
Targeting Glucose Metabolism in Cancer Cells as an Approach to Overcoming Drug Resistance





Abstract
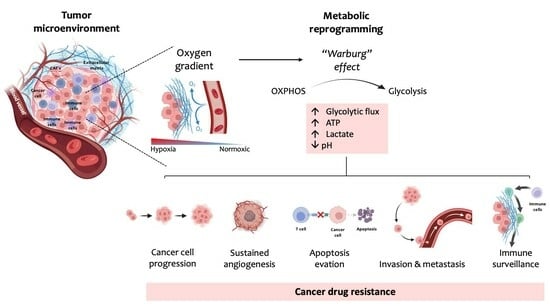
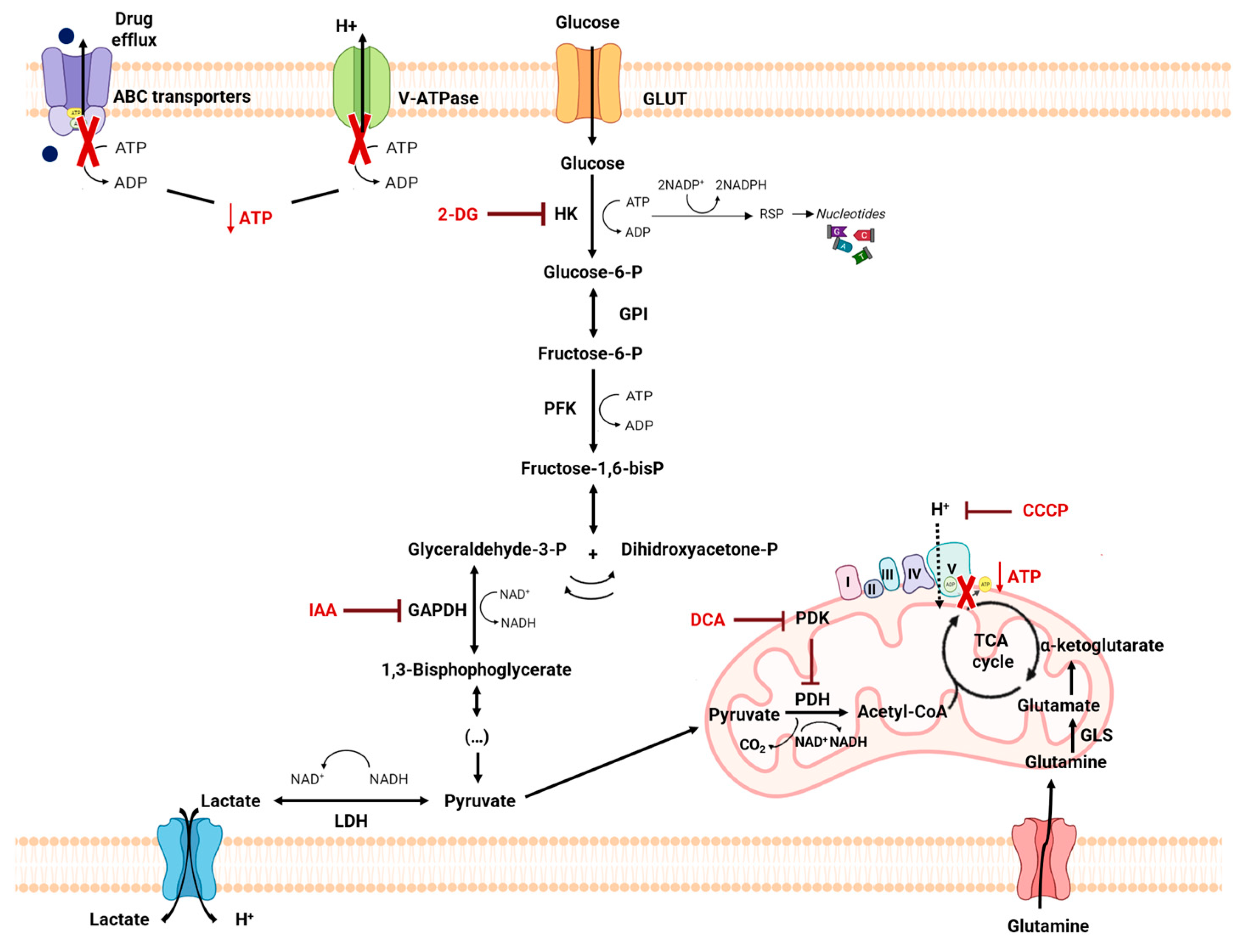
1. Introduction
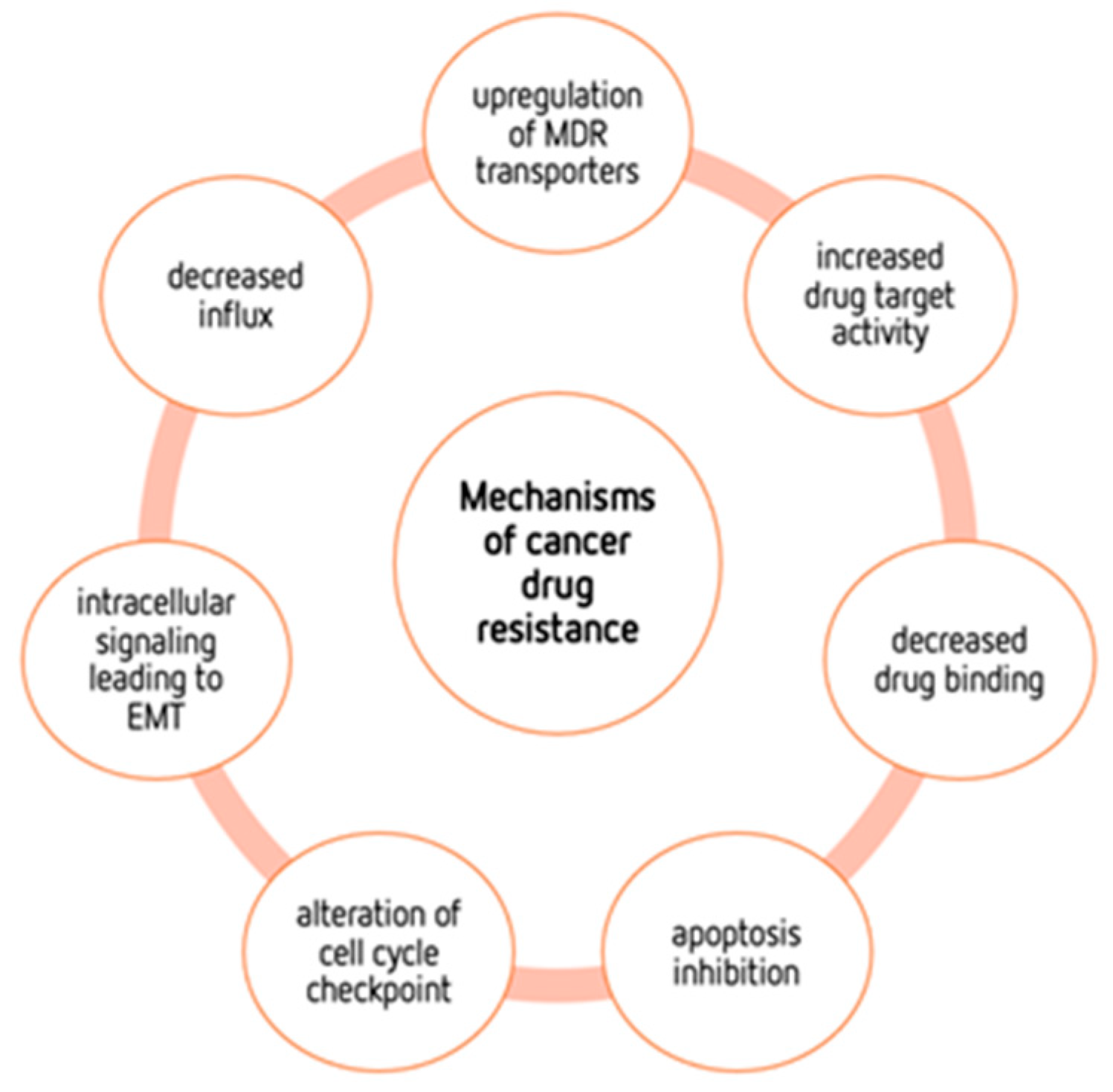
2. Glucose Metabolism
3. The Warburg Effect
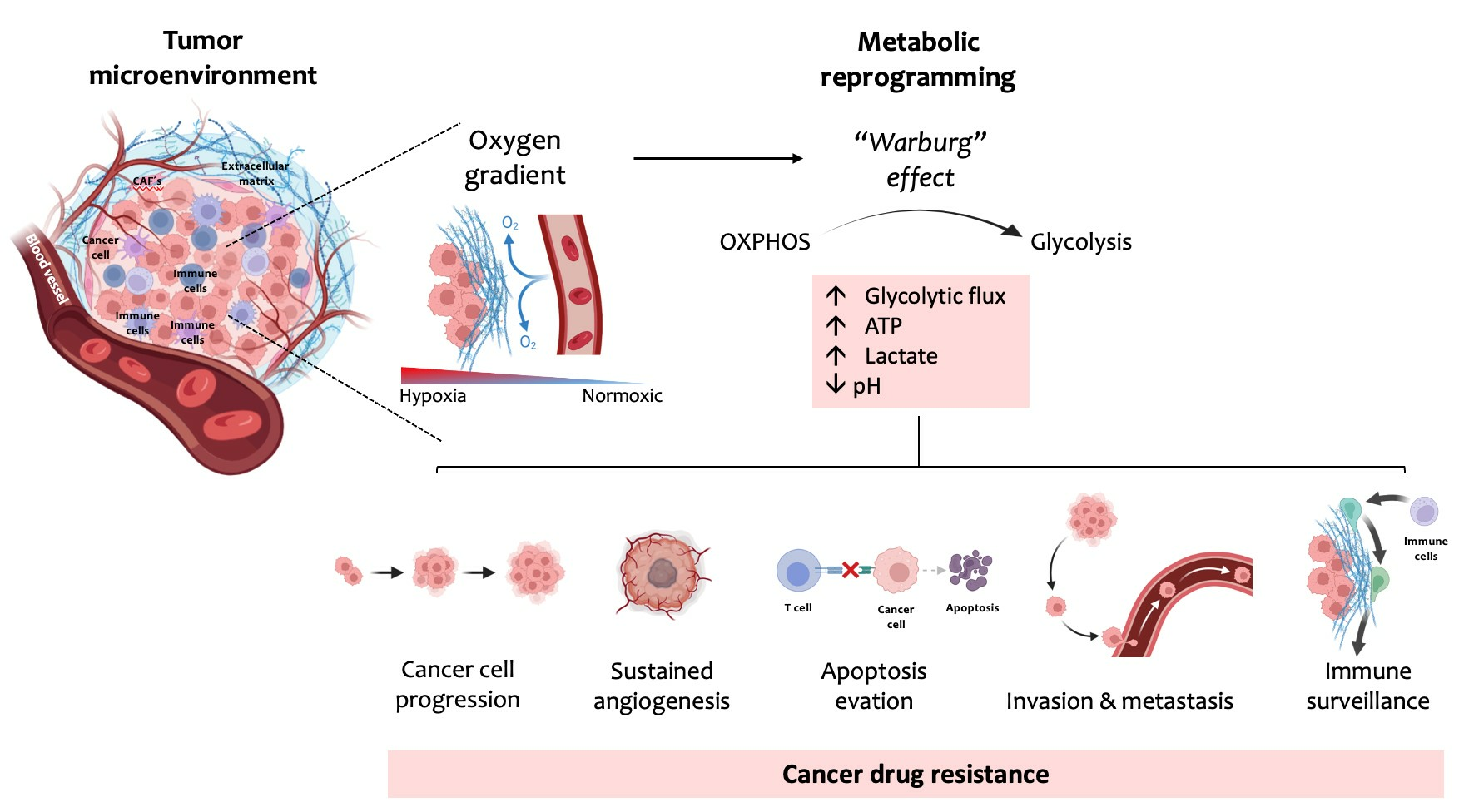
4. Mechanisms of Cancers’ Drug Resistance
4.1. ABC Transporters
4.1.1. MDR1 Transporter
4.1.2. MRP1 Transporter
4.1.3. BCRP Transporter
4.2. Metabolic Alterations Involved in Drug Resistance in Cancer
4.3. Metabolic Modulation as an Approach to Overcome Drug Resistance
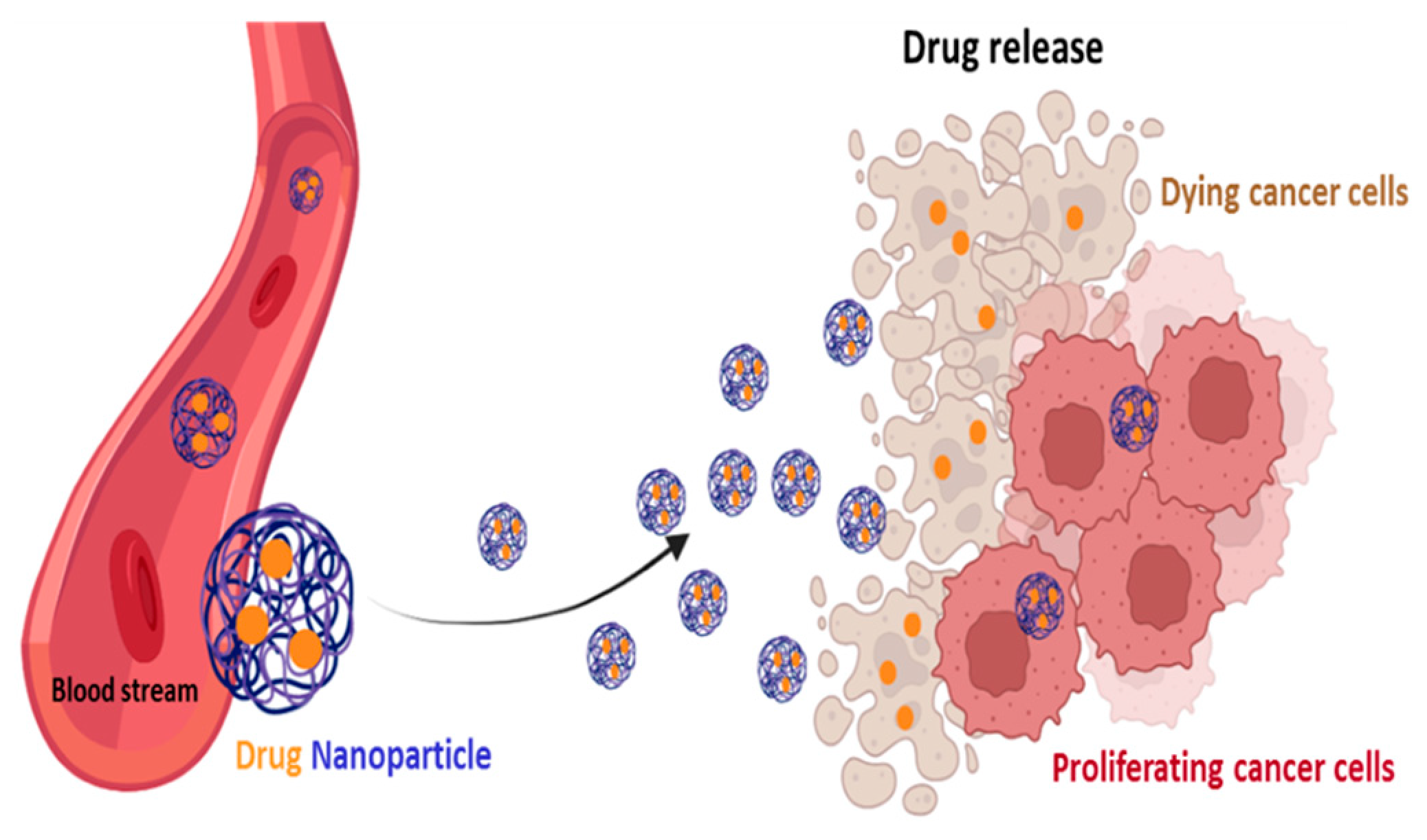
4.4. Self-Delivery of Nanomedicine to Overcome Drug Resistance
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Zaal, E.A.; Berkers, C.R. The Influence of Metabolism on Drug Response in Cancer. Front. Oncol. 2018, 8, 500. [Google Scholar] [CrossRef]
- Chen, X.; Chen, S.; Yu, D. Metabolic Reprogramming of Chemoresistant Cancer Cells and the Potential Significance of Metabolic Regulation in the Reversal of Cancer Chemoresistance. Metabolites 2020, 10, 289. [Google Scholar] [CrossRef]
- Ortega, A.D.; Sanchez-Arago, M.; Giner-Sanchez, D.; Sanchez-Cenizo, L.; Willers, I.; Cuezva, J.M. Glucose avidity of carcinomas. Cancer Lett. 2009, 276, 125–135. [Google Scholar] [CrossRef]
- Vanhove, K.; Graulus, G.J.; Mesotten, L.; Thomeer, M.; Derveaux, E.; Noben, J.P.; Guedens, W.; Adriaensens, P. The Metabolic Landscape of Lung Cancer: New Insights in a Disturbed Glucose Metabolism. Front. Oncol. 2019, 9, 1215. [Google Scholar] [CrossRef]
- Cameron, M.E.; Yakovenko, A.; Trevino, J.G. Glucose and Lactate Transport in Pancreatic Cancer: Glycolytic Metabolism Revisited. J. Oncol. 2018, 2018, 6214838. [Google Scholar] [CrossRef]
- Reckzeh, E.S.; Waldmann, H. Small-Molecule Inhibition of Glucose Transporters GLUT-1-4. Chembiochem 2020, 21, 45–52. [Google Scholar] [CrossRef]
- Holman, G.D. Structure, function and regulation of mammalian glucose transporters of the SLC2 family. Pflug. Arch. 2020, 472, 1155–1175. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Moreno-Sanchez, R.; Rodriguez-Enriquez, S.; Marin-Hernandez, A.; Saavedra, E. Energy metabolism in tumor cells. FEBS J. 2007, 274, 1393–1418. [Google Scholar] [CrossRef] [PubMed]
- Gillies, R.J.; Robey, I.; Gatenby, R.A. Causes and consequences of increased glucose metabolism of cancers. J. Nucl. Med. 2008, 49 (Suppl. S2), 24S–42S. [Google Scholar] [CrossRef]
- Sullivan, L.B.; Gui, D.Y.; Hosios, A.M.; Bush, L.N.; Freinkman, E.; Vander Heiden, M.G. Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 2015, 162, 552–563. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef]
- Yoo, H.C.; Yu, Y.C.; Sung, Y.; Han, J.M. Glutamine reliance in cell metabolism. Exp. Mol. Med. 2020, 52, 1496–1516. [Google Scholar] [CrossRef] [PubMed]
- Hui, S.; Ghergurovich, J.M.; Morscher, R.J.; Jang, C.; Teng, X.; Lu, W.; Esparza, L.A.; Reya, T.; Le, Z.; Yanxiang Guo, J.; et al. Glucose feeds the TCA cycle via circulating lactate. Nature 2017, 551, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Mashimo, T.; Pichumani, K.; Vemireddy, V.; Hatanpaa, K.J.; Singh, D.K.; Sirasanagandla, S.; Nannepaga, S.; Piccirillo, S.G.; Kovacs, Z.; Foong, C.; et al. Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell 2014, 159, 1603–1614. [Google Scholar] [CrossRef]
- Green, C.R.; Wallace, M.; Divakaruni, A.S.; Phillips, S.A.; Murphy, A.N.; Ciaraldi, T.P.; Metallo, C.M. Branched-chain amino acid catabolism fuels adipocyte differentiation and lipogenesis. Nat. Chem. Biol. 2016, 12, 15–21. [Google Scholar] [CrossRef]
- Amemiya, T.; Yamaguchi, T. Oscillations and Dynamic Symbiosis in Cellular Metabolism in Cancer. Front. Oncol. 2022, 12, 783908. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Peiris-Pages, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer metabolism: A therapeutic perspective. Nat. Rev. Clin. Oncol. 2017, 14, 11–31. [Google Scholar] [CrossRef]
- Zheng, J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Yang, Z.; Chen, Y. A Novel Oxidative Phosphorylation-Associated Gene Signature for Prognosis Prediction in Patients with Hepatocellular Carcinoma. Dis. Markers 2022, 2022, 3594901. [Google Scholar] [CrossRef]
- Miranda-Goncalves, V.; Honavar, M.; Pinheiro, C.; Martinho, O.; Pires, M.M.; Pinheiro, C.; Cordeiro, M.; Bebiano, G.; Costa, P.; Palmeirim, I.; et al. Monocarboxylate transporters (MCTs) in gliomas: Expression and exploitation as therapeutic targets. Neuro Oncol. 2013, 15, 172–188. [Google Scholar] [CrossRef] [PubMed]
- Suganuma, K.; Miwa, H.; Imai, N.; Shikami, M.; Gotou, M.; Goto, M.; Mizuno, S.; Takahashi, M.; Yamamoto, H.; Hiramatsu, A.; et al. Energy metabolism of leukemia cells: Glycolysis versus oxidative phosphorylation. Leuk. Lymphoma 2010, 51, 2112–2119. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Coelho, F.; Nunes, C.; Gouveia-Fernandes, S.; Rosas, R.; Silva, F.; Gameiro, P.; Carvalho, T.; Gomes da Silva, M.; Cabecadas, J.; Dias, S.; et al. Monocarboxylate transporter 1 (MCT1), a tool to stratify acute myeloid leukemia (AML) patients and a vehicle to kill cancer cells. Oncotarget 2017, 8, 82803–82823. [Google Scholar] [CrossRef]
- Evans, K.W.; Yuca, E.; Scott, S.S.; Zhao, M.; Paez Arango, N.; Cruz Pico, C.X.; Saridogan, T.; Shariati, M.; Class, C.A.; Bristow, C.A.; et al. Oxidative Phosphorylation Is a Metabolic Vulnerability in Chemotherapy-Resistant Triple-Negative Breast Cancer. Cancer Res. 2021, 81, 5572–5581. [Google Scholar] [CrossRef]
- Queiros, O.; Preto, A.; Pacheco, A.; Pinheiro, C.; Azevedo-Silva, J.; Moreira, R.; Pedro, M.; Ko, Y.H.; Pedersen, P.L.; Baltazar, F.; et al. Butyrate activates the monocarboxylate transporter MCT4 expression in breast cancer cells and enhances the antitumor activity of 3-bromopyruvate. J. Bioenerg. Biomembr. 2012, 44, 141–153. [Google Scholar] [CrossRef]
- Morais-Santos, F.; Granja, S.; Miranda-Goncalves, V.; Moreira, A.H.; Queiros, S.; Vilaca, J.L.; Schmitt, F.C.; Longatto-Filho, A.; Paredes, J.; Baltazar, F.; et al. Targeting lactate transport suppresses in vivo breast tumour growth. Oncotarget 2015, 6, 19177–19189. [Google Scholar] [CrossRef]
- Chen, E.; Wang, T.; Zhang, J.; Zhou, X.; Niu, Y.; Liu, F.; Zhong, Y.; Huang, D.; Chen, W. Mitochondrial Targeting and pH-Responsive Nanogels for Co-Delivery of Lonidamine and Paclitaxel to Conquer Drug Resistance. Front. Bioeng. Biotechnol. 2021, 9, 787320. [Google Scholar] [CrossRef]
- Jiang, H.; Zhang, L.; Kuo, J.; Kuo, K.; Gautam, S.C.; Groc, L.; Rodriguez, A.I.; Koubi, D.; Hunter, T.J.; Corcoran, G.B.; et al. Resveratrol-induced apoptotic death in human U251 glioma cells. Mol. Cancer Ther. 2005, 4, 554–561. [Google Scholar] [CrossRef]
- Adeshakin, F.O.; Adeshakin, A.O.; Liu, Z.; Cheng, J.; Zhang, P.; Yan, D.; Zhang, G.; Wan, X. Targeting Oxidative Phosphorylation-Proteasome Activity in Extracellular Detached Cells Promotes Anoikis and Inhibits Metastasis. Life 2021, 12, 42. [Google Scholar] [CrossRef]
- Tavares-Valente, D.; Granja, S.; Baltazar, F.; Queiros, O. Bioenergetic modulators hamper cancer cell viability and enhance response to chemotherapy. J. Cell. Mol. Med. 2018, 22, 3782–3794. [Google Scholar] [CrossRef]
- Vorobyev, P.O.; Kochetkov, D.V.; Chumakov, P.M.; Zakirova, N.F.; Zotova-Nefedorova, S.I.; Vasilenko, K.V.; Alekseeva, O.N.; Kochetkov, S.N.; Bartosch, B.; Lipatova, A.V.; et al. 2-Deoxyglucose, an Inhibitor of Glycolysis, Enhances the Oncolytic Effect of Coxsackievirus. Cancers 2022, 14, 5611. [Google Scholar] [CrossRef] [PubMed]
- Parczyk, J.; Ruhnau, J.; Pelz, C.; Schilling, M.; Wu, H.; Piaskowski, N.N.; Eickholt, B.; Kuhn, H.; Danker, K.; Klein, A. Dichloroacetate and PX-478 exhibit strong synergistic effects in a various number of cancer cell lines. BMC Cancer 2021, 21, 481. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Liu, Z.; Zou, X.; Lan, Y.; Sun, X.; Wang, X.; Zhao, S.; Jiang, C.; Liu, H. Mechanisms underlying 3-bromopyruvate-induced cell death in colon cancer. J. Bioenerg. Biomembr. 2015, 47, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Goncalves, V.; Granja, S.; Martinho, O.; Honavar, M.; Pojo, M.; Costa, B.M.; Pires, M.M.; Pinheiro, C.; Cordeiro, M.; Bebiano, G.; et al. Hypoxia-mediated upregulation of MCT1 expression supports the glycolytic phenotype of glioblastomas. Oncotarget 2016, 7, 46335–46353. [Google Scholar] [CrossRef]
- Cunha, A.; Rocha, A.C.; Barbosa, F.; Baiao, A.; Silva, P.; Sarmento, B.; Queiros, O. Glycolytic Inhibitors Potentiated the Activity of Paclitaxel and Their Nanoencapsulation Increased Their Delivery in a Lung Cancer Model. Pharmaceutics 2022, 14, 2021. [Google Scholar] [CrossRef]
- Herst, P.M.; Berridge, M.V. Cell surface oxygen consumption: A major contributor to cellular oxygen consumption in glycolytic cancer cell lines. Biochim. Biophys. Acta 2007, 1767, 170–177. [Google Scholar] [CrossRef]
- Ullah, M.S.; Davies, A.J.; Halestrap, A.P. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J. Biol. Chem. 2006, 281, 9030–9037. [Google Scholar] [CrossRef]
- Porporato, P.E.; Payen, V.L.; Perez-Escuredo, J.; De Saedeleer, C.J.; Danhier, P.; Copetti, T.; Dhup, S.; Tardy, M.; Vazeille, T.; Bouzin, C.; et al. A mitochondrial switch promotes tumor metastasis. Cell Rep. 2014, 8, 754–766. [Google Scholar] [CrossRef]
- De Saedeleer, C.J.; Porporato, P.E.; Copetti, T.; Perez-Escuredo, J.; Payen, V.L.; Brisson, L.; Feron, O.; Sonveaux, P. Glucose deprivation increases monocarboxylate transporter 1 (MCT1) expression and MCT1-dependent tumor cell migration. Oncogene 2014, 33, 4060–4068. [Google Scholar] [CrossRef] [PubMed]
- Kapur, A.; Mehta, P.; Simmons, A.D.; Ericksen, S.S.; Mehta, G.; Palecek, S.P.; Felder, M.; Stenerson, Z.; Nayak, A.; Dominguez, J.M.A.; et al. Atovaquone: An Inhibitor of Oxidative Phosphorylation as Studied in Gynecologic Cancers. Cancers 2022, 14, 2297. [Google Scholar] [CrossRef] [PubMed]
- Valera, V.F.M.; Prabharasuth, D.; Chaimowitz, M.; Choudhury, M.; Phillips, J.; Konno, S. Is targeting glycolysis with 2-deoxyglucose a viable therapeutic approach to bladder cancer? Int. J. Cancer Ther. Oncol. 2017, 5, 511. [Google Scholar]
- Ho, N.; Morrison, J.; Silva, A.; Coomber, B.L. The effect of 3-bromopyruvate on human colorectal cancer cells is dependent on glucose concentration but not hexokinase II expression. Biosci. Rep. 2016, 36, e00299. [Google Scholar] [CrossRef]
- Yu, H.; Zhang, H.; Dong, M.; Wu, Z.; Shen, Z.; Xie, Y.; Kong, Z.; Dai, X.; Xu, B. Metabolic reprogramming and AMPKalpha1 pathway activation by caulerpin in colorectal cancer cells. Int. J. Oncol. 2017, 50, 161–172. [Google Scholar] [CrossRef]
- Madhok, B.M.; Yeluri, S.; Perry, S.L.; Hughes, T.A.; Jayne, D.G. Dichloroacetate induces apoptosis and cell-cycle arrest in colorectal cancer cells. Br. J. Cancer 2010, 102, 1746–1752. [Google Scholar] [CrossRef]
- Olinger, A.M.P.; Tummala, H. Effect of 2-Deoxyglucose on Colorectal Cancer Cell Lines. J. Undergrad. Res. 2013, 11, 5. [Google Scholar]
- Miranda-Goncalves, V.; Goncalves, C.S.; Granja, S.; Vieira de Castro, J.; Reis, R.M.; Costa, B.M.; Baltazar, F. MCT1 Is a New Prognostic Biomarker and Its Therapeutic Inhibition Boosts Response to Temozolomide in Human Glioblastoma. Cancers 2021, 13, 3468. [Google Scholar] [CrossRef]
- Akers, L.J.; Fang, W.; Levy, A.G.; Franklin, A.R.; Huang, P.; Zweidler-McKay, P.A. Targeting glycolysis in leukemia: A novel inhibitor 3-BrOP in combination with rapamycin. Leuk. Res. 2011, 35, 814–820. [Google Scholar] [CrossRef]
- Saulle, E.; Spinello, I.; Quaranta, M.T.; Pasquini, L.; Pelosi, E.; Iorio, E.; Castelli, G.; Chirico, M.; Pisanu, M.E.; Ottone, T.; et al. Targeting Lactate Metabolism by Inhibiting MCT1 or MCT4 Impairs Leukemic Cell Proliferation, Induces Two Different Related Death-Pathways and Increases Chemotherapeutic Sensitivity of Acute Myeloid Leukemia Cells. Front. Oncol. 2020, 10, 621458. [Google Scholar] [CrossRef]
- Vital, P.D.S.; Bonatelli, M.; Dias, M.P.; de Salis, L.V.V.; Pinto, M.T.; Baltazar, F.; Maria-Engler, S.S.; Pinheiro, C. 3-Bromopyruvate Suppresses the Malignant Phenotype of Vemurafenib-Resistant Melanoma Cells. Int. J. Mol. Sci. 2022, 23, 15650. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Enriquez, S.; Carreno-Fuentes, L.; Gallardo-Perez, J.C.; Saavedra, E.; Quezada, H.; Vega, A.; Marin-Hernandez, A.; Olin-Sandoval, V.; Torres-Marquez, M.E.; Moreno-Sanchez, R. Oxidative phosphorylation is impaired by prolonged hypoxia in breast and possibly in cervix carcinoma. Int. J. Biochem. Cell Biol. 2010, 42, 1744–1751. [Google Scholar] [CrossRef] [PubMed]
- Tavares-Valente, D.; Baltazar, F.; Moreira, R.; Queiros, O. Cancer cell bioenergetics and pH regulation influence breast cancer cell resistance to paclitaxel and doxorubicin. J. Bioenerg. Biomembr. 2013, 45, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Korga, A.; Ostrowska, M.; Iwan, M.; Herbet, M.; Dudka, J. Inhibition of glycolysis disrupts cellular antioxidant defense and sensitizes HepG2 cells to doxorubicin treatment. FEBS Open Bio 2019, 9, 959–972. [Google Scholar] [CrossRef] [PubMed]
- Jardim-Messeder, D.; Moreira-Pacheco, F. 3-Bromopyruvic Acid Inhibits Tricarboxylic Acid Cycle and Glutaminolysis in HepG2 Cells. Anticancer Res. 2016, 36, 2233–2241. [Google Scholar]
- Jeon, J.Y.; Lee, M.; Whang, S.H.; Kim, J.W.; Cho, A.; Yun, M. Regulation of Acetate Utilization by Monocarboxylate Transporter 1 (MCT1) in Hepatocellular Carcinoma (HCC). Oncol. Res. 2018, 26, 71–81. [Google Scholar] [CrossRef]
- Sun, H.; Zhu, A.; Zhou, X.; Wang, F. Suppression of pyruvate dehydrogenase kinase-2 re-sensitizes paclitaxel-resistant human lung cancer cells to paclitaxel. Oncotarget 2017, 8, 52642–52650. [Google Scholar] [CrossRef]
- Danhier, P.; Banski, P.; Payen, V.L.; Grasso, D.; Ippolito, L.; Sonveaux, P.; Porporato, P.E. Cancer metabolism in space and time: Beyond the Warburg effect. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 556–572. [Google Scholar] [CrossRef]
- Vaupel, P.; Multhoff, G. Revisiting the Warburg effect: Historical dogma versus current understanding. J. Physiol. 2021, 599, 1745–1757. [Google Scholar] [CrossRef]
- Ghanbari Movahed, Z.; Rastegari-Pouyani, M.; Mohammadi, M.H.; Mansouri, K. Cancer cells change their glucose metabolism to overcome increased ROS: One step from cancer cell to cancer stem cell? Biomed. Pharmacother. 2019, 112, 108690. [Google Scholar] [CrossRef]
- Nadzialek, S.; Vanparys, C.; Van der Heiden, E.; Michaux, C.; Brose, F.; Scippo, M.L.; De Coen, W.; Kestemont, P. Understanding the gap between the estrogenicity of an effluent and its real impact into the wild. Sci. Total Environ. 2010, 408, 812–821. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Pouyssegur, J. Tumor cell metabolism: Cancer’s Achilles’ heel. Cancer Cell 2008, 13, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Kamarajugadda, S.; Stemboroski, L.; Cai, Q.; Simpson, N.E.; Nayak, S.; Tan, M.; Lu, J. Glucose oxidation modulates anoikis and tumor metastasis. Mol. Cell. Biol. 2012, 32, 1893–1907. [Google Scholar] [CrossRef] [PubMed]
- Jesser, E.A.; Brady, N.J.; Huggins, D.N.; Witschen, P.M.; O’Connor, C.H.; Schwertfeger, K.L. STAT5 is activated in macrophages by breast cancer cell-derived factors and regulates macrophage function in the tumor microenvironment. Breast Cancer Res. 2021, 23, 104. [Google Scholar] [CrossRef]
- Putney, L.K.; Barber, D.L. Expression profile of genes regulated by activity of the Na-H exchanger NHE1. BMC Genom. 2004, 5, 46. [Google Scholar] [CrossRef]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef]
- Mazurek, S. Pyruvate kinase type M2: A key regulator of the metabolic budget system in tumor cells. Int. J. Biochem. Cell Biol. 2011, 43, 969–980. [Google Scholar] [CrossRef]
- Murugan, A.K.; Alzahrani, A.S. Isocitrate Dehydrogenase IDH1 and IDH2 Mutations in Human Cancer: Prognostic Implications for Gliomas. Br. J. Biomed. Sci. 2022, 79, 10208. [Google Scholar] [CrossRef]
- Fu, Y.; Liu, S.; Yin, S.; Niu, W.; Xiong, W.; Tan, M.; Li, G.; Zhou, M. The reverse Warburg effect is likely to be an Achilles’ heel of cancer that can be exploited for cancer therapy. Oncotarget 2017, 8, 57813–57825. [Google Scholar] [CrossRef]
- Fan, T.W.; Kucia, M.; Jankowski, K.; Higashi, R.M.; Ratajczak, J.; Ratajczak, M.Z.; Lane, A.N. Rhabdomyosarcoma cells show an energy producing anabolic metabolic phenotype compared with primary myocytes. Mol. Cancer 2008, 7, 79. [Google Scholar] [CrossRef]
- Minemura, H.; Takagi, K.; Sato, A.; Yamaguchi, M.; Hayashi, C.; Miki, Y.; Harada-Shoji, N.; Miyashita, M.; Sasano, H.; Suzuki, T. Isoforms of IDH in breast carcinoma: IDH2 as a potent prognostic factor associated with proliferation in estrogen-receptor positive cases. Breast Cancer 2021, 28, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Lunetti, P.; Di Giacomo, M.; Vergara, D.; De Domenico, S.; Maffia, M.; Zara, V.; Capobianco, L.; Ferramosca, A. Metabolic reprogramming in breast cancer results in distinct mitochondrial bioenergetics between luminal and basal subtypes. FEBS J. 2019, 286, 688–709. [Google Scholar] [CrossRef] [PubMed]
- Pertega-Gomes, N.; Vizcaino, J.R.; Attig, J.; Jurmeister, S.; Lopes, C.; Baltazar, F. A lactate shuttle system between tumour and stromal cells is associated with poor prognosis in prostate cancer. BMC Cancer 2014, 14, 352. [Google Scholar] [CrossRef] [PubMed]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef]
- Arcucci, A.; Ruocco, M.R.; Granato, G.; Sacco, A.M.; Montagnani, S. Cancer: An Oxidative Crosstalk between Solid Tumor Cells and Cancer Associated Fibroblasts. Biomed. Res. Int. 2016, 2016, 4502846. [Google Scholar] [CrossRef]
- Chan, J.S.; Tan, M.J.; Sng, M.K.; Teo, Z.; Phua, T.; Choo, C.C.; Li, L.; Zhu, P.; Tan, N.S. Cancer-associated fibroblasts enact field cancerization by promoting extratumoral oxidative stress. Cell Death Dis. 2017, 8, e2562. [Google Scholar] [CrossRef]
- Faubert, B.; Li, K.Y.; Cai, L.; Hensley, C.T.; Kim, J.; Zacharias, L.G.; Yang, C.; Do, Q.N.; Doucette, S.; Burguete, D.; et al. Lactate Metabolism in Human Lung Tumors. Cell 2017, 171, 358–371.e359. [Google Scholar] [CrossRef]
- Roy, S.; Kumaravel, S.; Sharma, A.; Duran, C.L.; Bayless, K.J.; Chakraborty, S. Hypoxic tumor microenvironment: Implications for cancer therapy. Exp. Biol. Med. 2020, 245, 1073–1086. [Google Scholar] [CrossRef]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef]
- Rezayatmand, H.; Razmkhah, M.; Razeghian-Jahromi, I. Drug resistance in cancer therapy: The Pandora’s Box of cancer stem cells. Stem Cell Res. Ther. 2022, 13, 181. [Google Scholar] [CrossRef]
- De Las Rivas, J.; Brozovic, A.; Izraely, S.; Casas-Pais, A.; Witz, I.P.; Figueroa, A. Cancer drug resistance induced by EMT: Novel therapeutic strategies. Arch. Toxicol. 2021, 95, 2279–2297. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.I.; Haber, M.; Henderson, M.J.; Norris, M.D. ABC transporters in cancer: More than just drug efflux pumps. Nat. Rev. Cancer 2010, 10, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Ley, T.J.; Larson, D.E.; Miller, C.A.; Koboldt, D.C.; Welch, J.S.; Ritchey, J.K.; Young, M.A.; Lamprecht, T.; McLellan, M.D.; et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012, 481, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. Mechanisms of resistance to therapies targeting BRCA-mutant cancers. Nat. Med. 2013, 19, 1381–1388. [Google Scholar] [CrossRef]
- Kim, H.; Xu, H.; George, E.; Hallberg, D.; Kumar, S.; Jagannathan, V.; Medvedev, S.; Kinose, Y.; Devins, K.; Verma, P.; et al. Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models. Nat. Commun. 2020, 11, 3726. [Google Scholar] [CrossRef]
- Kaplon, J.; Zheng, L.; Meissl, K.; Chaneton, B.; Selivanov, V.A.; Mackay, G.; van der Burg, S.H.; Verdegaal, E.M.; Cascante, M.; Shlomi, T.; et al. A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature 2013, 498, 109–112. [Google Scholar] [CrossRef]
- Swanton, C. Intratumor heterogeneity: Evolution through space and time. Cancer Res. 2012, 72, 4875–4882. [Google Scholar] [CrossRef]
- Jia, D.; Park, J.H.; Kaur, H.; Jung, K.H.; Yang, S.; Tripathi, S.; Galbraith, M.; Deng, Y.; Jolly, M.K.; Kaipparettu, B.A.; et al. Towards decoding the coupled decision-making of metabolism and epithelial-to-mesenchymal transition in cancer. Br. J. Cancer 2021, 124, 1902–1911. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Seidel, M.G.; Wu, W.; Kamizono, S.; Ferrando, A.A.; Bronson, R.T.; Iwasaki, H.; Akashi, K.; Morimoto, A.; Hitzler, J.K.; et al. Slug, a highly conserved zinc finger transcriptional repressor, protects hematopoietic progenitor cells from radiation-induced apoptosis in vivo. Cancer Cell 2002, 2, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Olmeda, D.; Moreno-Bueno, G.; Flores, J.M.; Fabra, A.; Portillo, F.; Cano, A. SNAI1 is required for tumor growth and lymph node metastasis of human breast carcinoma MDA-MB-231 cells. Cancer Res. 2007, 67, 11721–11731. [Google Scholar] [CrossRef] [PubMed]
- Tavares-Valente, D.; Cannone, S.; Greco, M.R.; Carvalho, T.M.A.; Baltazar, F.; Queiros, O.; Agrimi, G.; Reshkin, S.J.; Cardone, R.A. Extracellular Matrix Collagen I Differentially Regulates the Metabolic Plasticity of Pancreatic Ductal Adenocarcinoma Parenchymal Cell and Cancer Stem Cell. Cancers 2023, 15, 3868. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.; Han, Z.; Zhang, S.; Liu, Y.; Wei, L. Epithelial-Mesenchymal Transition in tumor microenvironment. Cell Biosci. 2011, 1, 29. [Google Scholar] [CrossRef]
- Taki, M.; Abiko, K.; Ukita, M.; Murakami, R.; Yamanoi, K.; Yamaguchi, K.; Hamanishi, J.; Baba, T.; Matsumura, N.; Mandai, M. Tumor Immune Microenvironment during Epithelial-Mesenchymal Transition. Clin. Cancer Res. 2021, 27, 4669–4679. [Google Scholar] [CrossRef]
- Wang, Q.; Wu, M.; Li, H.; Rao, X.; Ao, L.; Wang, H.; Yao, L.; Wang, X.; Hong, X.; Wang, J.; et al. Therapeutic targeting of glutamate dehydrogenase 1 that links metabolic reprogramming and Snail-mediated epithelial-mesenchymal transition in drug-resistant lung cancer. Pharmacol. Res. 2022, 185, 106490. [Google Scholar] [CrossRef]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef]
- Brown, M.S.; Abdollahi, B.; Wilkins, O.M.; Lu, H.; Chakraborty, P.; Ognjenovic, N.B.; Muller, K.E.; Jolly, M.K.; Christensen, B.C.; Hassanpour, S.; et al. Phenotypic heterogeneity driven by plasticity of the intermediate EMT state governs disease progression and metastasis in breast cancer. Sci. Adv. 2022, 8, eabj8002. [Google Scholar] [CrossRef]
- Ochi, K.; Suzawa, K.; Tomida, S.; Shien, K.; Takano, J.; Miyauchi, S.; Takeda, T.; Miura, A.; Araki, K.; Nakata, K.; et al. Overcoming epithelial-mesenchymal transition-mediated drug resistance with monensin-based combined therapy in non-small cell lung cancer. Biochem. Biophys. Res. Commun. 2020, 529, 760–765. [Google Scholar] [CrossRef]
- Du, B.; Shim, J.S. Targeting Epithelial-Mesenchymal Transition (EMT) to Overcome Drug Resistance in Cancer. Molecules 2016, 21, 965. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, M. Epithelial-mesenchymal transition is regulated at post-transcriptional levels by transforming growth factor-beta signaling during tumor progression. Cancer Sci. 2015, 106, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Celia-Terrassa, T. Dynamics of Phenotypic Heterogeneity Associated with EMT and Stemness during Cancer Progression. J. Clin. Med. 2019, 8, 1542. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Tu, B.; Yao, J.; Gong, J.; Carugo, A.; Bristow, C.A.; Wang, Q.; Zhu, C.; Dai, B.; Kang, Y.; et al. Targeting Glucose Metabolism Sensitizes Pancreatic Cancer to MEK Inhibition. Cancer Res. 2021, 81, 4054–4065. [Google Scholar] [CrossRef]
- Guo, J.; Satoh, K.; Tabata, S.; Mori, M.; Tomita, M.; Soga, T. Reprogramming of glutamine metabolism via glutamine synthetase silencing induces cisplatin resistance in A2780 ovarian cancer cells. BMC Cancer 2021, 21, 174. [Google Scholar] [CrossRef]
- Guo, J.; Yu, J.; Peng, F.; Li, J.; Tan, Z.; Chen, Y.; Rao, T.; Wang, Y.; Peng, J.; Zhou, H. In vitro and in vivo analysis of metabolites involved in the TCA cycle and glutamine metabolism associated with cisplatin resistance in human lung cancer. Expert Rev. Proteom. 2021, 18, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Eu, J.Q.; Kong, L.R.; Wang, L.; Lim, Y.C.; Goh, B.C.; Wong, A.L.A. Targeting Metabolism in Cancer Cells and the Tumour Microenvironment for Cancer Therapy. Molecules 2020, 25, 4831. [Google Scholar] [CrossRef]
- Lotz, C.; Kelleher, D.K.; Gassner, B.; Gekle, M.; Vaupel, P.; Thews, O. Role of the tumor microenvironment in the activity and expression of the p-glycoprotein in human colon carcinoma cells. Oncol. Rep. 2007, 17, 239–244. [Google Scholar] [CrossRef]
- Skeberdyte, A.; Sarapiniene, I.; Aleksander-Krasko, J.; Stankevicius, V.; Suziedelis, K.; Jarmalaite, S. Dichloroacetate and Salinomycin Exert a Synergistic Cytotoxic Effect in Colorectal Cancer Cell Lines. Sci. Rep. 2018, 8, 17744. [Google Scholar] [CrossRef]
- Juan-Carlos, P.M.; Perla-Lidia, P.P.; Stephanie-Talia, M.M.; Monica-Griselda, A.M.; Luz-Maria, T.E. ABC transporter superfamily. An updated overview, relevance in cancer multidrug resistance and perspectives with personalized medicine. Mol. Biol. Rep. 2021, 48, 1883–1901. [Google Scholar] [CrossRef]
- Xiao, H.; Zheng, Y.; Ma, L.; Tian, L.; Sun, Q. Clinically-Relevant ABC Transporter for Anti-Cancer Drug Resistance. Front. Pharmacol. 2021, 12, 648407. [Google Scholar] [CrossRef] [PubMed]
- Robert, J.; Morvan, V.L.; Smith, D.; Pourquier, P.; Bonnet, J. Predicting drug response and toxicity based on gene polymorphisms. Crit. Rev. Oncol. Hematol. 2005, 54, 171–196. [Google Scholar] [CrossRef] [PubMed]
- Aye, I.L.; Singh, A.T.; Keelan, J.A. Transport of lipids by ABC proteins: Interactions and implications for cellular toxicity, viability and function. Chem. Biol. Interact. 2009, 180, 327–339. [Google Scholar] [CrossRef]
- Bugde, P.; Biswas, R.; Merien, F.; Lu, J.; Liu, D.X.; Chen, M.; Zhou, S.; Li, Y. The therapeutic potential of targeting ABC transporters to combat multi-drug resistance. Expert Opin. Ther. Targets 2017, 21, 511–530. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.A.; Young, M.J.; Wang, Y.C.; Chen, S.H.; Liu, C.Y.; Lo, Y.A.; Jen, H.H.; Hsu, K.C.; Hung, J.J. USP24 promotes drug resistance during cancer therapy. Cell Death Differ. 2021, 28, 2690–2707. [Google Scholar] [CrossRef]
- Fletcher, J.I.; Williams, R.T.; Henderson, M.J.; Norris, M.D.; Haber, M. ABC transporters as mediators of drug resistance and contributors to cancer cell biology. Drug Resist. Updates 2016, 26, 1–9. [Google Scholar] [CrossRef]
- Welte, Y.; Adjaye, J.; Lehrach, H.R.; Regenbrecht, C.R. Cancer stem cells in solid tumors: Elusive or illusive? Cell Commun. Signal. 2010, 8, 6. [Google Scholar] [CrossRef]
- Scharenberg, C.W.; Harkey, M.A.; Torok-Storb, B. The ABCG2 transporter is an efficient Hoechst 33342 efflux pump and is preferentially expressed by immature human hematopoietic progenitors. Blood 2002, 99, 507–512. [Google Scholar] [CrossRef]
- Zochbauer-Muller, S.; Filipits, M.; Rudas, M.; Brunner, R.; Krajnik, G.; Suchomel, R.; Schmid, K.; Pirker, R. P-glycoprotein and MRP1 expression in axillary lymph node metastases of breast cancer patients. Anticancer Res. 2001, 21, 119–124. [Google Scholar]
- Ambudkar, S.V.; Kimchi-Sarfaty, C.; Sauna, Z.E.; Gottesman, M.M. P-glycoprotein: From genomics to mechanism. Oncogene 2003, 22, 7468–7485. [Google Scholar] [CrossRef]
- Fung, K.L.; Gottesman, M.M. A synonymous polymorphism in a common MDR1 (ABCB1) haplotype shapes protein function. Biochim. Biophys. Acta 2009, 1794, 860–871. [Google Scholar] [CrossRef] [PubMed]
- Dulucq, S.; Bouchet, S.; Turcq, B.; Lippert, E.; Etienne, G.; Reiffers, J.; Molimard, M.; Krajinovic, M.; Mahon, F.X. Multidrug resistance gene (MDR1) polymorphisms are associated with major molecular responses to standard-dose imatinib in chronic myeloid leukemia. Blood 2008, 112, 2024–2027. [Google Scholar] [CrossRef] [PubMed]
- Robinson, L.J.; Roberts, W.K.; Ling, T.T.; Lamming, D.; Sternberg, S.S.; Roepe, P.D. Human MDR 1 protein overexpression delays the apoptotic cascade in Chinese hamster ovary fibroblasts. Biochemistry 1997, 36, 11169–11178. [Google Scholar] [CrossRef] [PubMed]
- Smyth, M.J.; Krasovskis, E.; Sutton, V.R.; Johnstone, R.W. The drug efflux protein, P-glycoprotein, additionally protects drug-resistant tumor cells from multiple forms of caspase-dependent apoptosis. Proc. Natl. Acad. Sci. USA 1998, 95, 7024–7029. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Xiang, D.; Wang, T.; Zhang, Y.; Pham, C.V.; Zhou, S.; Jiang, G.; Hou, Y.; Zhu, Y.; Han, Y.; et al. The inhibition of ABCB1/MDR1 or ABCG2/BCRP enables doxorubicin to eliminate liver cancer stem cells. Sci. Rep. 2021, 11, 10791. [Google Scholar] [CrossRef]
- Hattinger, C.M.; Stoico, G.; Michelacci, F.; Pasello, M.; Scionti, I.; Remondini, D.; Castellani, G.C.; Fanelli, M.; Scotlandi, K.; Picci, P.; et al. Mechanisms of gene amplification and evidence of coamplification in drug-resistant human osteosarcoma cell lines. Genes Chromosomes Cancer 2009, 48, 289–309. [Google Scholar] [CrossRef]
- Liu, X.; Yuan, J.; Zhang, X.; Li, L.; Dai, X.; Chen, Q.; Wang, Y. ATF3 Modulates the Resistance of Breast Cancer Cells to Tamoxifen through an N(6)-Methyladenosine-Based Epitranscriptomic Mechanism. Chem. Res. Toxicol. 2021, 34, 1814–1821. [Google Scholar] [CrossRef]
- Yusuf, R.Z.; Duan, Z.; Lamendola, D.E.; Penson, R.T.; Seiden, M.V. Paclitaxel resistance: Molecular mechanisms and pharmacologic manipulation. Curr. Cancer Drug Targets 2003, 3, 1–19. [Google Scholar] [CrossRef]
- Hadzic, T.; Aykin-Burns, N.; Zhu, Y.; Coleman, M.C.; Leick, K.; Jacobson, G.M.; Spitz, D.R. Paclitaxel combined with inhibitors of glucose and hydroperoxide metabolism enhances breast cancer cell killing via H2O2-mediated oxidative stress. Free Radic. Biol. Med. 2010, 48, 1024–1033. [Google Scholar] [CrossRef]
- Cole, S.P. Targeting multidrug resistance protein 1 (MRP1, ABCC1): Past, present, and future. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 95–117. [Google Scholar] [CrossRef]
- Emmanouilidi, A.; Casari, I.; Gokcen Akkaya, B.; Maffucci, T.; Furic, L.; Guffanti, F.; Broggini, M.; Chen, X.; Maxuitenko, Y.Y.; Keeton, A.B.; et al. Inhibition of the Lysophosphatidylinositol Transporter ABCC1 Reduces Prostate Cancer Cell Growth and Sensitizes to Chemotherapy. Cancers 2020, 12, 2022. [Google Scholar] [CrossRef] [PubMed]
- Schinkel, A.H.; Jonker, J.W. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: An overview. Adv. Drug Deliv. Rev. 2003, 55, 3–29. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.P. Multidrug resistance protein 1 (MRP1, ABCC1), a “multitasking” ATP-binding cassette (ABC) transporter. J. Biol. Chem. 2014, 289, 30880–30888. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Li, X.; Zeng, Y.; Zhuang, X.; Shen, H.; Zhu, H.; Liu, H.; Xiao, H. In vitro and in vivo inhibition of MRP gene expression and reversal of multidrug resistance by siRNA. Basic Clin. Pharmacol. Toxicol. 2011, 108, 177–184. [Google Scholar] [CrossRef]
- Sullivan, G.F.; Yang, J.M.; Vassil, A.; Yang, J.; Bash-Babula, J.; Hait, W.N. Regulation of expression of the multidrug resistance protein MRP1 by p53 in human prostate cancer cells. J. Clin. Investig. 2000, 105, 1261–1267. [Google Scholar] [CrossRef]
- Zhou, X.; Huang, J.M.; Li, T.M.; Liu, J.Q.; Wei, Z.L.; Lan, C.L.; Zhu, G.Z.; Liao, X.W.; Ye, X.P.; Peng, T. Clinical Significance and Potential Mechanisms of ATP Binding Cassette Subfamily C Genes in Hepatocellular Carcinoma. Front. Genet. 2022, 13, 805961. [Google Scholar] [CrossRef]
- Zander, S.A.; Kersbergen, A.; van der Burg, E.; de Water, N.; van Tellingen, O.; Gunnarsdottir, S.; Jaspers, J.E.; Pajic, M.; Nygren, A.O.; Jonkers, J.; et al. Sensitivity and acquired resistance of BRCA1;p53-deficient mouse mammary tumors to the topoisomerase I inhibitor topotecan. Cancer Res. 2010, 70, 1700–1710. [Google Scholar] [CrossRef]
- Deng, F.; Sjostedt, N.; Santo, M.; Neuvonen, M.; Niemi, M.; Kidron, H. Novel inhibitors of breast cancer resistance protein (BCRP, ABCG2) among marketed drugs. Eur. J. Pharm. Sci. 2023, 181, 106362. [Google Scholar] [CrossRef]
- Robey, R.W.; Massey, P.R.; Amiri-Kordestani, L.; Bates, S.E. ABC transporters: Unvalidated therapeutic targets in cancer and the CNS. Anticancer Agents Med. Chem. 2010, 10, 625–633. [Google Scholar] [CrossRef]
- Rabindran, S.K.; He, H.; Singh, M.; Brown, E.; Collins, K.I.; Annable, T.; Greenberger, L.M. Reversal of a novel multidrug resistance mechanism in human colon carcinoma cells by fumitremorgin C. Cancer Res. 1998, 58, 5850–5858. [Google Scholar]
- Nishiyama, M.; Kuga, T. Central effects of the neurotropic mycotoxin fumitremorgin A in the rabbit (I). Effects on the spinal cord. Jpn. J. Pharmacol. 1989, 50, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.D.; van Loevezijn, A.; Lakhai, J.M.; van der Valk, M.; van Tellingen, O.; Reid, G.; Schellens, J.H.; Koomen, G.J.; Schinkel, A.H. Potent and specific inhibition of the breast cancer resistance protein multidrug transporter in vitro and in mouse intestine by a novel analogue of fumitremorgin C. Mol. Cancer Ther. 2002, 1, 417–425. [Google Scholar] [PubMed]
- Weidner, L.D.; Zoghbi, S.S.; Lu, S.; Shukla, S.; Ambudkar, S.V.; Pike, V.W.; Mulder, J.; Gottesman, M.M.; Innis, R.B.; Hall, M.D. The Inhibitor Ko143 Is Not Specific for ABCG2. J. Pharmacol. Exp. Ther. 2015, 354, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Kita, D.H.; Guragossian, N.; Zattoni, I.F.; Moure, V.R.; Rego, F.G.M.; Lusvarghi, S.; Moulenat, T.; Belhani, B.; Picheth, G.; Bouacida, S.; et al. Mechanistic basis of breast cancer resistance protein inhibition by new indeno[1,2-b]indoles. Sci. Rep. 2021, 11, 1788. [Google Scholar] [CrossRef]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef]
- Maher, J.C.; Wangpaichitr, M.; Savaraj, N.; Kurtoglu, M.; Lampidis, T.J. Hypoxia-inducible factor-1 confers resistance to the glycolytic inhibitor 2-deoxy-D-glucose. Mol. Cancer Ther. 2007, 6, 732–741. [Google Scholar] [CrossRef]
- Reina-Campos, M.; Moscat, J.; Diaz-Meco, M. Metabolism shapes the tumor microenvironment. Curr. Opin. Cell Biol. 2017, 48, 47–53. [Google Scholar] [CrossRef]
- Liu, C.; Jin, Y.; Fan, Z. The Mechanism of Warburg Effect-Induced Chemoresistance in Cancer. Front. Oncol. 2021, 11, 698023. [Google Scholar] [CrossRef]
- Tavares-Valente, D.; Sousa, B.; Schmitt, F.; Baltazar, F.; Queiros, O. Disruption of pH Dynamics Suppresses Proliferation and Potentiates Doxorubicin Cytotoxicity in Breast Cancer Cells. Pharmaceutics 2021, 13, 242. [Google Scholar] [CrossRef]
- Kim, J.Y.; Lee, J.Y. Targeting Tumor Adaption to Chronic Hypoxia: Implications for Drug Resistance, and How It Can Be Overcome. Int. J. Mol. Sci. 2017, 18, 1854. [Google Scholar] [CrossRef]
- Li, X.; Zhong, Y.; Lu, J.; Axcrona, K.; Eide, L.; Syljuasen, R.G.; Peng, Q.; Wang, J.; Zhang, H.; Goscinski, M.A.; et al. MtDNA depleted PC3 cells exhibit Warburg effect and cancer stem cell features. Oncotarget 2016, 7, 40297–40313. [Google Scholar] [CrossRef] [PubMed]
- Schiliro, C.; Firestein, B.L. Mechanisms of Metabolic Reprogramming in Cancer Cells Supporting Enhanced Growth and Proliferation. Cells 2021, 10, 1056. [Google Scholar] [CrossRef] [PubMed]
- Kozal, K.; Jozwiak, P.; Krzeslak, A. Contemporary Perspectives on the Warburg Effect Inhibition in Cancer Therapy. Cancer Control 2021, 28, 10732748211041243. [Google Scholar] [CrossRef]
- Sonveaux, P.; Vegran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008, 118, 3930–3942. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Bryniarski, M.A.; Morris, M.E. In Vitro and In Vivo Efficacy of the Monocarboxylate Transporter 1 Inhibitor AR-C155858 in the Murine 4T1 Breast Cancer Tumor Model. AAPS J. 2018, 21, 3. [Google Scholar] [CrossRef]
- Curtis, N.J.; Mooney, L.; Hopcroft, L.; Michopoulos, F.; Whalley, N.; Zhong, H.; Murray, C.; Logie, A.; Revill, M.; Byth, K.F.; et al. Pre-clinical pharmacology of AZD3965, a selective inhibitor of MCT1: DLBCL, NHL and Burkitt’s lymphoma anti-tumor activity. Oncotarget 2017, 8, 69219–69236. [Google Scholar] [CrossRef]
- Noble, R.A.; Bell, N.; Blair, H.; Sikka, A.; Thomas, H.; Phillips, N.; Nakjang, S.; Miwa, S.; Crossland, R.; Rand, V.; et al. Inhibition of monocarboxyate transporter 1 by AZD3965 as a novel therapeutic approach for diffuse large B-cell lymphoma and Burkitt lymphoma. Haematologica 2017, 102, 1247–1257. [Google Scholar] [CrossRef]
- Halford, S.; Veal, G.J.; Wedge, S.R.; Payne, G.S.; Bacon, C.M.; Sloan, P.; Dragoni, I.; Heinzmann, K.; Potter, S.; Salisbury, B.M.; et al. A Phase I Dose-escalation Study of AZD3965, an Oral Monocarboxylate Transporter 1 Inhibitor, in Patients with Advanced Cancer. Clin. Cancer Res. 2023, 29, 1429–1439. [Google Scholar] [CrossRef]
- Akins, N.S.; Nielson, T.C.; Le, H.V. Inhibition of Glycolysis and Glutaminolysis: An Emerging Drug Discovery Approach to Combat Cancer. Curr. Top. Med. Chem. 2018, 18, 494–504. [Google Scholar] [CrossRef]
- Saunier, E.; Antonio, S.; Regazzetti, A.; Auzeil, N.; Laprevote, O.; Shay, J.W.; Coumoul, X.; Barouki, R.; Benelli, C.; Huc, L.; et al. Resveratrol reverses the Warburg effect by targeting the pyruvate dehydrogenase complex in colon cancer cells. Sci. Rep. 2017, 7, 6945. [Google Scholar] [CrossRef]
- Pranzini, E.; Pardella, E.; Paoli, P.; Fendt, S.M.; Taddei, M.L. Metabolic Reprogramming in Anticancer Drug Resistance: A Focus on Amino Acids. Trends Cancer 2021, 7, 682–699. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Copeland, C.; Le, A. Glutamine Metabolism in Cancer. Adv. Exp. Med. Biol. 2021, 1311, 17–38. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.W.; Jacene, H.; Song, D.; Vilardell, F.; Messersmith, W.A.; Laheru, D.; Wahl, R.; Endres, C.; Jimeno, A.; Pomper, M.G.; et al. [18F]fluorodeoxyglucose positron emission tomography correlates with Akt pathway activity but is not predictive of clinical outcome during mTOR inhibitor therapy. J. Clin. Oncol. 2009, 27, 2697–2704. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bhutia, Y.D.; Babu, E.; Ramachandran, S.; Ganapathy, V. Amino Acid transporters in cancer and their relevance to “glutamine addiction”: Novel targets for the design of a new class of anticancer drugs. Cancer Res. 2015, 75, 1782–1788. [Google Scholar] [CrossRef]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef]
- Poillet-Perez, L.; Xie, X.; Zhan, L.; Yang, Y.; Sharp, D.W.; Hu, Z.S.; Su, X.; Maganti, A.; Jiang, C.; Lu, W.; et al. Autophagy maintains tumour growth through circulating arginine. Nature 2018, 563, 569–573. [Google Scholar] [CrossRef]
- Gandhi, N.; Das, G.M. Metabolic Reprogramming in Breast Cancer and Its Therapeutic Implications. Cells 2019, 8, 89. [Google Scholar] [CrossRef]
- Kurihara-Shimomura, M.; Sasahira, T.; Nakashima, C.; Kuniyasu, H.; Shimomura, H.; Kirita, T. The Multifarious Functions of Pyruvate Kinase M2 in Oral Cancer Cells. Int. J. Mol. Sci. 2018, 19, 2907. [Google Scholar] [CrossRef]
- Li, T.E.; Wang, S.; Shen, X.T.; Zhang, Z.; Chen, M.; Wang, H.; Zhu, Y.; Xu, D.; Hu, B.Y.; Wei, R.; et al. PKM2 Drives Hepatocellular Carcinoma Progression by Inducing Immunosuppressive Microenvironment. Front. Immunol. 2020, 11, 589997. [Google Scholar] [CrossRef]
- Zhou, Y.; Huang, Z.; Su, J.; Li, J.; Zhao, S.; Wu, L.; Zhang, J.; He, Y.; Zhang, G.; Tao, J.; et al. Benserazide is a novel inhibitor targeting PKM2 for melanoma treatment. Int. J. Cancer 2020, 147, 139–151. [Google Scholar] [CrossRef]
- Nakano, A.; Tsuji, D.; Miki, H.; Cui, Q.; El Sayed, S.M.; Ikegame, A.; Oda, A.; Amou, H.; Nakamura, S.; Harada, T.; et al. Glycolysis inhibition inactivates ABC transporters to restore drug sensitivity in malignant cells. PLoS ONE 2011, 6, e27222. [Google Scholar] [CrossRef]
- Ma, S.; Jia, R.; Li, D.; Shen, B. Targeting Cellular Metabolism Chemosensitizes the Doxorubicin-Resistant Human Breast Adenocarcinoma Cells. Biomed. Res. Int. 2015, 2015, 453986. [Google Scholar] [CrossRef]
- Prieto-Vila, M.; Takahashi, R.U.; Usuba, W.; Kohama, I.; Ochiya, T. Drug Resistance Driven by Cancer Stem Cells and Their Niche. Int. J. Mol. Sci. 2017, 18, 2574. [Google Scholar] [CrossRef] [PubMed]
- Martins, J.P.; das Neves, J.; de la Fuente, M.; Celia, C.; Florindo, H.; Gunday-Tureli, N.; Popat, A.; Santos, J.L.; Sousa, F.; Schmid, R.; et al. The solid progress of nanomedicine. Drug Deliv. Transl. Res. 2020, 10, 726–729. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zheng, X.; Chen, L.; Gong, X.; Yang, H.; Duan, X.; Zhu, Y. Multifunctional Gold Nanoparticles in Cancer Diagnosis and Treatment. Int. J. Nanomed. 2022, 17, 2041–2067. [Google Scholar] [CrossRef] [PubMed]
- Yao, W.; Yao, J.; Qian, F.; Que, Z.; Yu, P.; Luo, T.; Zheng, D.; Zhang, Z.; Tian, J. Paclitaxel-loaded and folic acid-modified PLGA nanomedicine with glutathione response for the treatment of lung cancer. Acta Biochim. Biophys. Sin. 2021, 53, 1027–1036. [Google Scholar] [CrossRef]
- van den Boogaard, W.M.C.; Komninos, D.S.J.; Vermeij, W.P. Chemotherapy Side-Effects: Not All DNA Damage Is Equal. Cancers 2022, 14, 627. [Google Scholar] [CrossRef]
- Sousa, A.R.; Oliveira, M.J.; Sarmento, B. Impact of CEA-targeting Nanoparticles for Drug Delivery in Colorectal Cancer. J. Pharmacol. Exp. Ther. 2019, 370, 657–670. [Google Scholar] [CrossRef]
- Barenholz, Y. Doxil(R)—The first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef]
- Blair, H.A.; Deeks, E.D. Albumin-Bound Paclitaxel: A Review in Non-Small Cell Lung Cancer. Drugs 2015, 75, 2017–2024. [Google Scholar] [CrossRef]
- Zhao, M.; van Straten, D.; Broekman, M.L.D.; Preat, V.; Schiffelers, R.M. Nanocarrier-based drug combination therapy for glioblastoma. Theranostics 2020, 10, 1355–1372. [Google Scholar] [CrossRef] [PubMed]
- Zheng, R.R.; Zhao, L.P.; Liu, L.S.; Deng, F.A.; Chen, X.Y.; Jiang, X.Y.; Wang, C.; Yu, X.Y.; Cheng, H.; Li, S.Y. Self-delivery nanomedicine to overcome drug resistance for synergistic chemotherapy. Biomater. Sci. 2021, 9, 3445–3452. [Google Scholar] [CrossRef] [PubMed]
- Niculescu, A.G.; Grumezescu, A.M. Novel Tumor-Targeting Nanoparticles for Cancer Treatment-A Review. Int. J. Mol. Sci. 2022, 23, 5253. [Google Scholar] [CrossRef]
- Rezvantalab, S.; Drude, N.I.; Moraveji, M.K.; Guvener, N.; Koons, E.K.; Shi, Y.; Lammers, T.; Kiessling, F. PLGA-Based Nanoparticles in Cancer Treatment. Front. Pharmacol. 2018, 9, 1260. [Google Scholar] [CrossRef]
- Lu, L.; Peter, S.J.; Lyman, M.D.; Lai, H.L.; Leite, S.M.; Tamada, J.A.; Uyama, S.; Vacanti, J.P.; Langer, R.; Mikos, A.G. In vitro and in vivo degradation of porous poly(DL-lactic-co-glycolic acid) foams. Biomaterials 2000, 21, 1837–1845. [Google Scholar] [CrossRef]
- Luderer, F.; Lobler, M.; Rohm, H.W.; Gocke, C.; Kunna, K.; Kock, K.; Kroemer, H.K.; Weitschies, W.; Schmitz, K.P.; Sternberg, K. Biodegradable sirolimus-loaded poly(lactide) nanoparticles as drug delivery system for the prevention of in-stent restenosis in coronary stent application. J. Biomater. Appl. 2011, 25, 851–875. [Google Scholar] [CrossRef]
- Wu, L.; Ding, J. In vitro degradation of three-dimensional porous poly(D,L-lactide-co-glycolide) scaffolds for tissue engineering. Biomaterials 2004, 25, 5821–5830. [Google Scholar] [CrossRef]
- Zhang, B.; Sai Lung, P.; Zhao, S.; Chu, Z.; Chrzanowski, W.; Li, Q. Shape dependent cytotoxicity of PLGA-PEG nanoparticles on human cells. Sci. Rep. 2017, 7, 7315. [Google Scholar] [CrossRef]
- Cai, J.; Qian, K.; Zuo, X.; Yue, W.; Bian, Y.; Yang, J.; Wei, J.; Zhao, W.; Qian, H.; Liu, B. PLGA nanoparticle-based docetaxel/LY294002 drug delivery system enhances antitumor activities against gastric cancer. J. Biomater. Appl. 2019, 33, 1394–1406. [Google Scholar] [CrossRef]
- Zhang, L.; Zhai, B.Z.; Wu, Y.J.; Wang, Y. Recent progress in the development of nanomaterials targeting multiple cancer metabolic pathways: A review of mechanistic approaches for cancer treatment. Drug Deliv. 2023, 30, 1–18. [Google Scholar] [CrossRef]
- Ren, M.; Zheng, X.; Gao, H.; Jiang, A.; Yao, Y.; He, W. Nanomedicines Targeting Metabolism in the Tumor Microenvironment. Front. Bioeng. Biotechnol. 2022, 10, 943906. [Google Scholar] [CrossRef] [PubMed]
- Sa, P.; Sahoo, S.K.; Dilnawaz, F. Responsive Role of Nanomedicine in the Tumor Microenvironment and Cancer Drug Resistance. Curr. Med. Chem. 2023, 30, 3335–3355. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Nishina, S.; Yamauchi, A.; Fukuda, K.; Hara, Y.; Yamamura, M.; Egashira, K.; Hino, K. Nanoparticle-Mediated Delivery of 2-Deoxy-D-Glucose Induces Antitumor Immunity and Cytotoxicity in Liver Tumors in Mice. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 739–762. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Chen, Y.; Shi, J. Tumor-Specific Chemotherapy by Nanomedicine-Enabled Differential Stress Sensitization. Angew. Chem. Int. Ed. Engl. 2020, 59, 9693–9701. [Google Scholar] [CrossRef]
- Cui, L.; Gouw, A.M.; LaGory, E.L.; Guo, S.; Attarwala, N.; Tang, Y.; Qi, J.; Chen, Y.S.; Gao, Z.; Casey, K.M.; et al. Mitochondrial copper depletion suppresses triple-negative breast cancer in mice. Nat. Biotechnol. 2021, 39, 357–367. [Google Scholar] [CrossRef]
- Ding, X.L.; Liu, M.D.; Cheng, Q.; Guo, W.H.; Niu, M.T.; Huang, Q.X.; Zeng, X.; Zhang, X.Z. Multifunctional liquid metal-based nanoparticles with glycolysis and mitochondrial metabolism inhibition for tumor photothermal therapy. Biomaterials 2022, 281, 121369. [Google Scholar] [CrossRef]
- Dong, F.; Jiang, Q.; Li, L.; Liu, T.; Zuo, S.; Gao, L.; Fang, M.; Gao, Y.; Sun, B.; Luo, C.; et al. Synergetic lethal energy depletion initiated by cancer cell membrane camouflaged nano-inhibitor for cancer therapy. Nano Res. 2022, 15, 3422–3433. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Wu, P.; Han, J.; Gong, Y.; Liu, C.; Yu, H.; Xie, N. Nanoparticle-Based Drug Delivery Systems Targeting Tumor Microenvironment for Cancer Immunotherapy Resistance: Current Advances and Applications. Pharmaceutics 2022, 14, 1990. [Google Scholar] [CrossRef]
- Wu, S.; Zhang, K.; Liang, Y.; Wei, Y.; An, J.; Wang, Y.; Yang, J.; Zhang, H.; Zhang, Z.; Liu, J.; et al. Nano-enabled Tumor Systematic Energy Exhaustion via Zinc (II) Interference Mediated Glycolysis Inhibition and Specific GLUT1 Depletion. Adv. Sci. 2022, 9, e2103534. [Google Scholar] [CrossRef]
- Guimaraes, P.P.G.; Gaglione, S.; Sewastianik, T.; Carrasco, R.D.; Langer, R.; Mitchell, M.J. Nanoparticles for Immune Cytokine TRAIL-Based Cancer Therapy. ACS Nano 2018, 12, 912–931. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Luo, Q.; Li, H.; Liu, Q.; Ju, Y.; Song, G. Increased Oxidative Phosphorylation Is Required for Stemness Maintenance in Liver Cancer Stem Cells from Hepatocellular Carcinoma Cell Line HCCLM3 Cells. Int. J. Mol. Sci. 2020, 21, 5276. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Energetic Profile | Type of Cancer/ Cancer Cell Line | Antimetabolic Drug Effect | Expression and Regulation of MCTs | References |
|---|---|---|---|---|
| Mainly OXPHOS | Breast (MDA-MB-468) | IACS-010759 induced cell death and inhibited oxygen consumption rate | MCT1 expression at the plasma membrane. MCT4 is expressed on cytoplasm | [26,28] |
| Cervical (HeLa) | Metformin and Rotenone promoted anoikis | MCT1 expression > MCT4 expression Hypoxia induced the expression of MCT4 | [31,38,39] | |
| Cervical (siHa) | Rotenone decreased cell migration | 2DG and rotenone increased the expression of MCT1 and CD147 | [40,41] | |
| Leukemia (THP-1) | Resistant to 2DG and sensitive to oligomycin | MCT4 expression Lactate and VEGF increased the expression of MCT4, but not of MCT1 | [24,25] | |
| Lung (A549) | Resistant to 3BP, DCA and 2DG | No changes were observed in MCT1 and MCT4 upon treatment with 3BP, DCA and 2DG | [37] | |
| Melanoma (B16F10) | Metformin and Rotenone promoted anoikis | No data | [31] | |
| Ovarian (OVCAR-3) | Atovaquone slowed ovarian cancer growth | No data | [42] | |
| Mainly Glycolytic | Bladder (5637) | Sensitive to 2DG. 2DG depleted cellular ATP and potentiated the toxicity of conventional drugs | High expression of MCT1, MCT4 and CD147 Knockdown of MCT4 inhibited 5637 cancer cell line proliferation and clonogenic activity | [43] |
| Colon (SW480) | Sensitive to 3BP, 2DG and DCA | High expression of MCT1, MCT2 and MCT4 3BP decreased the expression of MCT1and MCT4, but not of MCT2 | [35,44,45,46,47] | |
| Glioma (U251) | Sensitive to DCA, 2DG, resveratrol and CHC | High plasma-membrane expression of GLUT1, MCT1, CD147 Silencing of MCT1 decreased the glycolytic phenotype | [23,30,32,33,34,48] | |
| Leukemia (NB4) | Sensitive to 2DG and 3BP | High expression of MCT1 and MCT4 | [24,49,50] | |
| Lung (NCI-H460) | Sensitive to 3BP, 2DG and DCA | No association was observed between MCT1 and MCT4 expression and treatment effect with 3BP, DCA and 2DG | [37] | |
| Melanoma (A375) | Sensitive to 3BP | High expression of MCT1 | [51] | |
| Both glycolytic and OXPHOS | Breast (MCF-7) | 2DG, IAA, DCA and CCP and 3BP induced cell death Pre-treatment with 2DG, IAA, DCA and CCCP enhanced PTX and DOX toxicity Lonidamine potentiated the effect of PTX | High plasma-membrane expression of MCT1 and MCT4. 3BP did not alter the expression | [27,29,31,52,53] |
| Glioma (SW1088) | Metformin and Rotenone promoted anoikis DCA, 2DG and phenformin led to a decrease in ATP content Resistent to CHC | Low plasma-membrane expression of MCT1, MCT4 and CD147 | [23,32,36] | |
| Liver (HepG2) | 2DG, 3BP and DCA induced cell death and potentiated the effect of DOX Phenphormin inhibited proliferation | High expression of MCT1 and MCT4 and lower expression of MCT2 | [54,55,56] |
| Metabolism Pathway | Nanoparticle | Advantages | Disadvantages | Future Perspectives | References |
|---|---|---|---|---|---|
| Mitochondrial respiration | DCA NP PLGA | Control drug delivery system of small drug molecules | Increased DCA in normal cells could lead to serious side effects | Functionalize NPs to specific tissue receptors | [37] |
| CDN polymersome NPs | Induce a metabolic shift toward glycolysis Low toxicity of CDNs in healthy mice | Not applicable to glycolytic cells | Apply to other types of cancer | [195] | |
| Aerobic glycolysis | 2DG-NPs-PLGA | Control drug-delivery system of small drug molecules | Extremely low loading rate of 2DG into the 2DG-PLGA-NPs | Combination therapy with 2DG-PLGA-NPs and other therapeutic agents | [193] |
| Nanoenabled Energy Interrupter | Sensitive to an acidic TME | Preferential inhibition of NPs on melanoma cells | Increase specificity for other tumor types | [200] | |
| Aerobic glycolysis and Mitochondrial respiration | Liposome NPs | Acidic TME favorable for the decomposition of NPs | No data | Combination therapy with nanolipossoma and antitumor agents | [196] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cunha, A.; Silva, P.M.A.; Sarmento, B.; Queirós, O. Targeting Glucose Metabolism in Cancer Cells as an Approach to Overcoming Drug Resistance. Pharmaceutics 2023, 15, 2610. https://doi.org/10.3390/pharmaceutics15112610
Cunha A, Silva PMA, Sarmento B, Queirós O. Targeting Glucose Metabolism in Cancer Cells as an Approach to Overcoming Drug Resistance. Pharmaceutics. 2023; 15(11):2610. https://doi.org/10.3390/pharmaceutics15112610
Chicago/Turabian StyleCunha, Andrea, Patrícia M. A. Silva, Bruno Sarmento, and Odília Queirós. 2023. "Targeting Glucose Metabolism in Cancer Cells as an Approach to Overcoming Drug Resistance" Pharmaceutics 15, no. 11: 2610. https://doi.org/10.3390/pharmaceutics15112610
APA StyleCunha, A., Silva, P. M. A., Sarmento, B., & Queirós, O. (2023). Targeting Glucose Metabolism in Cancer Cells as an Approach to Overcoming Drug Resistance. Pharmaceutics, 15(11), 2610. https://doi.org/10.3390/pharmaceutics15112610