
Sugars and Polyols of Natural Origin as Carriers for Solubility and Dissolution Enhancement


Abstract
1. Introduction
2. Solid Dispersions
2.1. Solid Dispersions of Sugar Carriers

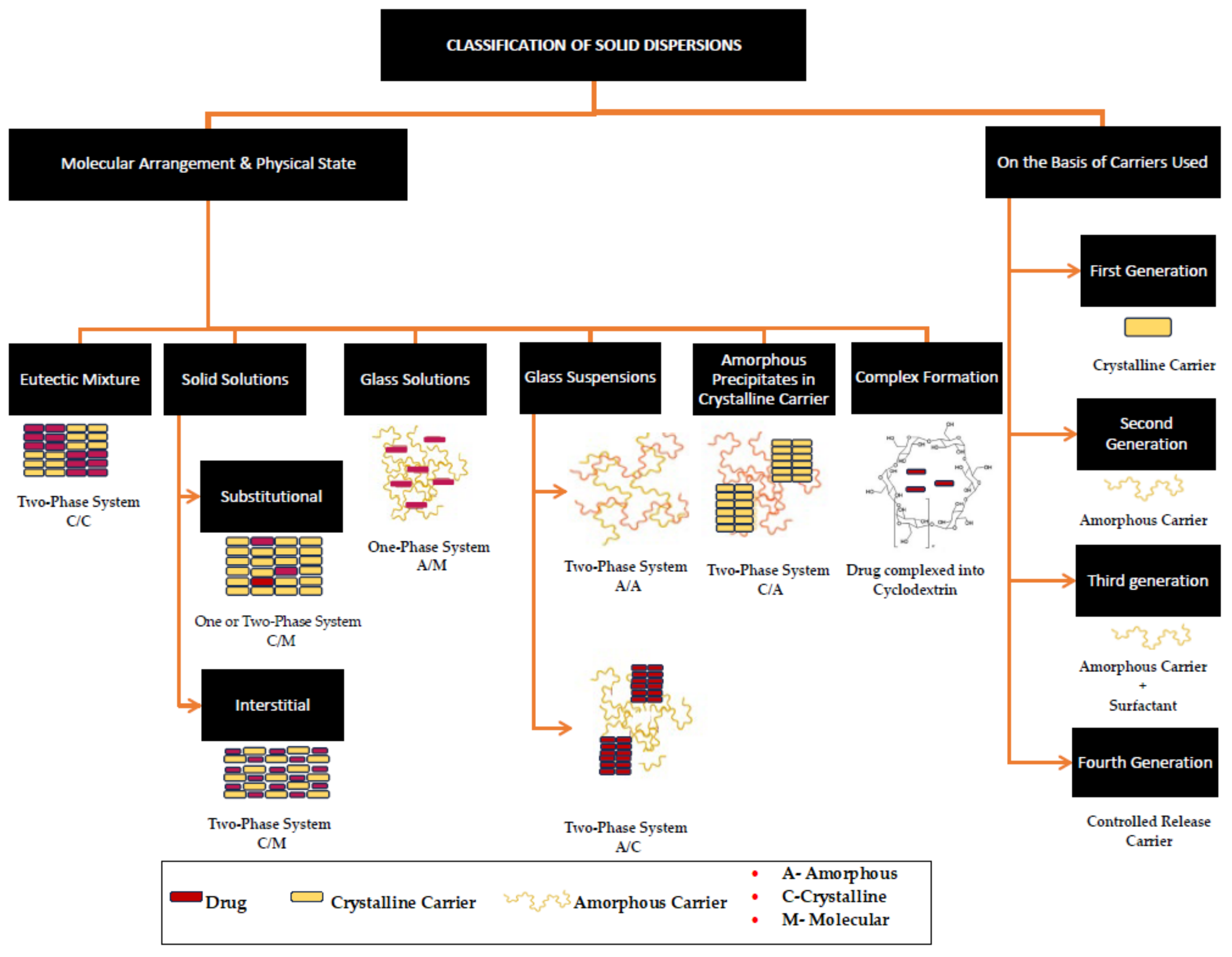{kind=link}
| Method | Sugar | Drug: Sugar (w/w) | Drug | Solubility | Dissolution | Remarks | Ref. |
|---|---|---|---|---|---|---|---|
| Fusion | Dextrose Fructose Maltose | 1:3, 1:1, and 3:1 | Clotrimazole | Fructose showed a slight increase at a 1:1 ratio | Increased dissolution rate for fructose SDs. Increased with an increase in sugar concentration. | Partial dispersion of drug at molecular level. | [7] |
| Dextrose Galactose Sucrose | 1:33 and 1:40 | Corticosteroids | N/A | Increased dissolution rate with bi-phasic drug release. | Partial dispersion of drug at molecular level. Hygroscopic and heating resulted in discolouration. | [35] | |
| Sucrose–mannitol (1:1) Sorbitol–mannitol (1:1) | 1:19 | Corticosteroids | N/A | Increased dissolution rate with bi-phasic drug release. Galactose showed smaller dissolution rate. | Sucrose-mannitol eutectic showed less hygroscopicity and no discolouration. | [36] | |
| Lactose Galactose | 1:3 | Carbamazepine Nitrazepam | N/A | Increased dissolution rate with bi-phasic drug release. Galactose showed slower dissolution rate. | Partial dispersion of drug at molecular level. | [63] | |
| Dextrose Icing Sugar Lactose | 4:1, 2:1, 1:1, and 1:4 | Ibuprofen | N/A | Only 80% of the drug was released within 60 min. | An increase in the sugar concentration had an insignificant effect on drug release. | [40] | |
| Quench Cooling | Glucose Galactose Maltose Sucrose | 1:1 | Sulfamethoxazole | Sugars with free carbonyl group showed slight increase in solubility. | A 100% drug release in 5 min (glucose and maltose). Galactose showed slower dissolution rate. | Partial dispersion of drug at molecular level. Hygroscopic and heating resulted in discolouration. | [37] |
| Lactose | 1:1, 1:3, 1:5, and 1:10 | Carbamazepine Ethenzamide | N/A | Increase in dissolution rate with increase in carrier concentration. Five- to eight-fold increase in dissolution rate. | Hydrogen bonding with amide and carboxyl groups of the CBZ. | [39] | |
| Glucose | 1:0.03 | Indomethacin | N/A | Eight-fold increase in dissolution rate. | Ultrasonication of the melt increased the miscibility between the drug and carrier. | [42] | |
| Solvent evaporation | Lactose Sucrose | 1:1 and 1:5 | Etoricoxib | 1.5 to 1.8-fold at 1:5 ratio | N/A | Intermolecular hydrogen bonding between S=O group of etoricoxib and O–H group of sugar carriers. | [44] |
| G-HCL | 4:1, 2:1,1:1, 1:2, and 1:4 | Carbamazepine | Solubility of solid dispersions is lower than pure drug and physical mixtures | Concentration of carrier and solvent system used affected the dissolution rate. | Presence of water in the binary solvent system reduced the dissolution rate because of the formation of the dihydrate form of the CBZ. | [45] | |
| G-HCL | 1:1, 1:2, 1:3, 1:4, and 1:5 | Acyclovir | 12-fold increase | Higher concentrations of carriers (1:4 and 1:5) showed reduced dissolution due to reduced access of the ACV to dissolution medium. | Decreased dissolution rate during storage. Hydrogen bond between amine group of ACV and O-H of G-HCL. | [46] | |
| Freeze drying | Trehalose, Sucrose, InulinDP11 InulinDP23 | Diazepam Nifedipine THC Cyclosporine A | N/A | Solution-mediated phase transition in the case of trehalose, sucrose, and inulinDP11 SDs. | The chain length of inulin affected the Tg of SDs, dissolution behaviour, and stability. | [50,51] | |
| Freeze drying followed by vacuum drying | Sucrose α-maltose Trehalose α-lactose | 1:10 | Fat-soluble flavours | N/A | A 100-times increase for α-maltose, trehalose and maltitol in methanol. | ASDs exhibited solution-mediated phase transition after 200 s. | [54] |
| Trehalose α-maltose Palatinose | 1 to 10% w/w | Indomethacin, Ibuprofen, Gliclazide, Nifedipine | 20–1000% increase | Palatinose and/or α-maltose showed superior dissolution. | ASDs exhibited solution-mediated phase transition because of low Tg. | [55] | |
| Trehalose α-maltose Palatinose | 0.1 to 10% w/w | Curcumin | N/A | α-maltose and trehalose showed superior dissolution. | ASDs exhibited solution-mediated phase transition. | [56] | |
| Kneading | Lactose Maltose Sucrose | 1:1, 1:3, and 1:5 | Allopurinol | N/A | No significant increase in dissolution rate. | No intermolecular interactions found. Slight increase in dissolution rate due to partial amorphization. | [58,59] |
| Roller compaction | Lactose Maltose | 1:4 | Griseofulvin | N/A | A 35- to 40-fold increase. | Processing problems due to sticking and physical instability. | [61] |
| Centrifugal spinning | Sucrose | 1:9 | Olanzapine Piroxicam | Increase in solubility is proportional to sucrose concentration | A 3- and 1.7-fold increase in dissolution rate of olanzapine and piroxicam, respectively. | Intermolecular interactions observed with olanzapine SDs. No solution-mediated phase transition was observed after 4 h of dissolution | [62] |
2.2. Solid Dispersions of Sugar Alcohol (Polyol) Carriers
2.2.1. Mannitol-Based SDs
2.2.2. Sorbitol-Based Solid Dispersions
2.2.3. Xylitol-Based Solid Dispersions
2.2.4. Erythritol-Based Solid Dispersions
2.2.5. Isomalt-Based Solid Dispersions
2.2.6. Maltitol-Based Solid Dispersions
3. Co-Amorphous and Co-Crystalline Systems
4. Natural Deep Eutectic Solvents
5. Conclusions and Future Perspectives
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gupta, S.; Kesarla, R.; Omri, A. Formulation Strategies to Improve the Bioavailability of Poorly Absorbed Drugs with Special Emphasis on Self-Emulsifying Systems Shweta. ISRN Pharm. 2013, 2013, 1–16. [Google Scholar] [CrossRef]
- Bhalani, D.V.; Nutan, B.; Kumar, A.; Singh Chandel, A.K. Bioavailability Enhancement Techniques for Poorly Aqueous Soluble Drugs and Therapeutics. Biomedicines 2022, 10, 2055. [Google Scholar] [CrossRef] [PubMed]
- Ainurofiq, A.; Putro, D.S.; Ramadhani, D.A.; Putra, G.M.; Do Espirito Santo, L.D.C. A Review on Solubility Enhancement Methods for Poorly Water-Soluble Drugs. J. Rep. Pharm. Sci. 2021, 10, 137–147. [Google Scholar] [CrossRef]
- Pešić, N.; Dapčević, A.; Ivković, B.; Kachrimanis, K.; Mitrić, M.; Ibrić, S.; Medarević, D. Potential Application of Low Molecular Weight Excipients for Amorphization and Dissolution Enhancement of Carvedilol. Int. J. Pharm. 2021, 608, 121033. [Google Scholar] [CrossRef] [PubMed]
- Tekade, A.R.; Yadav, J.N. A Review on Solid Dispersion and Carriers Used Therein for Solubility Enhancement of Poorly Water Soluble Drugs. Adv. Pharm. Bull. 2020, 10, 359–369. [Google Scholar] [CrossRef]
- Kumar Bandaru, R.; Rout, S.R.; Kenguva, G.; Gorain, B.; Alhakamy, N.A.; Kesharwani, P.; Dandela, R. Recent Advances in Pharmaceutical Cocrystals: From Bench to Market. Front. Pharmacol. 2021, 12, 780582. [Google Scholar] [CrossRef]
- Madgulkar, A.; Bandivadekar, M.; Shid, T.; Rao, S. Sugars as Solid Dispersion Carrier to Improve Solubility and Dissolution of the BCS Class II Drug: Clotrimazole. Drug Dev. Ind. Pharm. 2016, 42, 28–38. [Google Scholar] [CrossRef]
- Thayyil, A.R.; Juturu, T.; Nayak, S.; Kamath, S. Pharmaceutical Co-Crystallization: Regulatory Aspects, Design, Characterization, and Applications. Adv. Pharm. Bull. 2020, 10, 203–212. [Google Scholar] [CrossRef]
- White, J.R. Sugar. Clin. Diabetes 2018, 36, 74–76. [Google Scholar] [CrossRef]
- Priya, K.; Gupta, V.R.M.; Srikanth, K. Natural Sweeteners: A Complete Review. J. Pharm. Res. 2011, 4, 2034–2039. [Google Scholar]
- Bazeed, A.Y.; Nouh, A.; Essa, E.A.; El Maghraby, G.M. Hydrophilic Sugars for Enhancing Dissolution Rate of Cilostazol: Effect of Wet Co-Processing. Pharm. Sci. 2021, 27, 111–120. [Google Scholar] [CrossRef]
- Bindhani, S.; Mohapatra, S. Recent Approaches of Solid Dispersion: A New Concept toward Oral Bioavailability. Asian J. Pharm. Clin. Res. 2018, 11, 72–78. [Google Scholar] [CrossRef]
- Silva, L.P.; Fernandez, L.; Conceiçao, J.H.F.; Martins, M.A.R.; Sosa, A.; Ortega, J.; Pinho, S.P.; Coutinho, J.A.P. Design and Characterization of Sugar-Based Deep Eutectic Solvents Using Conductor-like Screening Model for Real Solvents. ACS Sustain. Chem. Eng. 2018, 6, 10724–10734. [Google Scholar] [CrossRef]
- Maugeri, Z.; De María, P.D. Novel Choline-Chloride-Based Deep-Eutectic-Solvents with Renewable Hydrogen Bond Donors: Levulinic Acid and Sugar-Based Polyols. RSC Adv. 2012, 2, 421–425. [Google Scholar] [CrossRef]
- Mousa, T.M.; Donia, A.A.; El Maghraby, G.M. Co-Crystallization of Sofosbuvir with Sugars for Enhanced Dissolution Rate. Indones. J. Pharm. 2023, 34, 140–152. [Google Scholar] [CrossRef]
- Arafa, M.F.; El-Gizawy, S.A.; Osman, M.A.; El Maghraby, G.M. Xylitol as a Potential Co-Crystal Co-Former for Enhancing Dissolution Rate of Felodipine: Preparation and Evaluation of Sublingual Tablets. Pharm. Dev. Technol. 2018, 23, 454–463. [Google Scholar] [CrossRef]
- Sun, T.; Watson, S. Sorbitol/Dexlansoprazole Co-Crystals and Method for Making Same. U.S. Patent 8,318,943 B1, 27 November 2012. Available online: https://patentimages.storage.googleapis.com/8b/19/bb/4167c62761fffc/US8318943.pdf (accessed on 26 July 2023).
- Simperler, A.; Kornherr, A.; Chopra, R.; Bonnet, P.A.; Jones, W.; Motherwell, W.D.S.; Zifferer, G. Glass Transition Temperature of Glucose, Sucrose, and Trehalose: An Experimental and in Silico Study. J. Phys. Chem. B 2006, 110, 19678–19684. [Google Scholar] [CrossRef]
- Malkawi, R.; Malkawi, W.I.; Al-Mahmoud, Y.; Tawalbeh, J. Current Trends on Solid Dispersions: Past, Present, and Future. Adv. Pharmacol. Pharm. Sci. 2022, 2022, 5916013. [Google Scholar] [CrossRef]
- Tambe, S.; Jain, D.; Meruva, S.K.; Rongala, G.; Juluri, A.; Nihalani, G.; Mamidi, H.K.; Nukala, P.K.; Bolla, P.K. Recent Advances in Amorphous Solid Dispersions: Preformulation, Formulation Strategies, Technological Advancements and Characterization. Pharmaceutics 2022, 14, 2203. [Google Scholar] [CrossRef]
- Zhang, J.; Guo, M.; Luo, M.; Cai, T. Advances in the Development of Amorphous Solid Dispersions: The Role of Polymeric Carriers. Asian J. Pharm. Sci. 2023, 18, 100834. [Google Scholar] [CrossRef]
- Pandi, P.; Bulusu, R.; Kommineni, N.; Khan, W.; Singh, M. Amorphous Solid Dispersions: An Update for Preparation, Characterization, Mechanism on Bioavailability, Stability, Regulatory Considerations and Marketed Products. Int. J. Pharm. 2020, 586, 119560. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, Y.L.; Parashar, B.; Ostwal, P.P.; Jain, M.S. Solid Dispersion: Solubility Enhancement for Poorly Water Soluble Drug. Res. J. Pharm. Technol. 2012, 5, 190–197. [Google Scholar]
- Meng, F.; Gala, U.; Chauhan, H. Classification of Solid Dispersions: Correlation to (i) Stability and Solubility (Ii) Preparation and Characterization Techniques. Drug Dev. Ind. Pharm. 2015, 41, 1401–1415. [Google Scholar] [CrossRef]
- Attia, M.S.; Hasan, A.A.; Ghazy, F.E.S.; Gomaa, E. Solid Dispersion as a Technical Solution to Boost the Dissolution Rate and Bioavailability of Poorly Water-Soluble Drugs. Indian J. Pharm. Educ. Res. 2021, 55, s327–s339. [Google Scholar] [CrossRef]
- Nikam, V.K.; Shete, S.K.; Khapare, J.P. Most Promising Solid Dispersion Technique of Oral Dispersible Tablet. Beni-Suef Univ. J. Basic Appl. Sci. 2020, 9, 1–16. [Google Scholar] [CrossRef]
- Sharma, K.S.; Sahoo, J.; Agrawal, S.; Kumari, A. Solid Dispersions: A Technology for Improving Bioavailability. J. Anal. Pharm. Res. Rev. 2019, 8, 127–133. [Google Scholar] [CrossRef]
- Gadade, D.D.; Pekamwar, S.S. Pharmaceutical Cocrystals: Regulatory and Strategic Aspects, Design and Development. Adv. Pharm. Bull. 2016, 6, 479–494. [Google Scholar] [CrossRef]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug Solubility: Importance and Enhancement Techniques. ISRN Pharm. 2012, 2012, 195727. [Google Scholar] [CrossRef]
- Devhare, L.D.; Kore, P.K. A Recent Review on Bioavailability and Solubility Enhancement of Poorly Soluble Drugs by Physical and Chemical Modifications. Res. Chron. Health Sci. 2016, 2, 299–308. [Google Scholar]
- Lewis, S.; Udupa, N. Solid Dispersions: A Review. Pak. J. Pharm. Sci. 2009, 22, 234–246. [Google Scholar]
- Singh, A.; Van den Mooter, G. Spray Drying Formulation of Amorphous Solid Dispersions. Adv. Drug Deliv. Rev. 2016, 100, 27–50. [Google Scholar] [CrossRef] [PubMed]
- Soyata, A.; Kenti, K.; Sutoro, M.; Sagita, N. Impact of Preparation Method in Co-Amorphous System. Sci. Pharm. 2022, 1, 47–55. [Google Scholar] [CrossRef]
- Vo, C.-L.N.; Park, C.; Lee, B.J. Current Trends and Future Perspectives of Solid Dispersions Containing Poorly Water-Soluble Drugs. Eur. J. Pharm. Biopharm. 2013, 85, 799–813. [Google Scholar] [CrossRef] [PubMed]
- Allen, L.V.; Yanchick, V.A.; Maness, D.D. Dissolution Rates of Corticosteroids Utilizing Sugar Glass Dispersions. J. Pharm. Sci. 1977, 66, 494–497. [Google Scholar] [CrossRef]
- Allen, L.V.; Levinson, R.S.; De Martono, D. Dissolution Rates of Hydrocortisone and Prednisone Utilizing Sugar Solid Dispersion Systems in Tablet Form. J. Pharm. Sci. 1978, 67, 979–981. [Google Scholar] [CrossRef]
- Ghanem, A.; Meshali, M.; Ibraheem, Y. Dissolution Rates of Sulfamethoxazole Utilizing Sugar Glass Dispersions. J. Pharm. Pharmacol. 1980, 32, 675–677. [Google Scholar] [CrossRef]
- Bouchard, A.; Hofland, G.W.; Witkamp, G.J. Properties of Sugar, Polyol, and Polysaccharide Water-Ethanol Solutions. J. Chem. Eng. Data 2007, 52, 1838–1842. [Google Scholar] [CrossRef]
- Hirasawa, N.; Okamoto, H.; Danjo, Z. Lactose as a Low Molecular Weight Carrier of Solid Dispersions for Carbamazepine and Ethenzamide. Chem. Pharm. Bull. 1999, 3, 417–420. [Google Scholar] [CrossRef]
- Saffoon, N.; Jhanker, Y.M.; Huda, N.H. Dissolution Profile of Ibuprofen Solid Dispersion Prepared with Cellulosic Polymers and Sugar by Fusion Method. Stamford J. Pharm. Sci. 2011, 4, 31–37. [Google Scholar] [CrossRef]
- Etman, M.A.; Naggar, V.F. Thermodynamics of Paracetamol Solubility in Sugar-Water Cosolvent Systems. Int. J. Pharm. 1990, 58, 177–184. [Google Scholar] [CrossRef]
- Martínez, L.M.; Videa, M.; Silva, T.L.; Castro, S.; Lara-díaz, V.J.; Castorena-torres, F. Two-Phase Amorphous-Amorphous Solid Drug Dispersion with Enhanced Stability, Solubility and Bioavailability Resulting from Ultrasonic Dispersion of an Immiscible System. Eur. J. Pharm. Biopharm. 2017, 119, 243–252. [Google Scholar] [CrossRef]
- Tran, P.; Pyo, Y.C.; Kim, D.H.; Lee, S.E.; Kim, J.K.; Park, J.S. Overview of the Manufacturing Methods of Solid Dispersion Technology for Improving the Solubility of Poorly Water-Soluble Drugs and Application to Anticancer Drugs. Pharmaceutics 2019, 11, 132. [Google Scholar] [CrossRef]
- Das, A.; Nayak, A.K.; Mohanty, B.; Panda, S. Solubility and Dissolution Enhancement of Etoricoxib by Solid Dispersion Technique Using Sugar Carriers. ISRN Pharm. 2011, 2011, 819765. [Google Scholar] [CrossRef] [PubMed]
- Al-Hamidi, H.; Edwards, A.A.; Mohammad, M.A.; Nokhodchi, A. To Enhance Dissolution Rate of Poorly Water-Soluble Drugs: Glucosamine Hydrochloride as a Potential Carrier in Solid Dispersion Formulations. Colloids Surf. B Biointerfaces 2010, 76, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Telange, D.R.; Bhagat, S.B.; Patil, A.T.; Umekar, M.J.; Pethe, A.M.; Raut, N.A.; Dave, V.S. Glucosamine HCL-Based Solid Dispersions to Enhance the Biopharmaceutical Properties of Acyclovir. J. Excipients Food Chem. 2019, 10, 65–81. [Google Scholar]
- Pagola, S. Outstanding Advantages, Current Drawbacks, and Significant Recent Developments in Mechanochemistry: A Perspective View. Crystals 2023, 13, 124. [Google Scholar] [CrossRef]
- Budiman, A.; Nurfadilah, N.; Muchtaridi, M.; Sriwidodo, S.; Aulifa, D.L.; Rusdin, A. The Impact of Water-Soluble Chitosan on the Inhibition of Crystal Nucleation of Alpha-Mangostin from Supersaturated Solutions. Polymers 2022, 14, 4370. [Google Scholar] [CrossRef]
- Siow, C.R.S.; Wan Sia Heng, P.; Chan, L.W. Application of Freeze-Drying in the Development of Oral Drug Delivery Systems. Expert. Opin. Drug Deliv. 2016, 13, 1595–1608. [Google Scholar] [CrossRef]
- Van Drooge, D.J.; Hinrichs, W.L.J.; Frijlink, H.W. Anomalous Dissolution Behaviour of Tablets Prepared from Sugar Glass-Based Solid Dispersions. J. Control. Release 2004, 97, 441–452. [Google Scholar] [CrossRef]
- Van Drooge, D.J.; Hinrichs, W.L.J.; Frijlink, H.W. Incorporation of Lipophilic Drugs in Sugar Glasses by Lyophilization Using a Mixture of Water and Tertiary Butyl Alcohol as Solvent. J. Pharm. Sci. 2004, 93, 713–725. [Google Scholar] [CrossRef]
- Srinarong, P.; Kouwen, S.; Visser, M.R.; Hinrichs, W.L.J.; Frijlink, H.W. Effect of Drug-Carrier Interaction on the Dissolution Behavior of Solid Dispersion Tablets. Pharm. Dev. Technol. 2010, 15, 460–468. [Google Scholar] [CrossRef]
- Nikghalb, L.A.; Singh, G.; Singh, G.; Kahkeshan, K.F. Solid Dispersion: Methods and Polymers to Increase the Solubility of Poorly Soluble Drugs. J. Appl. Pharm. Sci. 2012, 2, 170–175. [Google Scholar] [CrossRef]
- Satoh, T.; Hidaka, F.; Miyake, K.; Yoshiyama, N.; Takeda, K.; Matsuura, T.; Imanaka, H.; Ishida, N.; Imamura, K. Surfactant-Free Solid Dispersion of Fat-Soluble Flavour in an Amorphous Sugar Matrix. Food Chem. 2016, 197, 1136–1142. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Gotoda, Y.; Hirota, D.; Hidaka, F.; Sato, T.; Matsuura, T.; Imanaka, H.; Ishida, N.; Imamura, K. Surfactant-Free Solid Dispersions of Hydrophobic Drugs in an Amorphous Sugar Matrix Dried from an Organic Solvent. Mol. Pharm. 2017, 14, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Sekitoh, T.; Okamoto, T.; Fujioka, A.; Tramis, O.; Takeda, K.; Matsuura, T.; Imanaka, H.; Ishida, N.; Imamura, K. Sole-Amorphous-Sugar-Based Solid Dispersion of Curcumin and the Influence of Formulation Composition and Heat Treatment on the Dissolution of Curcumin. Dry. Technol. 2021, 39, 2065–2074. [Google Scholar] [CrossRef]
- Takeda, K.; Sekitoh, T.; Fujioka, A.; Yamamoto, K.; Okamoto, T.; Matsuura, T.; Imanaka, H.; Ishida, N.; Imamura, K. Physical Stability of an Amorphous Sugar Matrix Dried from Methanol as an Amorphous Solid Dispersion Carrier and the Influence of Heat Treatment. J. Pharm. Sci. 2019, 108, 2056–2062. [Google Scholar] [CrossRef]
- Kaljoriya, H.; Mann, M. Sugar Carriers. Int. J. Pharm. Life Sci. 2021, 12, 5–10. [Google Scholar]
- Jyoti, J.; Shikha, D.A. Solubility Enhancement of Allopurinol by Solid Dispersion Using Sugar Carriers. Int. J. Curr. Res. 2019, 11, 6524–6529. [Google Scholar]
- Dai, X.L.; Yao, J.; Wu, C.; Deng, J.H.; Mo, Y.H.; Lu, T.B.; Chen, J.M. Solubility and Permeability Improvement of Allopurinol by Cocrystallization. Cryst. Growth Des. 2020, 20, 5160–5168. [Google Scholar] [CrossRef]
- Saito, M.; Ugajin, T.; Nozawa, Y.; Sadzuka, Y.; Miyagishima, A.; Sonobe, T. Preparation and Dissolution Characteristics of Griseofulvin Solid Dispersions with Saccharides. Int. J. Pharm. 2002, 249, 71–79. [Google Scholar] [CrossRef]
- Marano, S.; Barker, S.A.; Raimi-Abraham, B.T.; Missaghi, S.; Rajabi-Siahboomi, A.; Craig, D.Q.M. Development of Micro-Fibrous Solid Dispersions of Poorly Water-Soluble Drugs in Sucrose Using Temperature-Controlled Centrifugal Spinning. Eur. J. Pharm. Biopharm. 2016, 103, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Attia, M.A.; Habib, F.S. Dissolution Rates of Carbamazepine and Nitrazepam Utilizing Sugar Solid Dispersion System. Drug Dev. Ind. Pharm. 1985, 11, 1957–1969. [Google Scholar] [CrossRef]
- Lenhart, A.; Chey, W.D. A Systematic Review of the Effects of Polyols on Gastrointestinal Health and Irritable Bowel Syndrome. Adv. Nutr. 2017, 8, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Grembecka, M. Sugar Alcohols. In Encyclopedia of Food Chemistry; Melton, L., Shahidi, F., Varelis, P., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; Volume 1, pp. 265–275. ISBN 9780128140451. [Google Scholar]
- Embuscado, M.E. Polyols. In Optimising Sweet Taste in Foods; Spillane, W.J., Ed.; Woodhead Publishing: Sawston, UK, 2006; pp. 153–174. ISBN 9781845690083. [Google Scholar]
- Hadjikinova, R.; Marudova, M.; Hadjikinova, R.; Marudova, M. Thermal Behaviour of Confectionary Sweeteners’ Blends. Bulg. Chem. Commun. 2016, 48, 446–450. [Google Scholar]
- Zumbé, A.; Lee, A.; Storey, D. Polyols in Confectionery: The Route to Sugar-Free, Reduced Sugar and Reduced Calorie Confectionery. Br. J. Nutr. 2001, 85, S31–S45. [Google Scholar] [CrossRef]
- Talja, R.A.; Roos, Y.H. Phase and State Transition Effects on Dielectric, Mechanical, and Thermal Properties of Polyols. Thermochim. Acta 2001, 380, 109–121. [Google Scholar] [CrossRef]
- Langer, M.; Höltje, M.; Urbanetz, N.A.; Brandt, B.; Höltje, H.D.; Lippold, B.C. Investigations on the Predictability of the Formation of Glassy Solid Solutions of Drugs in Sugar Alcohols. Int. J. Pharm. 2003, 252, 167–179. [Google Scholar] [CrossRef]
- Chawla, G.; Bansal, A.K. Improved Dissolution of a Poorly Water Soluble Drug in Solid Dispersions with Polymeric and Non-Polymeric Hydrophilic Additives. Acta Pharm. 2008, 58, 257–274. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, J.; Hu, A.; Nie, T.; Cheng, Z.; Liu, W. A Critical Review on Engineering of D-Mannitol Crystals: Properties, Applications, and Polymorphic Control. Crystals 2022, 12, 1080. [Google Scholar] [CrossRef]
- Punitha, S.; Bn, V.H.; Karthikeyan, D. Enhancement of Celecoxib Solubility by Solid Disperson Using Mannitol. Int. J. Pharm. Pharm. Sci. 2010, 2, 4–6. [Google Scholar]
- Bahmani, K.; Singla, Y. Enhanced Solubility of Antihypertensive Drug Using Hydrophilic Carrier-Based Potent Solid Dispersion Systems. Int. J. Pharm. Res. Technol. 2019, 9, 24–37. [Google Scholar]
- Yadav, P.S.; Kumar, V.; Pratap, U.; Raj, H.; Mazumder, B. Physicochemical Characterization and in Vitro Dissolution Studies of Solid Dispersions of Ketoprofen with PVP K30 and D-Mannitol. Saudi Pharm. J. 2013, 21, 77–84. [Google Scholar] [CrossRef]
- Singh, G.; Chhabra, G.; Pathak, K. Dissolution Behavior and Thermodynamic Stability of Fused-Sugar Dispersions of a Poorly Water-Soluble Drug. Dissolution Technol. 2011, 18, 62–70. [Google Scholar] [CrossRef]
- Shittu, A.O.; Njinga, N.S.; Orshio, S.D. Development and Characterization of Ibuprofen Solid Dispersion for Solubility and Dissolution Improvement Using a Binary Carrier System Consisting of D-Mannitol—Polyethylene Glycol 6000. Niger. J. Pharm. 2022, 56, 109–118. [Google Scholar]
- Zajc, N.; Obreza, A.; Bele, M.; Srčič, S. Physical Properties and Dissolution Behaviour of Nifedipine/Mannitol Solid Dispersions Prepared by Hot Melt Method. Int. J. Pharm. 2005, 291, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.; Patel, R.; Shah, H.; Purohit, S.; Pawar, M.; Pathan, A. Solubility Enhancement of Azithromycin by Solid Dispersion Technique Using Mannitol and β-Cyclodextrin. Acta Sci. Pharm. Sci. 2022, 5, 48–54. [Google Scholar] [CrossRef]
- Walke, P.S.; Khairnar, P.S.; Narkhede, M.R.; Nehete, J.Y. Solubility Enhancement of Diacerein by Mannitol Solid Dispersons. Int. J. Pharm. Pharm. Sci. 2011, 3, 261–264. [Google Scholar]
- Krishnamoorthy, V.; Priya, V.; Prasad, R. Physicochemical Characterization and in Vitro Dissolution Behavior of Olanzapine-Mannitol Solid Dispersions. Braz. J. Pharm. Sci. 2012, 48, 243–255. [Google Scholar] [CrossRef]
- Okonogi, S.; Oguchi, T.; Yonemochi, E.; Puttipipatkhachorn, S.; Yamamoto, K. Improved Dissolution of Ofloxacin via Solid Dispersion. Int. J. Pharm. 1997, 156, 175–180. [Google Scholar] [CrossRef]
- Dubey, A.; Road, R.; Pradesh, M. Enhancement of Aqueous Solubility and Dissolution of Telmisartan. Int. J. Pharm. Sci. Res. 2014, 5, 4478–4485. [Google Scholar] [CrossRef]
- Neupane, S.; Thapa, C. Formulation and Enhancement of Dissolution Rate of Poorly Aqueous Soluble Drug Aceclofenac by Solid Dispersion Method: In Vitro Study. Afr. J. Pharm. Pharmacol. 2020, 14, 1–8. [Google Scholar] [CrossRef]
- Rao, Y.S.; Vijaya, L.; Varalakshmi, T.; Chandana, R.; Chowdary, K.P.R. Formulation and Evaluation of Carvedilol Solid Dispersions for Dissolution Rate Enhancement. Int. J. Adv. Pharm. Biol. Chem. 2012, 1, 489–495. [Google Scholar]
- Kanaze, F.I.; Kokkalou, E.; Niopas, I.; Georgarakis, M.; Stergiou, A.; Bikiaris, D. Thermal Analysis Study of Flavonoid Solid Dispersions Having Enhanced Solubility. J. Therm. Anal. Calorim. 2006, 83, 283–290. [Google Scholar] [CrossRef]
- Krishnamoorthy, V.; Verma, P.; Sen, S. Studies on Clozapine-Mannitol Solid Dispersions, Physico Chemical Characterization and Characterization and Evaluation. Turk. J. Pharm. Sci. 2013, 10, 109–124. [Google Scholar]
- Duret, C.; Wauthoz, N.; Sebti, T.; Vanderbist, F.; Amighi, K. Solid Dispersions of Itraconazole for Inhalation with Enhanced Dissolution, Solubility and Dispersion Properties. Int. J. Pharm. 2012, 428, 103–113. [Google Scholar] [CrossRef]
- Kauppinen, A.; Broekhuis, J.; Grasmeijer, N.; Tonnis, W.; Ketolainen, J. Efficient Production of Solid Dispersions by Spray Drying Solutions of High Solid Content Using a 3-Fluid Nozzle. Eur. J. Pharm. Biopharm. 2018, 123, 50–58. [Google Scholar] [CrossRef]
- El-maradny, H.; Saab, M. Spray-Dried Co-Amorphous Tadalafil Ternary Mixtures: A Promising Platform towards the Enhancement of Solubility and Bioavailability. Braz. J. Pharm. Sci. 2022, 58, 1–14. [Google Scholar]
- Thakur, P.S.; Thakore, S.D.; Bansal, A.K. Role of Surface Characteristics of Mannitol in Crystallization of Feno Fi Brate During Spray Drying. J. Pharm. Sci. 2020, 109, 1105–1114. [Google Scholar] [CrossRef]
- Thakral, S.; Sonje, J.; Munjal, B.; Bhatnagar, B.; Suryanarayanan, R. Mannitol as an Excipient for Lyophilized Injectable Formulations. J. Pharm. Sci. 2023, 112, 19–35. [Google Scholar] [CrossRef]
- Verma, U.; Naik, J.B.; Mokale, V.J. Preparation of Freeze-Dried Solid Dispersion Powder Using Mannitol to Enhance Solubility of Lovastatin and Development of Sustained Release Tablet Dosage Form. Am. J. Pharm. Sci. Nanotechnol. 2014, 1, 11–26. [Google Scholar]
- Kulthe, V.; Chaudhari, P.; Aboul-Enein, H. Freeze-Dried Amorphous Dispersions for Solubility Enhancement of Thermosensitive API Having Low Molecular Lipophilicity. Drug Res. 2014, 64, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Muehlenfeld, C.; Kann, B.; Windbergs, M.; Thommes, M. Solid Dispersions Prepared by Continuous Cogrinding in an Air Jet Mill. J. Pharm. Sci. 2013, 102, 4132–4139. [Google Scholar] [CrossRef] [PubMed]
- Lenschow, I.C.S.; Bazzo, G.C.; Zétola, M.; Stulzer, H.K.; Soares, L.; Pezzini, B.R. Ball-Milled Valsartan and Its Combination with Mannitol: The Case of Drug Polyamorphism. J. Therm. Anal. Calorim. 2022, 147, 8765–8777. [Google Scholar] [CrossRef]
- Zaini, E.; Umar, S.; Firdaus, N. Improvement of Dissolution Rate of Valsartan by Solid Dispersion System Using D(−) Mannitol. Asian J. Pharm. Clin. Res. 2017, 10, 288–290. [Google Scholar] [CrossRef]
- Deis, R.C.; Kearsley, M.W. Sorbitol and Mannitol. In Sweeteners and Sugar Alternatives in Food Technology; Kay O’Donnell, M.W.K., Ed.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2012; pp. 331–346. ISBN 9780470659687. [Google Scholar]
- Dash, R.P.; Srinivas, N.R.; Babu, R.J. Use of Sorbitol as Pharmaceutical Excipient in the Present Day Formulations–issues and Challenges for Drug Absorption and Bioavailability. Drug Dev. Ind. Pharm. 2019, 45, 1421–1429. [Google Scholar] [CrossRef]
- Hasan, A.; El, A.; Elghany, M.A.; Sabry, S. Design and Characterization of Intra-Oral Fast Dissolving Tablets Containing Diacerein-Solid Dispersion. J. Appl. Pharm. Sci. 2020, 10, 44–53. [Google Scholar] [CrossRef]
- Sinha, S.; Ali, M.; Baboota, S.; Ahuja, A.; Kumar, A.; Ali, J. Solid Dispersion as an Approach for Bioavailability Enhancement of Poorly Water-Soluble Drug Ritonavir. AAPS PharmSciTech 2010, 11, 518–527. [Google Scholar] [CrossRef]
- Kaialy, W.; Maniruzzaman, M.; Shojaee, S.; Nokhodchi, A. Antisolvent Precipitation of Novel Xylitol-Additive Crystals to Engineer Tablets with Improved Pharmaceutical Performance. Int. J. Pharm. 2014, 477, 282–293. [Google Scholar] [CrossRef]
- Sirenius, I.; Krogerus, V.E.; Leppänen, T. Dissolution Rate of P-aminobenzoates from Solid Xylitol Dispersions. J. Pharm. Sci. 1979, 68, 791–792. [Google Scholar] [CrossRef]
- Mummaneni, V.; Vasavada, R.C. Solubilization and Dissolution of Famotidine from Solid Glass Dispersions of Xylitol. Int. J. Pharm. 1990, 66, 71–77. [Google Scholar] [CrossRef]
- Pawar, J.N.; Fule, R.A.; Maniruzzaman, M.; Amin, P.D. Solid Crystal Suspension of Efavirenz Using Hot Melt Extrusion: Exploring the Role of Crystalline Polyols in Improving Solubility and Dissolution Rate. Mater. Sci. Eng. C 2017, 78, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Semjonov, K.; Kogermann, K.; Laidmäe, I.; Antikainen, O.; Strachan, C.J.; Ehlers, H.; Yliruusi, J.; Heinämäki, J. The Formation and Physical Stability of Two-Phase Solid Dispersion Systems of Indomethacin in Supercooled Molten Mixtures with Different Matrix Formers. Eur. J. Pharm. Sci. 2017, 97, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Narala, S.; Komanduri, N.; Nyavanandi, D.; Adel, A.; Youssef, A.; Mandati, P.; Alzahrani, A.; Kolimi, P.; Narala, N. Hard Gelatin Capsules Containing Hot Melt Extruded Solid Crystal Suspension of Carbamazepine for Improving Dissolution: Preparation and in Vitro Evaluation. J. Drug Deliv. Sci. Technol. 2023, 82, 104384. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.J.; Jeya, M.; Kim, I.W.; Lee, J.K. Biotechnological Production of Erythritol and Its Applications. Appl. Microbiol. Biotechnol. 2010, 86, 1017–1025. [Google Scholar] [CrossRef]
- Mazi, T.A.; Stanhope, K.L. Erythritol: An In-Depth Discussion of Its Potential to Be a Beneficial Dietary Component. Nutrients 2023, 15, 204. [Google Scholar] [CrossRef]
- Tyapkova, O.; Bader-Mittermaier, S.; Schweiggert-Weisz, U. Factors Influencing Crystallization of Erythritol in Aqueous Solutions: A Preliminary Study. J. Food Res. 2012, 1, 207–217. [Google Scholar] [CrossRef][Green Version]
- Nishimoto, Y.; Hattori, Y.; Otsuka, M. Characterization of Ternary Amorphous Solid Dispersion Containing Hypromellose Phthalate and Erythritol Prepared by Hot Melt Extrusion Using Melting Point Depression. J. Drug Deliv. Sci. Technol. 2020, 58, 101797. [Google Scholar] [CrossRef]
- Liu, X.; Hu, Y.; Yang, W.; Liu, Y.; Liang, M. Solubility of Erythritol in Methanol, Ethanol and Methanol+ethanol: Experimental Measurement and Thermodynamic Modeling. Fluid Phase Equilib. 2013, 360, 134–138. [Google Scholar] [CrossRef]
- Msomi, N.Z.; Erukainure, O.L.; Islam, M.S. Suitability of Sugar Alcohols as Antidiabetic Supplements: A Review. J. Food Drug Anal. 2021, 29, 1–14. [Google Scholar] [CrossRef]
- Ghanavati, R.; Taheri, A.; Homayouni, A. Anomalous Dissolution Behavior of Celecoxib in PVP/Isomalt Solid Dispersions Prepared Using Spray Drier. Mater. Sci. Eng. C 2017, 72, 501–511. [Google Scholar] [CrossRef]
- Khodaverdi, E.; Khalili, N.; Zangiabadi, F.; Homayouni, A. Preparation, Characterization and Stability Studies of Glassy Solid Dispersions of Indomethacin Using PVP and Isomalt as Carriers. Iran. J. Basic Med. Sci. 2012, 15, 820–832. [Google Scholar] [PubMed]
- Moura Ramos, J.J.; Viciosa, M.T.; Diogo, H.P. Thermal Behaviour of Two Anti-Inflammatory Drugs (Celecoxib and Rofecoxib) and Slow Relaxation Dynamics in Their Amorphous Solid State. Comparison between the Dynamic Fragility Obtained by Dielectric Spectroscopy and by Thermostimulated Currents. Mol. Phys. 2019, 117, 644–660. [Google Scholar] [CrossRef]
- Svoboda, R.; Košťálová, D.; Krbal, M.; Komersová, A. Indomethacin: The Interplay between Structural Relaxation, Viscous Flow and Crystal Growth. Molecules 2022, 27, 5668. [Google Scholar] [CrossRef] [PubMed]
- Kearsley, M.W.; Deis, R.C. Maltitol Powder. In Sweeteners and Sugar Alternatives in Food Technology; Wiley: Hoboken, NJ, USA, 2012; pp. 295–308. ISBN 9780470659687. [Google Scholar]
- Magan Montoto, Y. Sugar Alcohols and Other Organic Compounds as Phase Change Materials; Vienna University of Technology: Vienna, Austria, 2018. [Google Scholar]
- Izutsu, K.; Koide, T.; Takata, N.; Ikeda, Y.; Ono, M. Characterization and Quality Control of Cocrystals. Chem. Pharm. Bull. 2016, 64, 1421–1430. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Grohganz, H.; Löbmann, K.; Rades, T.; Hempel, N.J. Co-Amorphous Drug Formulations in Numbers: Recent Advances in Co-Amorphous Drug Formulations with Focus on Co-Formability, Molar Ratio, Preparation Methods, Physical Stability, in Vitro and in Vivo Performance, and New Formulation Strategies. Pharmaceutics 2021, 13, 389. [Google Scholar] [CrossRef] [PubMed]
- Löbmann, K.; Laitinen, R.; Strachan, C.; Rades, T.; Grohganz, H. Amino Acids as Co-Amorphous Stabilizers for Poorly Water-Soluble Drugs—Part 2: Molecular Interactions. Eur. J. Pharm. Biopharm. 2013, 85, 882–888. [Google Scholar] [CrossRef] [PubMed]
- Kuminek, G.; Cao, F.; Da Rocha, A.B.D.O.; Cardoso, S.G.; Rodríguez-Hornedo, N. Cocrystals to Facilitate Delivery of Poorly Soluble Compounds Beyond-Rule-of-5 Graphical Abstract HHS Public Access. Adv. Drug Deliv. Rev. 2016, 101, 143–166. [Google Scholar] [CrossRef]
- Bavishi, D.D.; Borkhataria, C.H. Spring and Parachute: How Cocrystals Enhance Solubility. Prog. Cryst. Growth Charact. Mater. 2016, 62, 1–8. [Google Scholar] [CrossRef]
- Guo, M.; Sun, X.; Chen, J.; Cai, T. Pharmaceutical Cocrystals: A Review of Preparations, Physicochemical Properties and Applications. Acta Pharm. Sin. B 2021, 11, 2537–2564. [Google Scholar] [CrossRef]
- Kiyonga, E.M.; Kekani, L.N.; Chidziwa, T.V.; Kahwenga, K.D.; Bronkhorst, E.; Milne, M.; Poka, M.S.; Mokhele, S.; Demana, P.H.; Witika, B.A. Nano- and Crystal Engineering Approaches in the Development of Therapeutic Agents for Neoplastic Diseases. Crystals 2022, 12, 926. [Google Scholar] [CrossRef]
- Karagianni, A.; Malamatari, M.; Kachrimanis, K. Pharmaceutical Cocrystals: New Solid Phase Modification Approaches for the Formulation of APIs. Pharmaceutics 2018, 10, 18. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.H.; Augis, L.; Fourmentin, S.; Barratt, G.; Legrand, F.X. Deep Eutectic Solvents for Innovative Pharmaceutical Formulations. In Deep Eutectic Solvents for Medicine, Gas Solubilization and Extraction of Natural Substances; Fourmentin, S., Gomes, M.C., Lichtfouse, E., Eds.; Springer: Berlin/Heidelberg, Germany, 2020; pp. 41–102. ISBN 9783540228608. [Google Scholar]
- Paiva, A.; Craveiro, R.; Aroso, I.; Martins, M.; Reis, R.L.; Duarte, A.R.C. Natural Deep Eutectic Solvents—Solvents for the 21st Century. ACS Sustain. Chem. Eng. 2014, 2, 1063–1071. [Google Scholar] [CrossRef]
- Vanda, H.; Verpoorte, R.; Klinkhamer, P.G.L.; Choi, Y.H. Natural Deep Eutectic Solvents: From Their Discovery to Their Applications. In Deep Eutectic Solvents: Synthesis, Properties, and Applications; Ramón, D.J., Guillena, G., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2019; pp. 61–81. ISBN 9783527818471. [Google Scholar]
- Jeliński, T.; Przybyłek, M.; Cysewski, P. Solubility Advantage of Sulfanilamide and Sulfacetamide in Natural Deep Eutectic Systems: Experimental and Theoretical Investigations. Drug Dev. Ind. Pharm. 2019, 45, 1120–1129. [Google Scholar] [CrossRef] [PubMed]
- Jeliński, T.; Przybyłek, M.; Cysewski, P. Natural Deep Eutectic Solvents as Agents for Improving Solubility, Stability and Delivery of Curcumin. Pharm. Res. 2019, 36, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; van Spronsen, J.; Witkamp, G.-J.; Verpoorte, R.; Choi, Y.H. Natural Deep Eutectic Solvents as New Potential Media for Green Technology. Anal. Chim. Acta 2013, 766, 61–68. [Google Scholar] [CrossRef]
- Liu, Y.; Friesen, J.B.; McAlpine, J.B.; Lankin, D.C.; Chen, S.N.; Pauli, G.F. Natural Deep Eutectic Solvents: Properties, Applications, and Perspectives. J. Nat. Prod. 2018, 81, 679–690. [Google Scholar] [CrossRef]

| Method | Advantages | Limitations | Ref. |
|---|---|---|---|
| Fusion |
|
| [29,30] |
| HME |
|
| [19,29,30,31] |
| KM |
|
| [25] |
| SE |
|
| [19,27,29] |
| Co-precipitation |
|
| [25] |
| SCF |
|
| [19,24] |
| SD |
|
| [25,32] |
| FD |
|
| [25,31] |
| ES |
|
| [25,31] |
| Milling |
|
| [33] |
| Polyol | Structure | MW (g/mol) | HBD/HBA | MP (°C) | Tg (°C) | Aq.S (% at 20 °C) | Ref. |
|---|---|---|---|---|---|---|---|
| Erythritol |  | 122.12 | 4/4 | 118–126 | −45.0–−42.50 | 37 | [67,68,69] |
| Isomalt |  | 344.31 | 9/11 | 98 and 155 | 61.5 | 25 | [67,68,70] |
| Mannitol |  | 182.17 | 6/6 | 164–176 | 10.7 | 20 | [68,70,71] |
| Maltitol |  | 344.31 | 9/11 | 148–151 | 43.1–49.5 | 60 | [67,68,70] |
| Sorbitol |  | 182.17 | 6/6 | 95–97 | −9.20–−6.0 | 73 | [67,68,69] |
| Xylitol |  | 152.15 | 5/5 | 92.7 | −24.10 | 63 | [68,69] |
| Polyol | Method | Drug: Carrier (w/w) | Drug | Solubility | Dissolution | Remarks | Ref. |
|---|---|---|---|---|---|---|---|
| Mannitol | Fusion | 1:3, 1:1, and 3:1 | Clotrimazole | Significant increase at 1:13 ratio. | Increase with increase in mannitol concentration. | Partial dispersion of drug at molecular level. | [7] |
| Fusion | 1:1 | Sulfamethoxazole | No significant change with increase in carrier concentration. | A 100% drug release in 5 min. | Partial dispersion of drug at molecular level. | [37] | |
| Fusion | 4:1, 2:1, 1:1, and 1:4 | Ibuprofen | N/A | A 100% drug release within 60 min at 1:4 ratio. | SDs of hydrophilic polymers released 100% within 15 min | [40] | |
| SE | 1:1 and 1:5 | Etoricoxib | 1.4 to 1.8-fold at 1:5 ratio. | N/A | Intermolecular hydrogen bonding between S=O group of etoricoxib and O–H group of mannitol. | [44] | |
| Quench Cooling | N/A | Irbesartan | Increased solubility reported. | Lower dissolution compared with pure drug due to formation of hard plug by SDs. | Mannitol does not affect the pH of the dissolution medium. | [71] | |
| Fusion and SE | 1:1, 1:3, and 1:5 | Celecoxib | Increase of 1.3- and 1.2-fold at a 1:5 ratio for fusion and SE, respectively. | Increases of 9- and 6-fold in drug release by 60 min for fusion and SE, respectively. | No intermolecular interactions formed. | [73] | |
| Fusion | 1:1, 1:2, 1:3, and 1:5 | Carvedilol | N/A | Close to 100% drug release by 30 min for SDs as compared to 53% for pure drug. | No intermolecular interactions formed. | [74] | |
| Fusion and Kneading | 1:1, 1:3, and 1:5 | Ketoprofen | Increase in solubility with an increase in carrier concentration. | Six-fold increase in comparison with the pure drug by 30 min. | No intermolecular interactions formed. | [75] | |
| Fusion | 1:1 and 1:9 | Nifedipine | N/A | SDs at 1:9 ratio showed better dissolution rate. | No intermolecular interactions formed. | [78] | |
| Fusion, Kneading and SE | 1:1, 1:2, 1:3, 1:4, and 1:5 | Azithromycin | Solubility in the order of fusion > SE > kneading | N/A | Solubility decreased at 1:4 ratio. Possible reason could be increased viscosity at high sugar concentrations. | [79] | |
| Fusion and SE | 1:1, 1:3, and 1:5 | Diacerein | Increase of 27- and 26-fold at a 1:5 ratio for fusion and SE, respectively. | A 100% and 87% drug release by 60 min for fusion and SE, respectively, in comparison with 53% for pure drug. | Possible hydrogen bonding between the carrier and drug with fusion method. | [80] | |
| SE and SD | 1:1, 1:2, 1:4, 1:6,1:8, and 1:10 | Olanzapine | N/A | Increase in dissolution rate with increase in carrier concentration. | Increased dissolution is associated with the decreased crystallinity of the drug. | [81] | |
| SE | 1:2 and 1:3 | Aceclofenac | N/A | Mannitol > dextrose > HPMC > PVA | None | [84] | |
| 1:1, 1:2, and 1:4 | Carvedilol | PEG > lactose > mannitol | PEG > lactose > mannitol | Increase in solubility and dissolution with increase in carrier concentration. | [85] | ||
| SE | 1:1, 1:2, 1:4, 1:6,1:8, and 1:10 | Clozapine | N/A | Increase in dissolution rate with increase in carrier concentration and no significant change beyond 1:2. | Increased dissolution is associated with the decreased crystallinity of the drug. | [87] | |
| SD | N/A | Diazepam | N/A | Dissolution rate increased with increasing water/organic solvent ratio. | Crystallinity of diazepam decreased when the water/organic solvent ratio increased. | [89] | |
| SD | 1:1, 1:2, and 1:4 | Tadalafil | 3 to 6-fold increases | Three- to ten-fold increase. | ASD formed were stable for 6 months at 40 °C. | [90] | |
| FD | Various ratios (between 1:1 and 1:2) | Lovastatin | 5–6-fold increase | Tablets of SDs mixed with hydrophilic polymers provided 1.4-fold increase. | SDs are amorphous in nature with particle sizes between 100 to 1000 nm. | [93] | |
| FD | 1:0.5, 1:1, and 1:2 | Acetazolamide | 5 to 6-fold increase at 1:1 ratio | Superior performance by ASD at a 1:1 ratio with >90% drug release by 60 min. | ASD with 1:1 ratio was stable for 6 months with no significant change in the crystallinity. | [94] | |
| Milling | 1:1, 1:3, and 1:9 | Griseofulvin | N/A | Significant increase in % drug release for all the SDs. | No significant difference in % drug release between physical mixtures and SDs. | [95] | |
| Milling | 1:1 and 1:3 | Valsartan | N/A | Tablets of SDs showed >90% drug release in 30 min in comparison with <20% pure drug. | No significant difference in % drug release between physical mixtures and SDs. | [96] | |
| Milling | 1:1, 1:3, and 1:5 | Valsartan | N/A | SDs showed >90% drug release in 60 min in comparison with <40% pure drug. | No significant difference in % drug release between SDs with various ratios of mannitol. | [97] | |
| Sorbitol | SE | 1:1, 1:3, and 1:5 | Diacerein | N/A | Increase in dissolution rate over pure drug and physical mixtures in the order of sorbitol > PEG400 > PVPk25. | Tablets prepared with SDs were stable at 30 °C/75% RH and 40 °C/75% RH for 12 weeks. | [100] |
| Fusion and SE | 1:1, 1:2, 1:3, and 1:4 | Ritonavir | 2000-fold increase in solubility at a 1:4 ratio | Two-fold and four-fold increases for SDs prepared with SE and fusion methods, respectively. | Hydrogen bond formation through interaction with amide and carbonyl groups of drug and hydroxyl groups of sorbitol. | [101] | |
| Xylitol | Fusion | 1:9 | Etoricoxib | N/A | Two-fold increase | Ternary and quaternary systems performed better than the binary systems. | [76] |
| Fusion | 5% | p-Aminobenzoates | N/A | A 200-fold increase. | N/A | [103] | |
| Fusion | 1:20 and 1:40 | Famotidine | A 32% increase in solubility over pure drug for 1:40 SD. | A 100% drug release in less than a minute for 1:20 SD, compared to 50% for pure drug. | Eutectic mixture formed at 1:40. | [104] | |
| HME | 1:1 and 1:4 | Efavirenz | An 81-fold increase for 1:4 SD. | A 4.1-fold increase in dissolution rate. | Reduced dissolution rate during storage during storage. | [105] | |
| HME | 1:4 and 2:3 | Carbamazepine | Aqueous solubility is increased by 50-fold. | A 100% drug release in 15 min in comparison with 40% for the pure drug. | Stable for 3 months under accelerated conditions. | [107] | |
| Erythritol | HME | Various | Griseofulvin | A 10-fold increase in solubility. | N/A | Substantial reduction in melting point of the drug, resulting in reduced processing temperature. | [111] |
| Isomalt | SD | Various | Celecoxib | A 5-fold increase in solubility. | A 94% drug release after 5 min for ternary systems in comparison with 31% for the pure drug. | Binary systems of drug-to-isomalt ratio showed recrystallisation after 1 week. Ternary systems with PVP were stable. | [114] |
| SD | 2%, 10%, and 30% | Indomethacin | N/A | Rate of dissolution decreased with increase in carrier concertation. | Isomalt SDs showed better physical stability compared with PVP SDs. | [115] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poka, M.S.; Milne, M.; Wessels, A.; Aucamp, M. Sugars and Polyols of Natural Origin as Carriers for Solubility and Dissolution Enhancement. Pharmaceutics 2023, 15, 2557. https://doi.org/10.3390/pharmaceutics15112557
Poka MS, Milne M, Wessels A, Aucamp M. Sugars and Polyols of Natural Origin as Carriers for Solubility and Dissolution Enhancement. Pharmaceutics. 2023; 15(11):2557. https://doi.org/10.3390/pharmaceutics15112557
Chicago/Turabian StylePoka, Madan Sai, Marnus Milne, Anita Wessels, and Marique Aucamp. 2023. "Sugars and Polyols of Natural Origin as Carriers for Solubility and Dissolution Enhancement" Pharmaceutics 15, no. 11: 2557. https://doi.org/10.3390/pharmaceutics15112557
APA StylePoka, M. S., Milne, M., Wessels, A., & Aucamp, M. (2023). Sugars and Polyols of Natural Origin as Carriers for Solubility and Dissolution Enhancement. Pharmaceutics, 15(11), 2557. https://doi.org/10.3390/pharmaceutics15112557