and one was still enrolling participants for Phase 3. All completed clinical trials showed a favorable response and increased survival compared to the control group [
9].
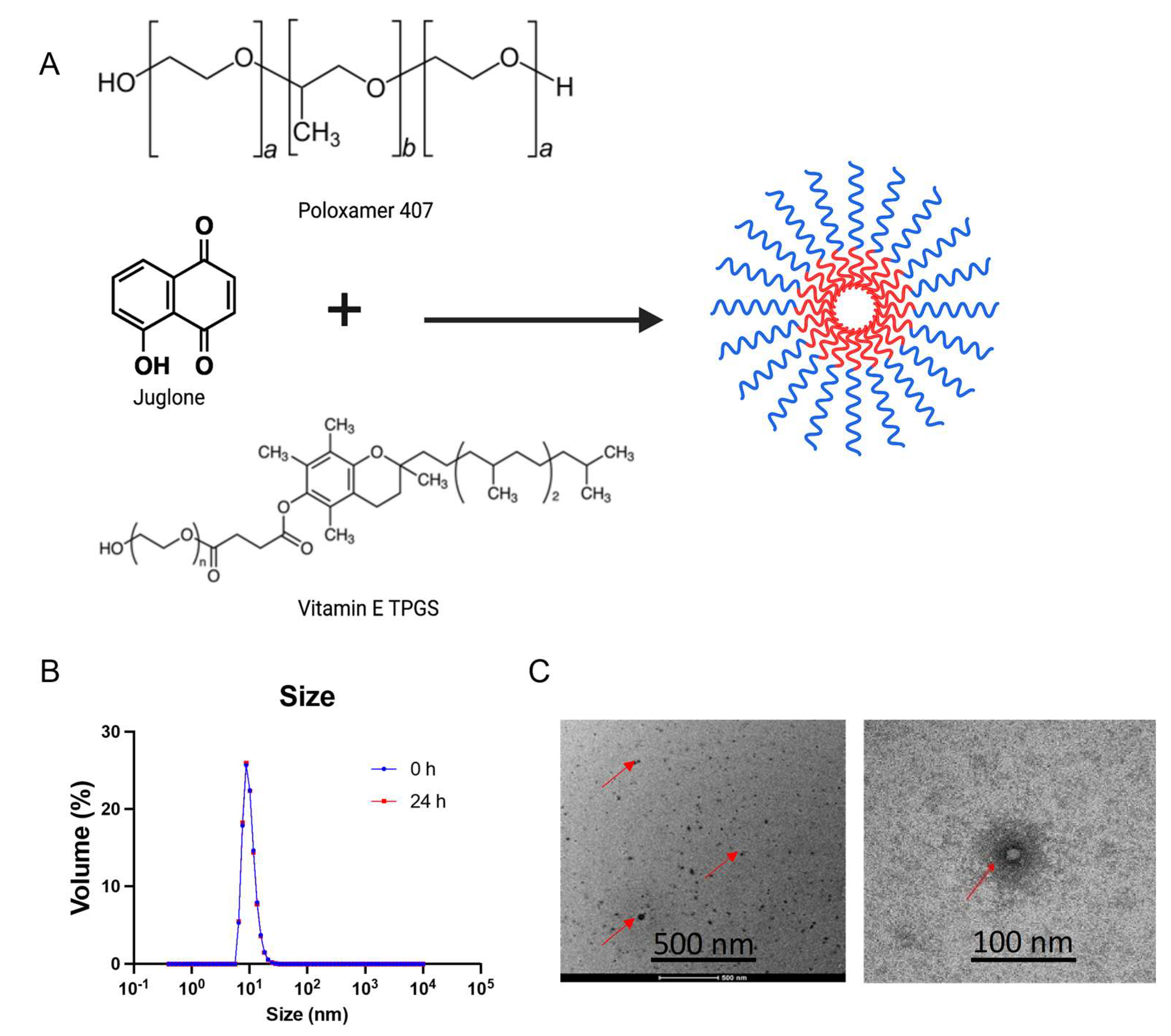
Juglone (5-hydroxy-1,4-naphthoquinone) is a natural 1,4-naphthoquinone found in walnut trees. Juglone has been reported to possess cytotoxic properties against human gastric, leukemia, cervical, colon, and pancreatic cancer cell lines confirmed through in vitro assays [
10,
11,
12,
13,
14,
15,
16,
17]. The studies have reported that juglone exhibits cytotoxic properties by inducing apoptosis through mitochondria-dependent pathways [
10,
12,
15,
16]. Although the antitumor activity of juglone is quite robust, in vivo efficacy studies on it are limited due to its poor water solubility (0.04 mg/mL). Secondly, such in-vivo studies were conducted for a short time < 14 days and dosed every two days, perhaps due to the compound’s toxicity. In the present study, nano-scaled polymeric micelles were designed for the delivery of juglone to pancreatic tumors in vivo.
Polymeric micelles have gained extensive attention as a delivery system for poorly water-soluble drugs as they increase solubility and promote accumulation and retention into tumor tissue via enhanced permeability and retention effects [
18,
19]. Micelles have a diameter typically ranging from 10 to 200 nm [
18,
19,
20]. They form spontaneously due to the aggregation of amphiphilic molecules, in which the hydrophobic chain of the polymers forms the core, and the hydrophilic ends form the structural shell [
18,
19]. Pluronics are block copolymers, which are widely used as micellar carriers [
21]. Pluronic comprises hydrophilic polyethylene oxide (PEO) and hydrophobic poly propylene oxide (PPO) segments arranged in a basic tri-block structure: PEO–PPO–PEO [
22]. Pluronic F127(F127) (PEO100-PPO69-PEO100) possesses an extended PEO block that improves the encapsulation efficacy, stability, and circulation time of drugs [
21]. The Food and Drug Administration acknowledges F127 as a safe pharmaceutical adjuvant, and its biocompatibility contributes to its widespread application as a nanodrug delivery polymer [
22].
F127 generates micelles at a low critical micelle concentration (CMC) of 0.0031%
w/
w and does not inhibit P-glycoprotein, a multidrug resistance (MDR) protein and an essential efflux drug transporter protein [
23,
24]. As a result, F127 micelles frequently have a low loading efficiency and a larger particle size. Published work [
25] indicates that each of these characteristics can potentially limit both the dose and the ability of micelles to cross biological barriers. Micelles containing an additional lipophilic copolymer may provide a superior platform by increasing loading and decreasing particle size to deliver hydrophobic drugs, such as juglone.
D-α-Tocopherol polyethylene glycol 1000 succinate (TPGS) is a non-ionic, water-soluble vitamin E derivative formed by conjugating vitamin E succinate with polyethylene glycol [
26]. It has been authorized by the Food and Drug Administration as a safe pharmaceutical adjuvant and is used in various drug formulations [
26]. In addition, TPGS has other advantages that make it an ideal nanocarrier for poorly soluble drug delivery. First, unlike F127, TPGS enhances the cellular uptake of the drugs [
27] by reversing MDR and inhibiting a P-glycoprotein-mediated efflux of drugs [
28]. Second, TPGS has been used as a drug carrier to form prodrugs for chemotherapeutical drugs, such as doxorubicin, paclitaxel, and gemcitabine, to promote drug delivery to tumors and to reverse MDR in cancer therapy. Third, TPGS possesses antioxidant activity [
29], which can protect pharmaceuticals from oxidative degradation during storage, thereby enhancing formulation stability. However, the CMC of TPGS is relatively high (0.02%,
w/
w), which may accelerate the dissociation of micelles in plasma. Therefore, we formulated mixed micelles with TPGS and F127 to deliver juglone as a nanoformulation suitable for in vivo evaluation, circumventing its poor water solubility and potentially enhancing its stability and circulation time for treating pancreatic cancer.
In this work, we prepared juglone micelles (JMs) using a solvent-casting method. The micelles were characterized for particle size distribution, morphological observation, zeta potential, and encapsulation efficiency. Then, we evaluated JM for its in vitro effects on pancreatic cancer cell lines and in vivo effects on tumor growth in both immunocompromised and immunocompetent pancreatic tumor mouse models. Bulk RNA sequencing was performed on pancreatic tumors treated with the JM to understand changes to the global transcriptomics following juglone delivery. Taking advantage of the therapeutic properties of juglone, we developed a polymeric micelle formulation that induces a significant reduction in tumor growth in both immunocompetent and immunocompromised mice, thus making the evaluation and therapeutic application of juglone feasible in in vivo pancreatic tumor models.
2. Materials and Methods
2.1. Materials
Juglone (Fischer Scientific, Waltham, MA, USA) (CAS 48139-0), Pluronics F-127 (Letco Medical, Decatur, AL, USA) (CAS 9003-11-6), D-alpha-Tocopherol polyethylene glycol 1000 succinate (TPGS), BioXtra, and water-soluble vitamin E conjugate (Sigma Aldrich, Burlington, MA, USA) (CAS 9002-96-4) were used for the in vitro micelle preparation.
2.2. Cell Culture
Human pancreatic ductal adenocarcinoma cell lines: Panc-1 and MiaPaCa-2 cells were purchased from ATCC and were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (HyClone), supplemented with 10% (v/v) fetal bovine serum (FBS) (Gibco, Waltham, MA, USA) and 1% (v/v) penicillin-streptomycin (HyClone, Logan, UT, USA). All cells were routinely maintained at 37 °C under 5% CO2 in a humidified incubator.
2.3. Preparation of Juglone Micelles (JMs) and Characterization
Polymeric micelle formulation of juglone was prepared using the solvent casting method using Pluronic F127 and TPGS. Briefly, 45 mg of Pluronic F-127 and 11 mg of TPGS were weighed and mixed in 1 mL of 100% ethanol. Approximately 2.8 mg of juglone from a stock solution of 8 mg/mL was spiked into the polymer mix, vortexed gently, and subjected to solvent evaporation using a rotary evaporator to form a thin film. Once the formed film cooled at room temperature, 1 mL of saline was added, and a round-bottom flask was allowed to rotate in the water bath to ensure complete hydration of the film. The micelles were spun down at 6000 rpm for 6 min at room temperature. The supernatant was filtered and collected using a 0.2 µm syringe filter. Drug loading and encapsulation efficiency were quantified using reverse-phase, high-performance liquid chromatography (RP-HPLC), (Shimadzu, Canby, OR, USA). The analysis was performed using a Zorbax C18 column (4.6 × 75 mm, 3.5 μm) (Agilent Technologies, Santa Clara, CA, USA) in an isocratic mode with acetonitrile/water (50/50) containing 0.1% phosphoric acid and 1% methanol at a flow rate of 1.5 mL/min, and an injection volume of 10 μL. Column temperature was maintained at 40 °C with a run time of 3 min. The juglone peak is monitored at 250 nm and has a retention time of 1.5 min. The micelles were dissolved in ethanol to extract the encapsulated juglone for quantification. The encapsulation efficiency (EE%) was calculated using the formula below: %EE = ((drug amount entrapped in the micelles/total amount of drug added) × 100%). The size distribution of micelles was measured using a dynamic light-scattering instrument (Malvern Zetasizer Nano ZS; Malvern Instruments, Worcestershire, UK). The data are presented as %EE, Zave ± SD, and PDI ± SD for four replicates.
The morphology of the micelles was observed under a transmission electron microscope (TEM; Philips CM 120, Amsterdam, the Netherlands). A drop of micellar solution was placed on a copper grid, covered with a carbon film, and stained using uranyl acetate solution (2%, w/v). After dyeing at room temperature, the sample was observed under the TEM.
To assess the physical stability of the juglone-loaded F127/TPGS mixed micelles, we incubated the micelles at room temperature for 24 h. At 24 h, the micelles were centrifuged at 6000 rpm for 6 min at room temperature. The supernatant was collected and filtered through a 0.22 μm filter membrane, followed by dilution with ethanol. The diluted supernatant was used to measure the concentration of juglone retained in micelles using high-performance liquid chromatography (Cary Elipse, Agilent, Santa Clara, CA, USA).
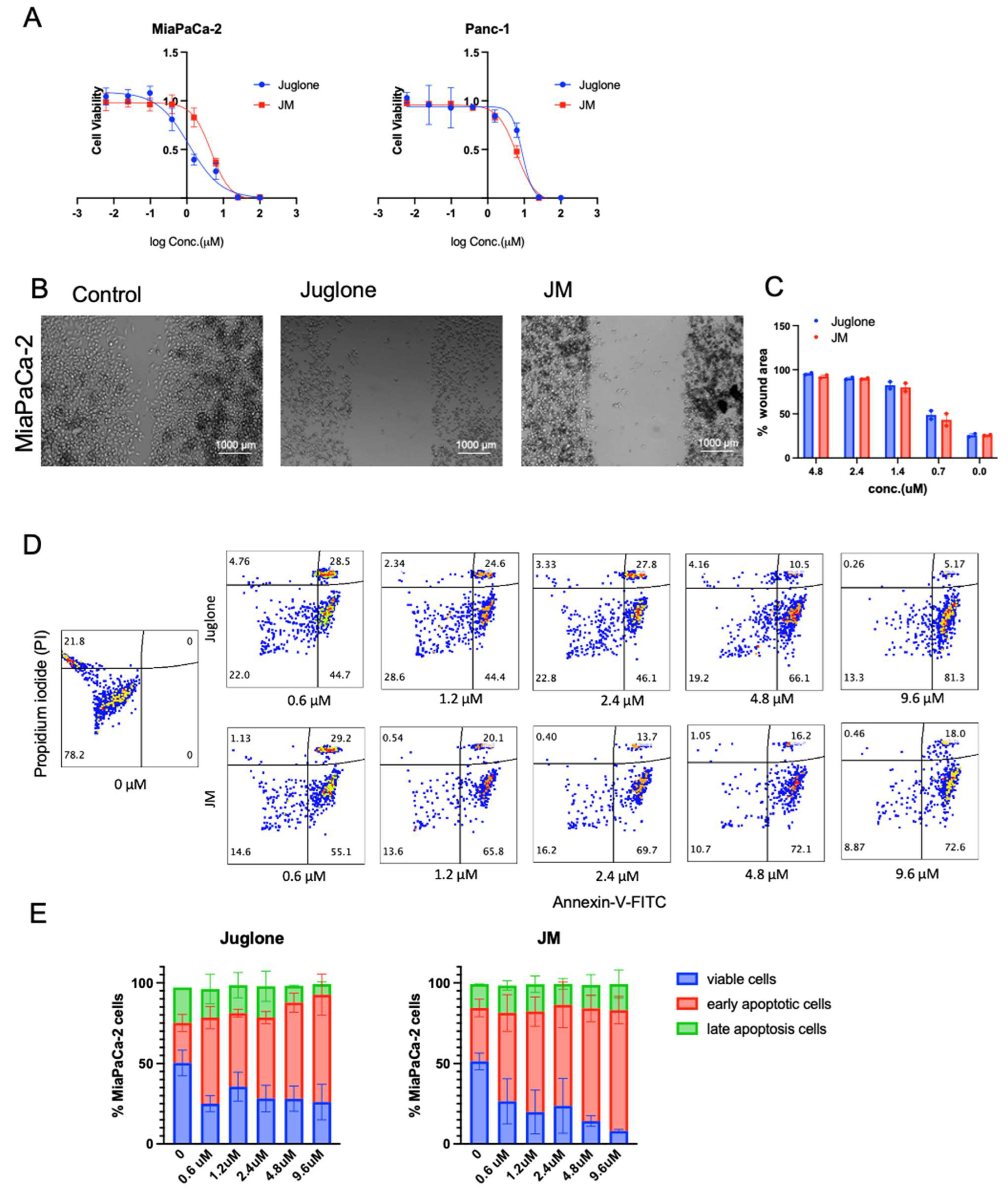
2.4. In Vitro Cell Viability
Panc-1 and MiaPaCa-2 cells were seeded on 96-well plates at a density of 2000 cells/well and allowed to grow as a monolayer for 24 h before drug treatment. Cells were then treated with blank micelle particles, free juglone, and JM particles for 48 h. Untreated cells served as a control. At the start and end of the 48 h period, cell viability was measured using a 3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay. Briefly, 10 μL of MTS (10 mg/mL) was added to 96-well plates and cultured for an additional 1 h. The absorbance value (OD) was determined using a BioSpa Cytation5 (BioTek) reader (Agilent Technologies, Santa Clara, CA, USA) at 490 nm. The following equation calculated the percentage of cell viability:
The half maximal inhibitory concentration (IC50) of free juglone and JM particles were further calculated from their inhibition rates on cell viability by using GraphPad Prism version 9.1 (La Jolla, CA, USA).
2.5. Cell Migration Assay
MiaPaca-2 cells (2 × 105 cells/well) were seeded in 24-well plates and allowed to adhere overnight. At 80–90% confluence, a “reference line” was scratched at the bottom of the plate using a sterile 200 μL pipette tip. After being washed with phosphate buffer saline (PBS) thrice, cells were further incubated with juglone and JMs (0.6, 1.2, 2.4, and 4.8 μM) or a vehicle (DMSO). Images of cells migrating across the reference line were taken in different fields with an Evos after treatment at 0 and 24 h, respectively. The wound closure was calculated by measuring the wound area using ImageJ software; the % area at 24 h was normalized to the area at 0 h and plotted as a function of both juglone and JM concentration.
2.6. Apoptosis Assay
Apoptosis was evaluated using the Annexin V/PI Kit (BD Biosciences, San Jose, CA, USA). Exponentially growing cells were seeded on 6-well plates and then treated with juglone and JM (0.6, 1.2, 2.4, 4.8, and 9.6 μM) or DMSO (vehicle control, final concentration of 0.1%) for 24 h. The treated cells were harvested and stained with Annexin V and propidium iodide (PI). Flow cytometry was conducted using Cytek Aurora (Cytek Biosciences, Fremont, CA, USA).
2.7. Mouse Models
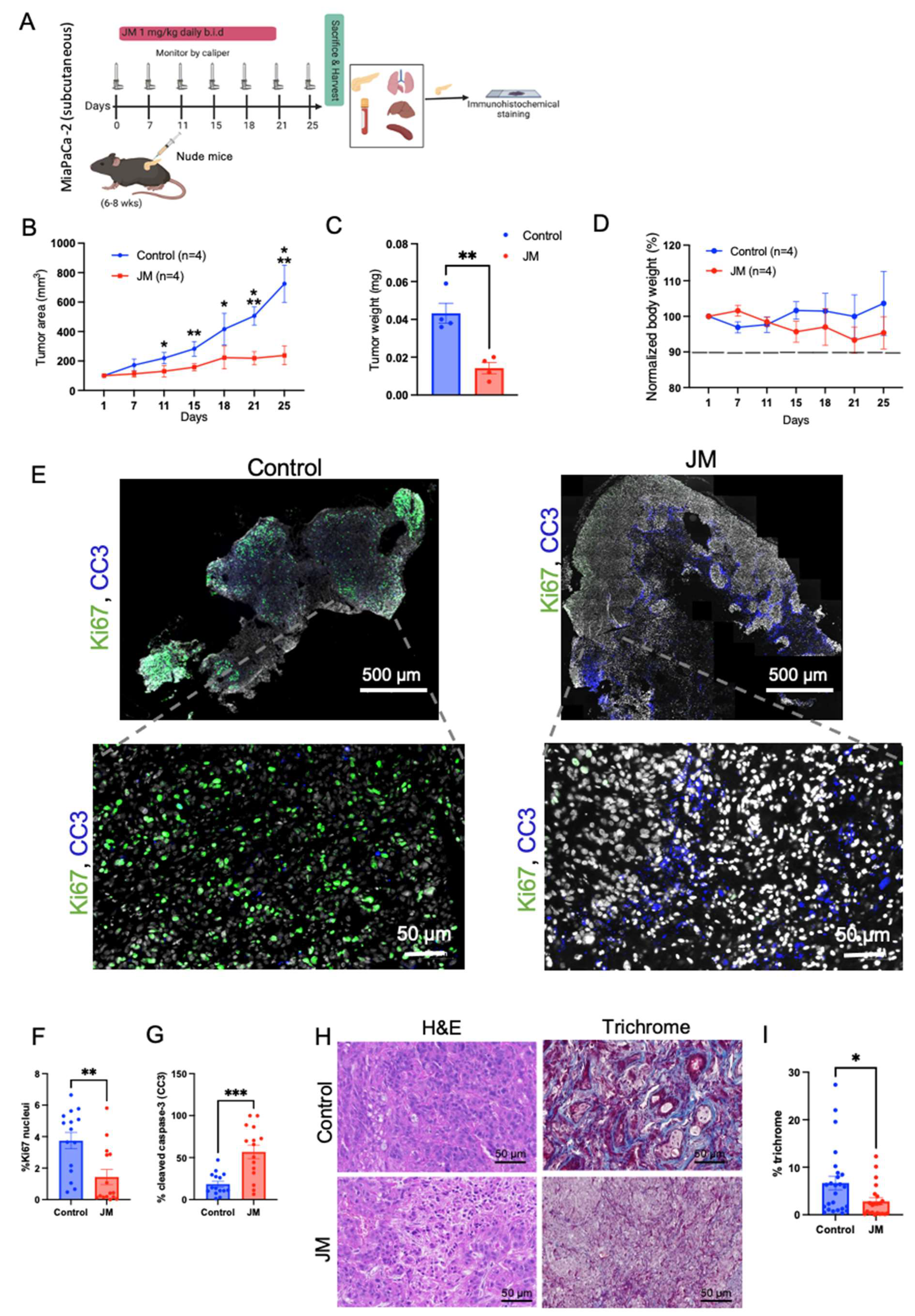
Six-to-eight-week-old male and female nude mice and C57BL/6 were purchased from Jackson Laboratory (Farmington, CT, USA) and housed in a pathogen-free barrier room. All animal studies complied with Oregon Health & Science University (OHSU) animal use guidelines and were approved by the OHSU Institutional Animal Care and Use Committee (protocol number TR1_IP00001014).
For the JM treatment of MiaPaCa-2 cell line subcutaneous xenografts, 100 µL of 1 million cells resuspended in 50% Matrigel/50% DMEM were implanted subcutaneously into the flank of 6–8-week-old nude mice. Once tumors were palpable, mice were randomized into treatment groups (control (4 mice) and JM (4 mice)) and treated intraperitoneally, with 1 mg/kg of JM twice daily for five times per week. Tumor size was monitored using caliper measurement. Each mouse was euthanized, and its tumor was harvested when it became moribund.
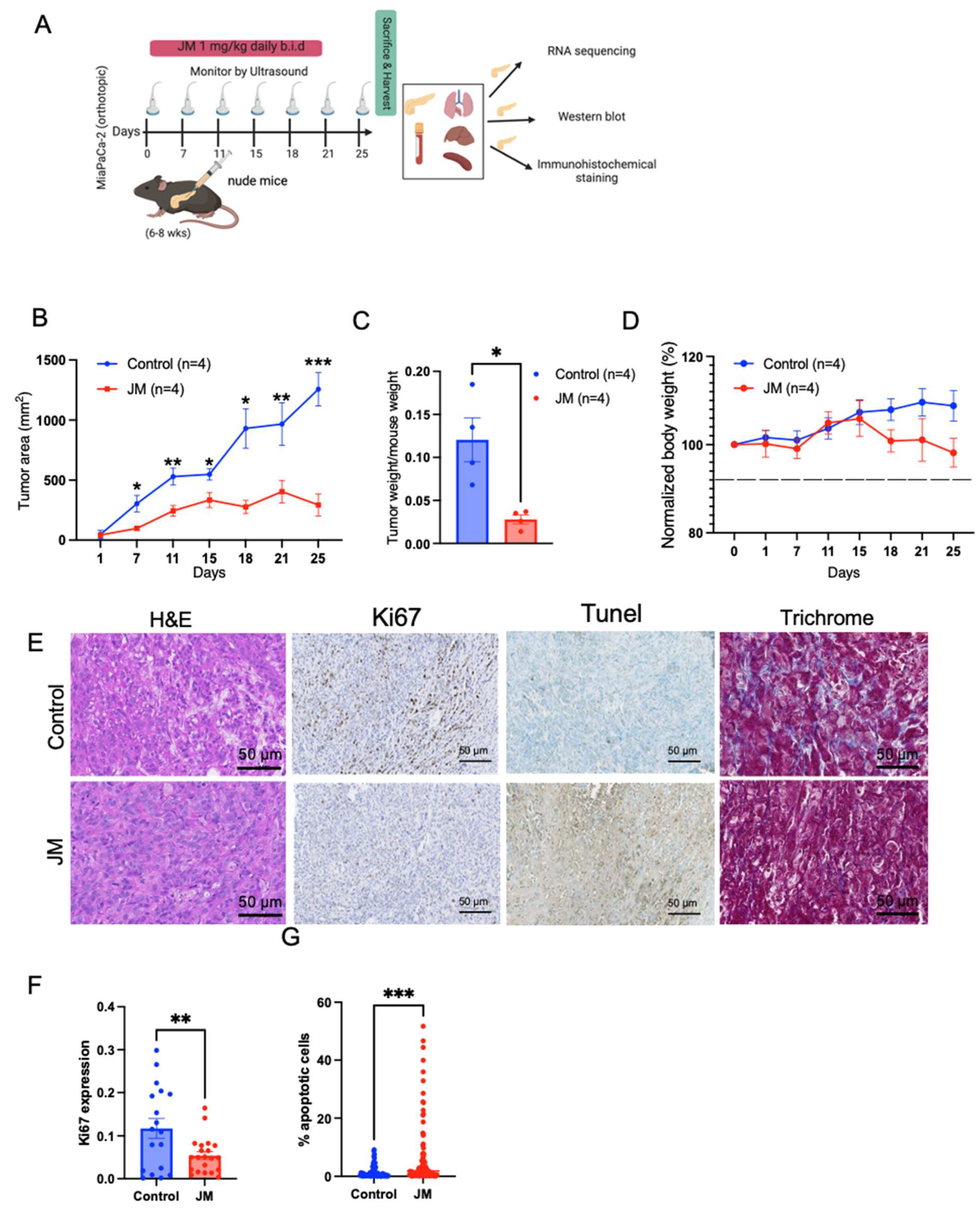
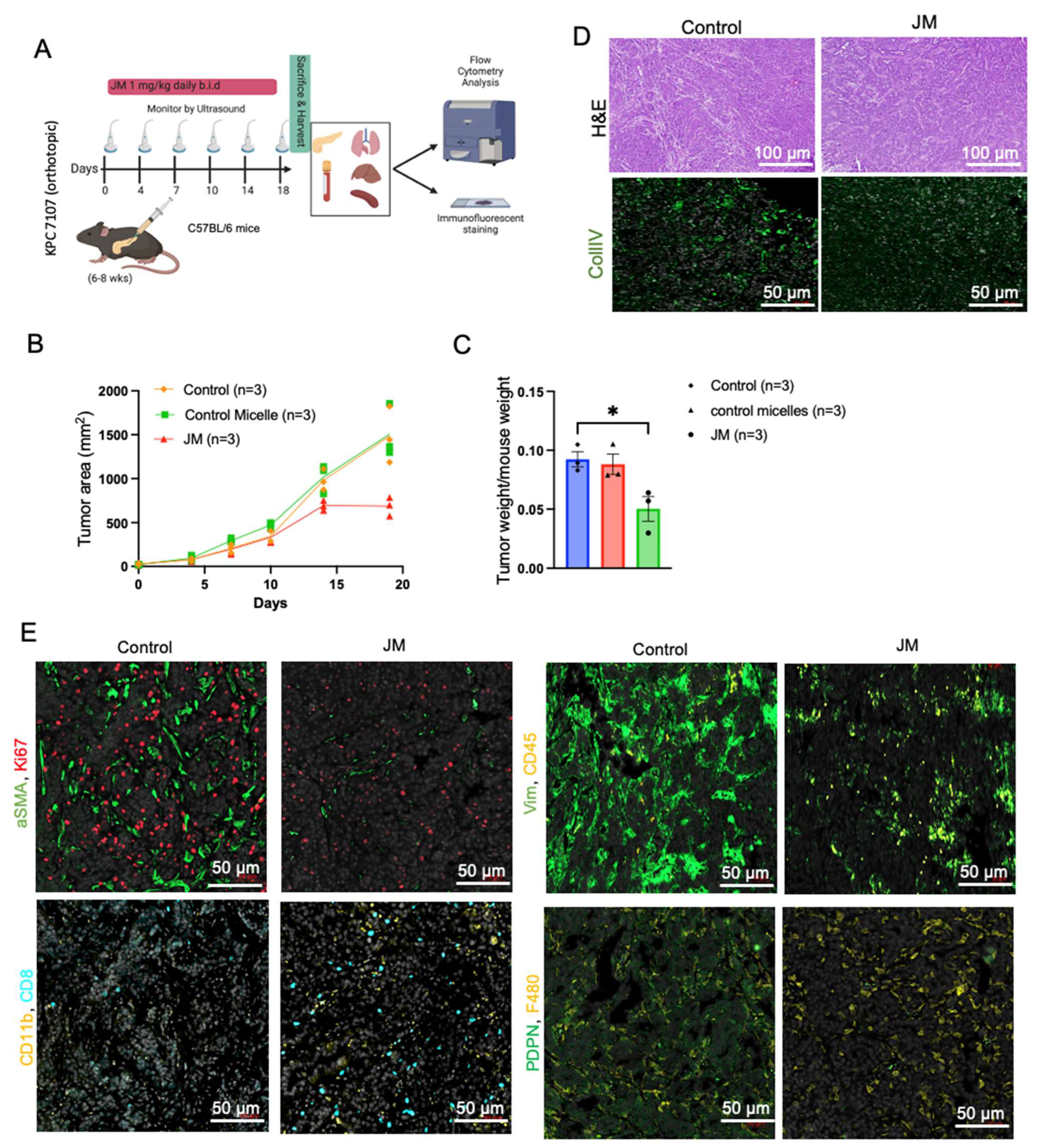
For the JM treatment of MiaPaCa-2 pancreatic orthotopic xenografts, mice were anesthetized with isoflurane, and meloxicam oral analgesia was used. A small abdominal flank incision was made and, using the spleen as the anchor, the pancreas was exteriorized. Next, 40 µL of 2 million cells resuspended in 50% DMEM/50% Matrigel were injected into the tail of the pancreas using a 28-gauge syringe needle. The needle was held in place for 30 s, and the injection site was swabbed with sterile gauze to prevent the leaking of tumor suspension. The pancreas was placed back into the peritoneal cavity, and the incision was sutured. Animals were monitored daily for 1 week post-injection to assess their overall health and wound healing. Mice were then imaged by ultrasound once per week with a FujiFilm VisualSonics Vevo 2100 high-frequency ultrasound (Toronto, ON, Canada). For KPC7107, 40 µL of 5000 cells was injected into C57BL/6. In the orthotopic MiPaCa-2 and KPC 7107 studies, the animals were divided into two treatment groups: control (4 mice) and JM (4 mice).
2.8. Western Blots
Cells were lysed in AB Lysis buffer, and Western blot analysis was performed. Blots were visualized and bands quantified using an Odyssey imaging system (LI-COR Biosciences, Lincoln, NE, USA). Primary antibodies’ total MYC (Y69 1:1000, Abcam, Waltham, MA, USA; ab32072), pMYC (1:500, Abcam, Waltham, MA, USA; ab78318), and GAPDH (6C5; 1:5000) were diluted in blocking buffer (1:1 Odyssey Blocking Buffer (LI-COR Biosciences, Lincoln, NE, USA)) PBS with 0.05% Tween20. Primary antibodies were detected with secondary antibodies labeled with the near-infrared fluorescent dyes IRDye800 (Rockland, Philadelphia, PA, USA) and Alexa Fluor 680 (Molecular Probes, Eugene, OR, USA) diluted 1:10,000 in a blocking buffer.
2.9. Quantitative RT-PCR (qRT-PCR)
RNA was isolated using an RNeasy kit with on-column DNase treatment (Qiagen, Germantown, MD, USA). cDNA was generated using the Multiscribe Reverse Transcriptase kit (Thermo Fisher, Waltham, MA, USA). qPCR analysis was performed with Fast SYBR Green reagent (Invitrogen, Waltham, MA, USA) using a StepOne machine (Applied Biosystems, Waltham, MA, USA). Primers were validated by performing a standard melt curve analysis, and the results are listed in the Key Resources Table.
2.10. Immunohistochemistry, Immunofluorescence, and Imaging
H&E staining was performed using standard methods with hematoxylin (Vector Laboratories, Burlingame, CA, USA) and Eosin Y solution (MilliporeSigma, Burlington, MA, USA). According to the manufacturer’s protocol, Masson’s Trichrome staining was performed with a Trichrome Stain Kit (MilliporeSigma).
For immunohistochemistry, formalin-fixed, paraffin-embedded (FFPE) sections were de-paraffinized and rehydrated, and antigen retrieval was performed in either pH 6 citric or pH 9 Tris-EDTA buffer (Dako) in a pressure cooker for 10 min. After cooling for 20 min, slides were quenched in 3% hydrogen peroxide for 10 min and blocked with 5% goat serum and 2.5% BSA for 1 h at room temperature. Primary antibodies Ki67 (G8; 1:400), TUNEL(MilliporeSigma™ Chemicon™ ApopTag™ Plus Peroxidase In Situ Apoptosis Kit: S7101), and pMYC (4B12 1:300) [
30] were diluted in blocking buffer and tissues incubated overnight at 4 °C. Sections were incubated with anti-biotin secondary antibodies (1:1000) for 1 h, the Vectastain ABC kit (Vector Laboratories) for 1 h, and then exposed to the DAB substrate for 5–10 min for color development. Slides were counterstained with hematoxylin for 5 min and then mounted using Vectamount mounting media (Vector Laboratories).
For IF on FFPE tissue sections, slides were de-paraffinized and rehydrated, and antigen retrieval was performed using either a pH 6 citric or pH 9 Tris-EDTA-based buffer (Dako) in a pressure cooker for 10 min. After cooling for 20 min, slides were blocked with 10% goat serum and 1% BSA. Conjugated Primary antibodies Ki67 (CST:D3B5, 1:400), cleaved caspase-3 (CST:9661S, 1:50), Vimentin (CST:9854, 1:100), aSMA (SC:32251, 1:200), CD45 (CST:D3F8Q, 1:50), F480 (Biolegend:916104, 1:50), CollV (BDBiosciences:203003;1:100), PDPN (Biolegend:916606,1:100), CD8 (eBio:4SM15,1:100), CD11b (abcam:EPR1344,1:100), CD11c (CST:D4E3M,1:100), and pMYC (Sears lab, 4B12; 1:300) were diluted in blocking buffer and tissues incubated overnight at 4 °C. Slides were mounted with a SlowFade mounting reagent using DAPI. H&E and Trichrome images were acquired using an Aperio AT Scanscope with ImageScope (Leica Biosystems) software or a Zeiss AxioScan with Zeiss Zen software (Zeiss Microscopy, Thornwood, NY, USA). Images were acquired using a Zeiss AxioScan with Zeiss Zen software for the immunostaining of tissue sections. Tiled images were digitally stitched on Zen software to generate full scan images and analyzed with FIJI (ImageJ).
The quantification of IHC staining for pMYC and total MYC in tumors was performed through the deconvolution of the IHC image using ImageJ Fiji software version 2.1.0/1.53c.Color Deconvolution with the “H DAB” vector option to separate the hematoxylin staining (blue/purple) from the DAB staining (brown). The threshold was adjusted for the DAB staining image, and the % positive area was calculated by dividing the DAB positive area by the total area of the image. For the MiaPaCa-2 xenografts, 4 tumors of each type were analyzed. For the KPC tumors, 3 tumors were analyzed. At least 8 randomly selected ROIs were analyzed in each tumor.
2.11. Flow Cytometry
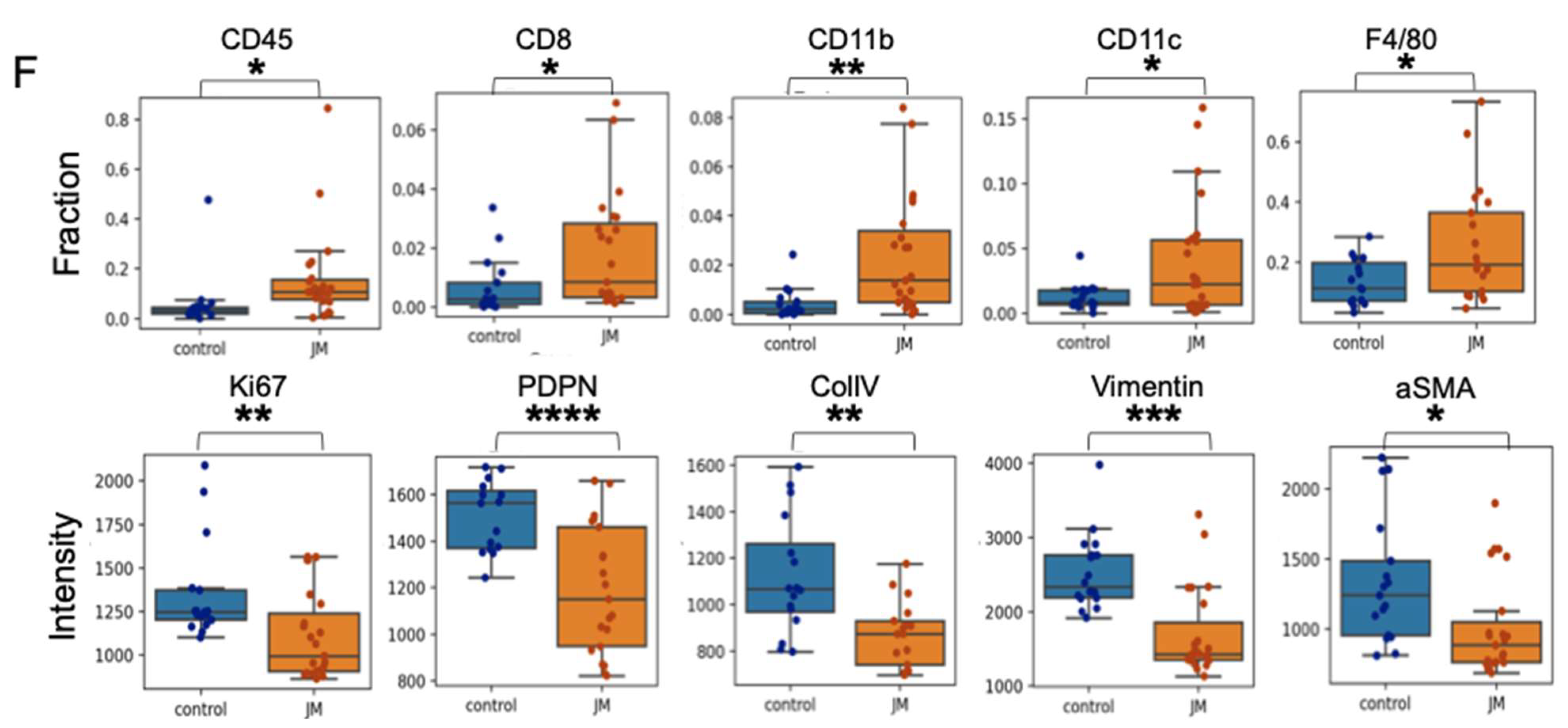
On day 18 of treatment, KPC7107 tumors (n = 3 per treatment group) were harvested in 1× PBS, minced to fine fragments, and incubated with Collagenase IV (Millipore Sigma, St. Louis, MO, USA), DNase I, Hyaluronidase, and a trypsin inhibitor (Sigma Aldrich, St. Louis, MO, USA) for 45 min at 37 °C on a rotating shaker. Enzymatic digestion was ceased by adding DMEM media containing 10% FBS. The resulting tissue homogenates were filtered through 70 μm cell strainers, and single-cell suspensions were collected and counted. Cell suspensions were then incubated with fixable viability dye-live dead Aqua (Thermo Fischer, L34966, 1:500) to gate viable cells. Non-specific antibody binding was blocked following incubation with the rat anti-mouse CD16/CD32 mAb (BD Bioscience, 553142, 12.4G2, 10 μg/mL) for 10 min at room temperature. Approximately 1 × 106 cells per sample were labeled with the various fluorochrome-conjugated antibodies, washed, and resuspended in 2% FBS, 1× PBS buffer. The anti-mouse antibodies used in the experiment are the following; CD4-BUV496 (BioLegend, San Diego, CA, USA, BDB612952, GK1.5), CD8-BUV395 (BioLegend, 135011, A7R34), B220-APC (BioLegend, 400511, RTK2758), CD8a-BUV395 (Biolegend, BDB563786, 53–6.7), Ly6C-PerCP (BD Bioscience, 12808, HK1.4), CD45-BV421 (Fischer Scientific, 50-604-825, 30-F11), Ly6G-APC (Fischer Scientific, 50-164-399, 1A8), CD3-APC-efluor 780 (BD Bioscience, 100347, 145-2C1), CD11b-Superbright 600 (eBioscience, San Diego, CA, USA, 63-0112-80, M1/70), CD11c-PE (BioLegend, 108407, RB6-8C5), F4/80-APC (BioLegend, 123116, BM8), CD45-FITC (BioLegend, 141720, C068C2), and MHCII-AF700 (eBioscience, eB14-5321, M5/114.15.2). Flow cytometry data were obtained using a Cytek Aurora flow cytometer and analyzed using FlowJo software. The data presented are representative of singlet live cells.
2.12. RNA-Sequencing
For RNA-sequencing, RNA was isolated from MiaPaCa-2 pancreatic orthotopic xenograft tumors using an RNeasy kit with on-column DNase treatment (Qiagen), and RNA integrity was assessed using an Agilent 2100 Bioanalyzer with an RNA 6000 Nano Chip. RNA integrity numbers (RINs) were calculated from Bioanalyzer electropherograms using the “Eukaryotic Total RNA Nano” program of the Bioanalyzer 2100 Expert software (B.02.08.SI648). RIN values were in the 8.5–10 range, indicating high-quality RNA. Library preparation and sequencing were performed using the OHSU Massively Parallel Sequencing Shared Resource (RRID SCR_009984). Briefly, cDNA libraries were prepared from poly(A)-selected RNA using an Illumina TruSeq Stranded mRNA library preparation kit (Illumina, San Diego, CA, USA). Poly(A)+ RNA was isolated from 100 ng of total RNA (per sample) using oligo-dT-coated magnetic beads, which were then chemically fragmented. This is followed by the generation of first-strand cDNA using random hexamers as primers for reverse transcriptase and the subsequent synthesis of strand-specific second-strand cDNA with the addition of a single ‘A’ nucleotide to each end. Illumina adaptors were then ligated to the cDNAs. A 15-cycle PCR was used to amplify the material to yield the final libraries. Library concentration was determined using real-time PCR with primers complementary to the Illumina adaptors. Sample libraries were diluted and applied to an Illumina HiSeq 2500 Sequencer with a target of 50 M reads per sample, which was then used to assemble the reads into standard FastQ formatted data. Trimmomatic (version 0.39) [
31] was used to trim low-quality reads and adapters (ILLUMINACLIP:2:30:10 SLIDINGWINDOW:5:25 LEADING:20 TRAILING:20 MINLEN:50). Fastqc (version 0.11.9) was used to assess the quality of trimmed reads. Only reads with forward and reverse pairs surviving were used for mapping. Mouse (GRCM39, refseq GCF_000001635.2) and human (GRCH38, refseq GCF_000001405.40) genomes were downloaded from NCBI and concatenated. This merged genome was then indexed and used to map trimmed reads using STAR (version 2.7.6). Reads aligning to mouse genes were removed from the count matrix. DESeq2 (version 1.34.0) [
32] was used to examine the differential expression of human-mapping genes. Visualizations were produced with ggplot2 (version 3.4.2) and EnhancedVolcano (version 1.12.0).
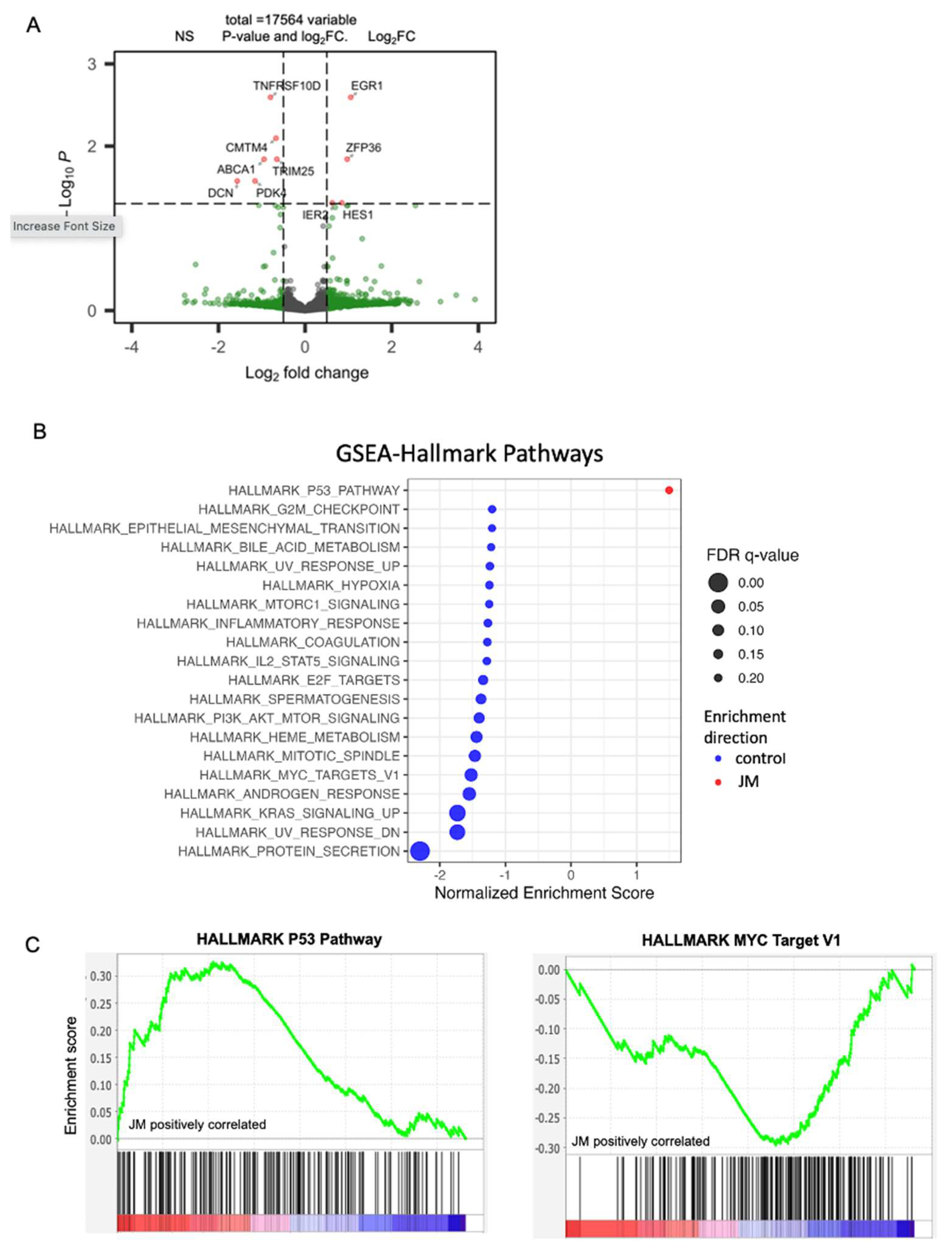
We used gene set enrichment analysis (GSEA) [
33] with default settings (1000 permutations) on the gene list obtained from DESeq data [
32] to compare enrichment to gene ontology and hallmark gene sets in the MSigDB database (version 7.5.1) [
34] to identify the pathways enriched by JM treatment. GSEA analysis was performed using the standalone GSEA application (version 4.3.2) using default parameters with normalized counts produced by using DESeq2 as the input. The hallmark gene set from MSigDB (version 2023.1) was used for pathway annotation. The statistically significant pathways were defined with a cutoff FDR < 0.25. In the setting of exploratory discovery, an FDR of 25% indicates that the result is likely to be valid 3 times out of 4, which is reasonable when one is interested in finding a candidate hypothesis to be further validated as a result of future research.
To find pathways and genes correlated with EGR1 expression in human PDAC data, we analyzed a pre-existing RNA-seq dataset [
35] that includes 218 primary human pancreatic tumor profiles. Sample collection, processing, and sequencing is described in the dataset [
35], and samples were obtained from the Oregon Pancreas Tissue Registry under IRB study # IRB00003609. Transcript abundances for the 218 primary tumor RNA-seq profiles were first summed into gene-level expression data using the R package tximport (v1.22.0) [
36], then organized into a DESeq2 object to which we applied variance-stabilizing transformation (VST). To perform GSVA [
37], we imported MSigDB hallmark gene sets via msigdbr (v.7.5.1) [
34] and applied GSVA on VST count data with the GSVA R package (v.1.42.0).
2.13. Data and Statistics
Three or more independent biological replicate experiments were performed in all cases except those indicated in the figure legend. All standard errors were calculated from biological replicates. For all work except sequencing analyses, GraphPad Prism software was used for statistical analysis; individual tests and p values are described in the figure legends. For all experiments, the sample size was estimated based on a two-sample t-test with 1% significance level. The selected sample size was expected to provide 80% power to detect a mean difference of a 1.0 standard deviation unit.
2.14. Data Accessibility
RNA-seq reads have been deposited in NCBI BioProject under the accession number PRJNA1014468.
4. Discussion
PDAC is characterized by a significantly low 5-year survival rate that has just recently reached 12%, indicating a poor prognosis for patients affected by this condition. This alarming statistic highlights the necessity for an innovative therapeutic approach. A growing body of research has compiled findings on the potential application of natural products as agents for inhibiting tumor growth. Natural products refer to the components derived from animals, plants, marine organisms, or microorganisms [
48]. Furthermore, various chemotherapeutic drugs and their analogs, including vincristine and paclitaxel, have been artificially produced using natural substances. Juglone, a naturally occurring compound, has demonstrated cytotoxic effects against various cancer types. Mechanistically, juglone inhibits cancer cell proliferation by inhibiting stem cell properties, epithelial-to-mesenchymal transition, and angiogenesis [
49], inhibiting cell cycle progression [
14], and/or by inducing apoptosis via mitochondrial-dependent pathways [
17]. Moreover, in vivo, juglone inhibited the progression of tumors in rodents by increasing apoptosis and cell cycle blockage [
50,
51].
While previous research, primarily conducted through in vitro experiments, has indicated that juglone has an impact on the proliferation, apoptosis, and metastasis of multiple cancer cell lines, there remains a need for further investigation into the role of juglone in vivo and in pancreatic cancer. In pancreatic cancer, juglone has been shown to exhibit inhibitory effects on the proliferation of PDAC cell lines through the induction of cell cycle arrest and apoptosis [
11]. Clinical use of juglone is hampered by its physicochemical properties, particularly its low water solubility, despite its potential therapeutic usefulness as an anticancer therapy. Prior research has examined the effects of juglone on animal tumor models such as liver metastasis [
13], melanoma [
39,
52], murine mammary adenocarcinoma [
50], and colorectal cancer [
53] utilizing free drug and liposomal formulations [
54]. However, these studies were affected by toxicity that limited them to a small number of administered doses (a total of three or four doses) every other day. Furthermore, no studies have been performed to evaluate the in vivo effect of juglone in pancreatic mouse models.
To overcome the inherent toxicity issue, we formulated a mixed micelle formulation of juglone. The data showed that the micelle formulation did not alter juglone’s cytotoxic properties but increased its water solubility. Upon evaluating its effect on proliferation, migration, and apoptosis in pancreatic cancer cells, the micelles inhibited proliferation, migration, and increased apoptosis in pancreatic cell lines, which is consistent with the published effect of juglone in pancreatic cell lines and other cancer cell lines. Herein, we performed in vivo experiments on both flank xenografts in the orthotopic PDAC models in immunocompetent and immunocompromised mice. We show tumor burden reduction compared to the control animals, which is consistent with the abovementioned studies. Hence, juglone micelles resolved the in vivo application of juglone and enhanced antitumor efficacy. This effect was correlated with increased apoptosis and decreased proliferation in all models. We also increased the antitumor effect and the tolerability of juglone as no observed side effects, such as death or weight loss, existed in mice with JM.
PDAC exhibits remarkable resistance to conventional therapy compared to other cancers and possesses a highly immunosuppressive tumor microenvironment. To interrogate if the antitumor effect of juglone is mediated by immune function, we used the syngeneic mouse model with a KPC cell line. Our data shows that juglone micelles induced an increased level of CD45+ compared to the control group. Moreover, we observed an increase in CD8+ cells in the treatment group compared to the control group. Meanwhile, we also showed an increase in CD11b+ cells, suggesting an effect on myeloid cells. We also observed decreased fibroblast activation markers such as vimentin, αSMA, and ColIV, suggesting JM modulated the stromal compartment of pancreatic cancer. Our data indicate that juglone exerts antitumor activity by directly acting on the cancer cells and modulating the immune and stromal microenvironments of PDAC.
TME is a complex and dynamic ecosystem of cancer cells, stromal cells, immune cells, and extracellular matrix components. The interactions between these various cellular and molecular elements play a crucial role in tumor growth, progression, and response to therapy. Beyond its direct antitumor effects, the treatment of JM has demonstrated a unique capacity to influence TME in immunocompetent mice, adding a dimension to its therapeutic potential. The immune system plays a pivotal role in recognizing and eliminating cancer cells. However, tumors often employ various mechanisms to evade immune surveillance. We found JM to promote the infiltration of immune cells, such as T cells, into the tumor microenvironment. This phenomenon indicates that JM can modulate the immunosuppressive TME, creating a more favorable environment for the immune system to mount an antitumor response. The increased immune cell infiltration in response to JM treatment indicates an immunomodulatory role. By promoting the presence and activity of immune effector cells within the tumor, JM can enhance the recognition and elimination of cancer cells, leading to an improved antitumor immune response. The stromal components of the TME, such as CAFs and ECM proteins, contribute to tumor growth and progression. These elements create a physical barrier that hinders drug penetration and immune cell infiltration. JM treatment was associated with a reduced ECM deposition, suggesting that it may remodel the tumor stroma and improve drug and immune cell accessibility to cancer cells. Activation markers on immune and stromal cells are indicative of their functional state.
JM treatment downregulates specific pro-tumor activation markers in the TME, potentially indicating a reduction in these cells’ supportive or immunosuppressive activity. This downregulation could contribute to the establishment of a more immune-permissive microenvironment. The immunomodulatory effects of JM could potentially complement immunotherapeutic approaches, such as immune checkpoint inhibitors or adoptive T-cell therapies. Combining JM with immunotherapies may enhance their efficacy by sensitizing the TME and augmenting the antitumor immune response. Overall, the observed influence of JM on the tumor microenvironment provides a novel perspective on its therapeutic potential. By reshaping the immune landscape and stromal composition within the tumor, JM can create a more permissive environment for antitumor immune responses and improve the overall effectiveness of cancer therapy. These findings underscore the importance of considering the immunomodulatory properties of JM and its potential for combination therapies with immunotherapeutic agents in the quest for more effective and personalized cancer treatments.
Numerous studies have furnished findings in favor of the notion that specific chemotherapeutic agents, as well as their nanoformulations, possess the capacity to induce cytotoxic effects and play a role in the restructuring of the tumor microenvironment. For instance, the therapeutic potential of a nanoparticle formulation containing oxaliplatin is believed to be enhanced by its increased cytotoxicity, the elevated response of dendritic cells, and the more significant infiltration of CD8+ T cells. Additionally, using this nanoparticle formulation reduces toxicity compared to administering free oxaliplatin [
55].
Moreover, incorporating a nitric oxide donor and recombinant tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) into the nanogel formulation can modify the desmoplastic stroma and reduce activity in the antiapoptotic pathway. The proposed alteration can increase the tumor-penetrating ability of TRAIL and significantly improve the effectiveness of TRAIL therapy in tumor treatment [
56]. While the exact mechanism behind the observed remodeling of the tumor microenvironment in response to JM requires further investigation, it is plausible that this phenomenon may contribute to the observed antitumor activity of this formulation. In summary, the data indicate that JM exhibits efficacy in inhibiting the advancement of PDAC by modulating the expression of markers linked to fibroblast activation and immune cells.
PDAC is a complex disease with various molecular alterations. Combining juglone with agents that target different signaling pathways can address the heterogeneity of the tumor and increase the likelihood of hitting multiple vulnerable points within cancer cells. Resistance to single-agent therapies is a common challenge in cancer treatment. With combination therapies, we can target multiple pathways simultaneously, making it more difficult for cancer cells to develop resistance. Combinatorial strategies can involve combining juglone with conventional chemotherapy, targeted therapies, immunotherapies, or other experimental agents. The choice of combination partners should be based on a solid understanding of the underlying molecular pathways driving pancreatic cancer growth, progression, and drug resistance. Based on our work, juglone’s immunomodulatory effects, when combined with immunotherapies or drugs that target the stromal components of the tumor, could enhance the overall antitumor response and promote a more favorable microenvironment for treatment. Identifying drugs that complement juglone’s mechanism of action could improve its ability to inhibit tumor growth, induce apoptosis, and suppress drug resistance. Combining juglone and other targeted agents or chemotherapy could help overcome resistance mechanisms and improve treatment outcomes. Moreover, based on our transcriptomics data, MYC might be a potential biomarker/target that can predict responses to juglone or other agents, enabling clinicians to design more effective and personalized treatment regimens.

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}