
Testing the Protective Effects of Sulfobutylether-Βeta-Cyclodextrin (SBECD) and Sugammadex against Chlorpromazine-Induced Acute Toxicity in SH-SY5Y Cell Line and in NMRI Mice


 and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Experiments
2.3. Animal Experiments
3. Results
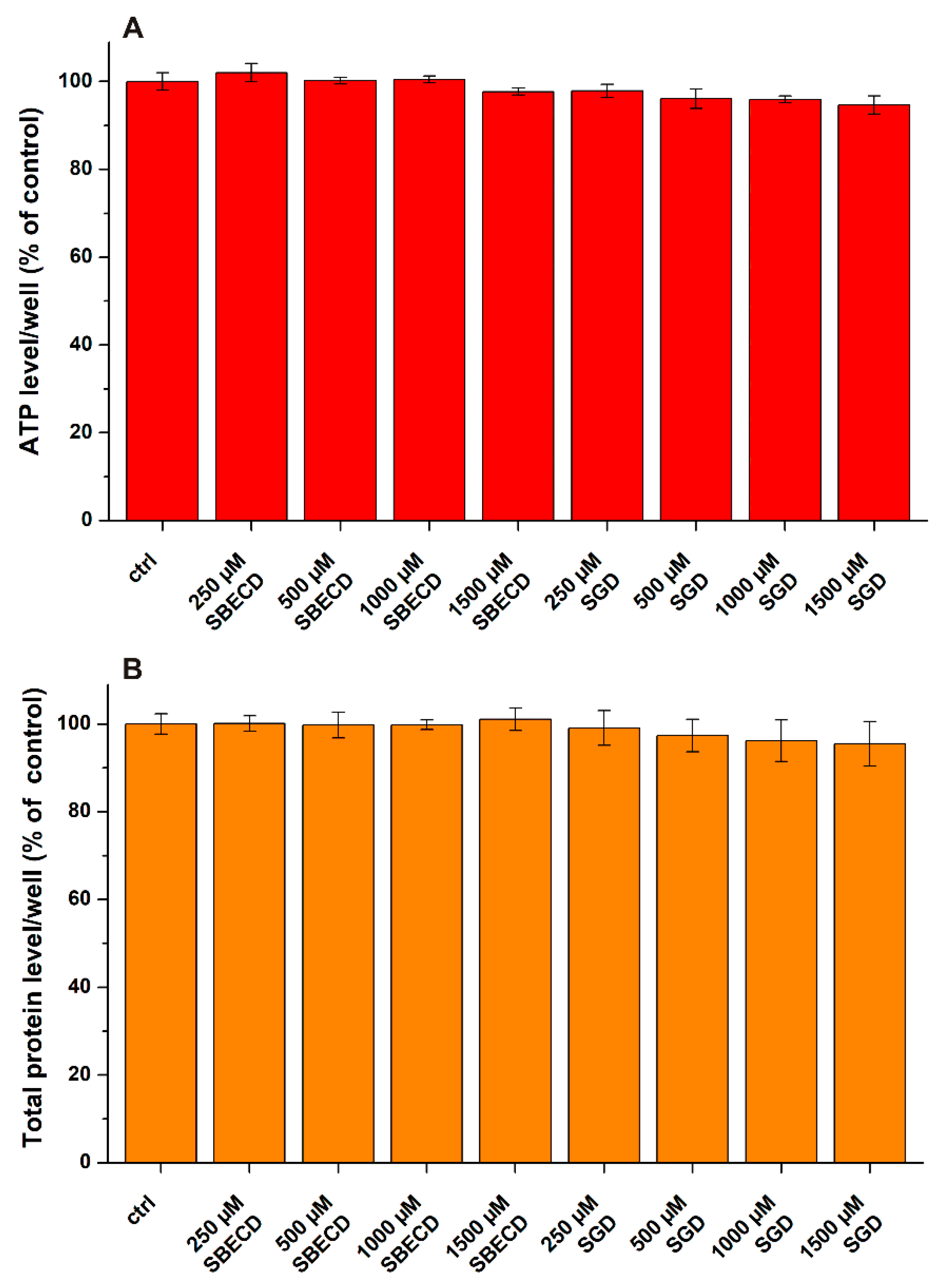
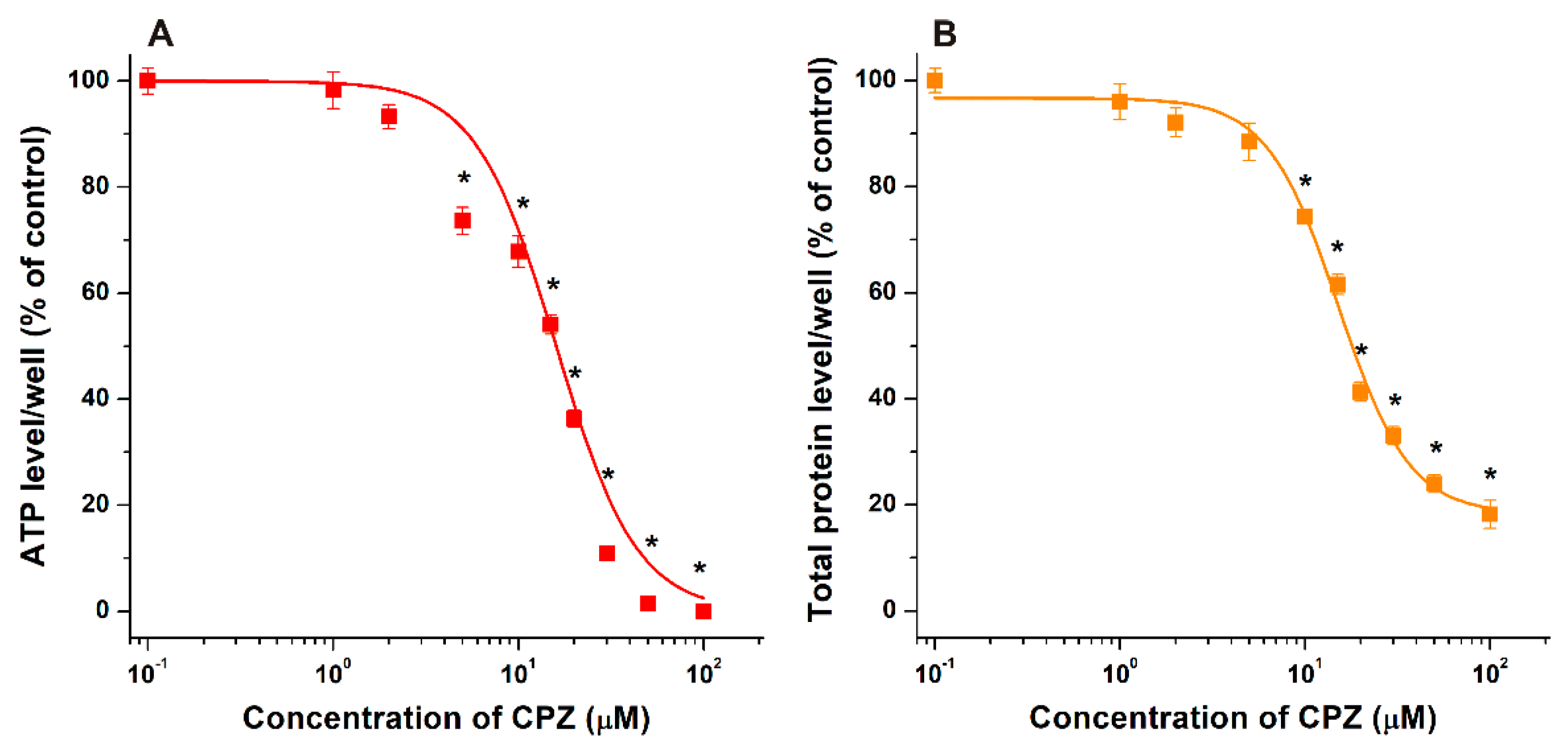
3.1. Effects of CDs on CPZ-Induced Decrease in Cell Viability
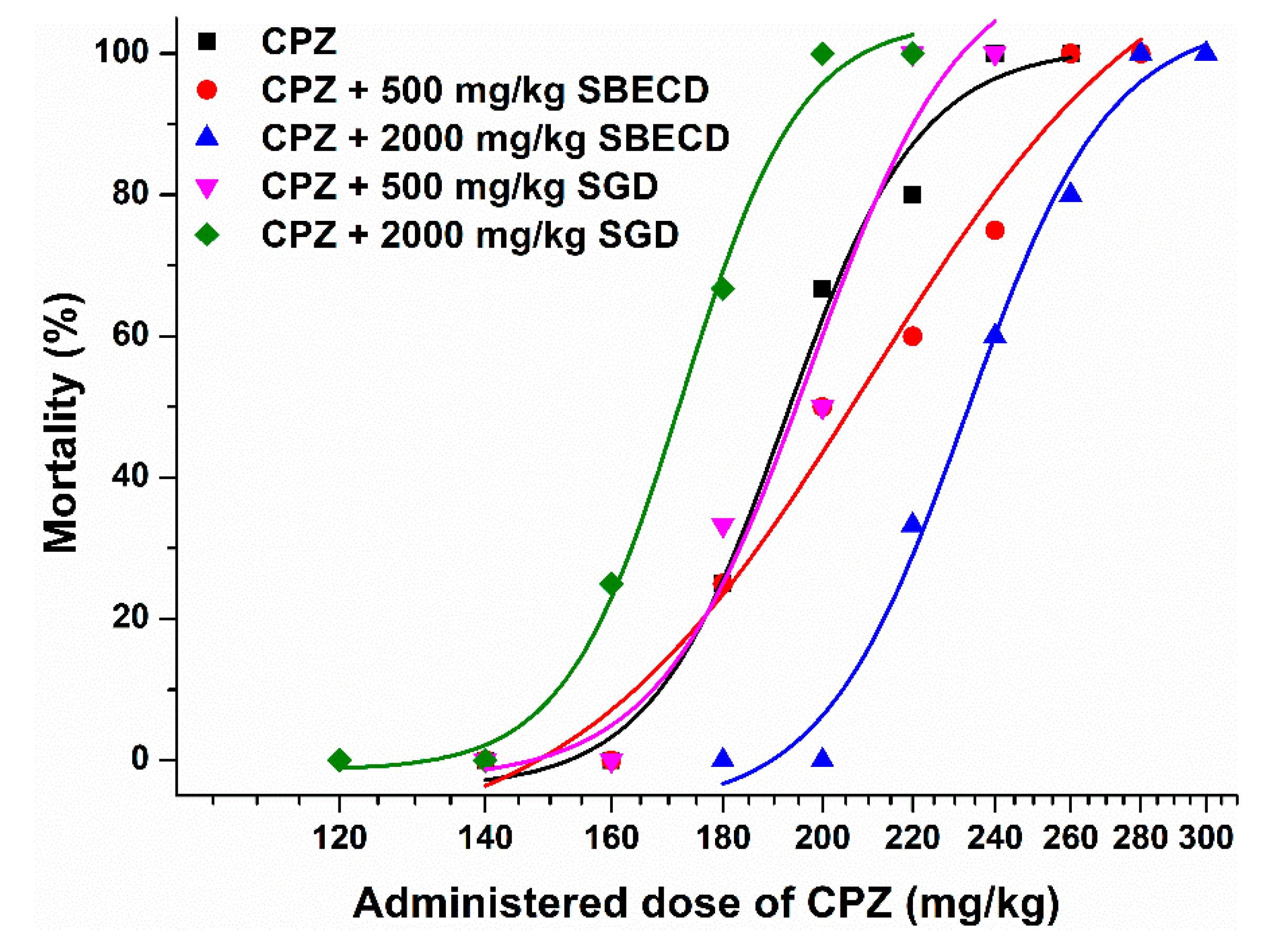
3.2. Effects of CDs on CPZ-Induced 24 h Mortality in Female NMRI Mice
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boyd-Kimball, D.; Gonczy, K.; Lewis, B.; Mason, T.; Siliko, N.; Wolfe, J. Classics in Chemical Neuroscience: Chlorpromazine. ACS Chem. Neurosci. 2019, 10, 79–88. [Google Scholar] [CrossRef]
- López-Muñoz, F.; Alamo, C.; Cuenca, E.; Shen, W.W.; Clervoy, P.; Rubio, G. History of the discovery and clinical introduction of chlorpromazine. Ann. Clin. Psychiatry 2005, 17, 113–135. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. World Health Organization Model List of Essential Medicines: 22nd List (2021); World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- Saha, K.B.; Bo, L.; Zhao, S.; Xia, J.; Sampson, S.; Zaman, R.U. Chlorpromazine versus atypical antipsychotic drugs for schizophrenia. Cochrane Database Syst. Rev. 2016, 4, CD010631. [Google Scholar] [CrossRef] [PubMed]
- Samara, M.T.; Cao, H.; Helfer, B.; Davis, J.M.; Leucht, S. Chlorpromazine versus every other antipsychotic for schizophrenia: A systematic review and meta-analysis challenging the dogma of equal efficacy of antipsychotic drugs. Eur. Neuropsychopharmacol. 2014, 24, 1046–1055. [Google Scholar] [CrossRef] [PubMed]
- Otręba, M.; Kośmider, L.; Rzepecka-Stojko, A. Antiviral activity of chlorpromazine, fluphenazine, perphenazine, prochlorperazine, and thioridazine towards RNA-viruses. A review. Eur. J. Pharmacol. 2020, 887, 173553. [Google Scholar] [CrossRef] [PubMed]
- Stip, E.; Rizvi, T.A.; Mustafa, F.; Javaid, S.; Aburuz, S.; Ahmed, N.N.; Aziz, K.A.; Arnone, D.; Subbarayan, A.; Al Mugaddam, F.; et al. The Large Action of Chlorpromazine: Translational and Transdisciplinary Considerations in the Face of COVID-19. Front. Pharmacol. 2020, 11, 577678. [Google Scholar] [CrossRef] [PubMed]
- Solmi, M.; Murru, A.; Pacchiarotti, I.; Undurraga, J.; Veronese, N.; Fornaro, M.; Stubbs, B.; Monaco, F.; Vieta, E.; Seeman, M.V.; et al. Safety, tolerability, and risks associated with first-and secondgeneration antipsychotics: A state-of-the-art clinical review. Therapeut. Clin. Risk Manag. 2017, 13, 757–777. [Google Scholar] [CrossRef]
- Szente, L.; Szejtli, J. Highly soluble cyclodextrin derivatives: Chemistry, properties, and trends in development. Adv. Drug Deliv. Rev. 1999, 36, 17–28. [Google Scholar] [CrossRef]
- Szente, L.; Singhal, A.; Domokos, A.; Song, B. Cyclodextrins: Assessing the impact of cavity size, occupancy, and substitutions on cytotoxicity and cholesterol homeostasis. Molecules 2018, 23, 1228. [Google Scholar] [CrossRef]
- Challa, R.; Ahuja, A.; Ali, J.; Khar, R.K. Cyclodextrins in drug delivery: An updated review. AAPS PharmSciTech 2005, 6, E329–E357. [Google Scholar] [CrossRef]
- Jambhekar, S.S.; Breen, P. Cyclodextrins in pharmaceutical formulations II: Solubilization, binding constant, and complexation efficiency. Drug. Discov. Today 2016, 21, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Szejtli, J.; Osa, T. Preparation of Cyclodextrin Complexes. In Comprehensive Supramolecular Chemistry, (Cyclodextrins); Szejtli, J., Osa, T., Eds.; Pergamon: Oxford, UK, 1996; Volume 3, pp. 243–251. [Google Scholar]
- Daruházi, A.E.; Szente, L.; Balogh, B.; Mátyus, P.; Béni, S.; Takács, M.; Gergely, A.; Horváth, P.; Szoke, E.; Lemberkovics, E. Utility of cyclodextrins in the formulation of genistein part 1. Preparation and physicochemical properties of genistein complexes with native cyclodextrins. J. Pharm. Biomed. Anal. 2008, 48, 636–640. [Google Scholar] [CrossRef] [PubMed]
- Franco, P.; De Marco, I. Preparation of non-steroidal anti-inflammatory drug/β-cyclodextrin inclusion complexes by supercritical antisolvent process. J. CO2 Util. 2021, 44, 101397. [Google Scholar] [CrossRef]
- Irie, T.; Uekama, K. Cyclodextrins in peptide and protein delivery. Adv. Drug Deliv. Rev. 1999, 36, 101–123. [Google Scholar] [CrossRef]
- Redenti, E.; Pietra, C.; Gerloczy, A.; Szente, L. Cyclodextrins in oligonucleotide delivery. Adv. Drug Deliv. Rev. 2001, 53, 235–244. [Google Scholar] [CrossRef]
- Poór, M.; Kunsági-Máté, S.; Sali, N.; Kőszegi, T.; Szente, L.; Peles-Lemli, B. Interactions of zearalenone with native and chemically modified cyclodextrins and their potential utilization. J. Photochem. Photobiol. B 2015, 151, 63–68. [Google Scholar] [CrossRef]
- Faisal, Z.; Garai, E.; Csepregi, R.; Bakos, K.; Fliszár-Nyúl, E.; Szente, L.; Balázs, A.; Cserháti, M.; Kőszegi, T.; Urbányi, B.; et al. Protective effects of beta-cyclodextrins vs. zearalenone-induced toxicity in HeLa cells and Tg(vtg1:mCherry) zebrafish embryos. Chemosphere 2020, 240, 124948. [Google Scholar] [CrossRef]
- Keating, G.M. Sugammadex: A Review of Neuromuscular Blockade Reversal. Drugs 2016, 76, 1041–1052. [Google Scholar] [CrossRef]
- Bom, A.; Bradley, M.; Cameron, K.; Clark, J.K.; Van Egmond, J.; Feilden, H.; MacLean, E.J.; Muir, A.W.; Palin, R.; Rees, D.C.; et al. A novel concept of reversing neuromuscular block: Chemical encapsulation of rocuronium bromide by a cyclodextrin-based synthetic host. Angew. Chem. Int. Ed. Engl. 2002, 41, 266–270. [Google Scholar] [CrossRef]
- Davidson, J.; Molitor, E.; Moores, S.; Gale, S.E.; Subramanian, K.; Jiang, X.; Sidhu, R.; Kell, P.; Zhang, J.; Fujiwara, H.; et al. 2-Hydroxypropyl-b-cyclodextrin is the active component in a triple combination formulation for treatment of Niemann-Pick C1 disease. Biochim. Biophys. Acta 2019, 1864, 1545–1561. [Google Scholar] [CrossRef]
- Tanaka, Y.; Yamada, Y.; Ishitsuka, Y.; Matsuo, M.; Shiraishi, K.; Wada, K.; Uchio, Y.; Kondo, Y.; Takeo, T.; Nakagata, N.; et al. Efficacy of 2-Hydroxypropyl-β-cyclodextrin in Niemann-Pick Disease Type C Model Mice and Its Pharmacokinetic Analysis in a Patient with the Disease. Biol. Pharm. Bull. 2015, 38, 844–851. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Chipot, C.; Cai, W.; Shao, X. Molecular dynamics study of the inclusion of cholesterol into cyclodextrins. J. Phys. Chem. B 2006, 110, 6372–6378. [Google Scholar] [CrossRef] [PubMed]
- Zidovetzki, R.; Levitan, I. Use of cyclodextrins to manipulate plasma membrane cholesterol content: Evidence, misconceptions and control strategies. Biochim. Biophys. Acta 2007, 1768, 1311–1324. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.W.; Gray, J.E.; Weaver, R.N. Cyclodextrin Nephrosis in the Rat. Am. J. Pathol. 1976, 83, 367–382. [Google Scholar]
- Gould, S.; Scott, R.C. 2-Hydroxypropyl-beta-cyclodextrin (HP-beta-CD): A toxicology review. Food Chem. Toxicol. 2005, 43, 1451–1459. [Google Scholar] [CrossRef] [PubMed]
- Stella, V.J.; He, Q. Cyclodextrins. Toxicol. Pathol. 2008, 36, 30–42. [Google Scholar] [CrossRef]
- Jansook, P.; Ogawa, N.; Loftsson, T. Cyclodextrins: Structure, physicochemical properties and pharmaceutical applications. Int. J. Pharm. 2018, 535, 272–284. [Google Scholar] [CrossRef]
- Fliszár-Nyúl, E.; Bock, I.; Csepregi, R.; Szente, L.; Szabó, I.; Csenki, Z.; Poór, M. Testing the protective effects of cyclodextrins vs. alternariol-induced acute toxicity in HeLa cells and in zebrafish embryos. Environ. Toxicol. Pharmacol. 2022, 95, 103965. [Google Scholar] [CrossRef]
- Uribe, L.A.; Leonardo, S.; Nielsen, T.T.; Steinmann, C.; Campàs, M.; Fragoso, A. Supramolecular Complexes of Plant Neurotoxin Veratridine with Cyclodextrins and Their Antidote-like Effect on Neuro-2a Cell Viability. Pharmaceutics 2022, 14, 598. [Google Scholar] [CrossRef]
- da Silva, M.C.G.; da Silva, J.F.; Santos, T.P.; da Silva, N.P.C.; dos Santos, A.R.; de Andrade, A.L.C.; da Silva Souza, E.H.L.; Cadena, M.R.S.; de Sá, F.B.; da Silva Junior, V.A.; et al. The complexation of steroid hormones into cyclodextrin alters the toxic effects on the biological parameters of zebrafish (Danio rerio). Chemosphere 2019, 214, 330–340. [Google Scholar] [CrossRef]
- Weiss-Errico, M.; Berry, J.; O’Shea, K. β-Cyclodextrin Attenuates Perfluorooctanoic Acid Toxicity in the Zebrafish Embryo Model. Toxics 2017, 5, 31. [Google Scholar] [CrossRef] [PubMed]
- Nam, Y.H.; Le, H.T.; Rodriguez, I.; Kim, E.Y.; Kim, K.; Jeong, S.Y.; Woo, S.H.; Lee, Y.R.; Castaneda, R.; Hong, J.; et al. Enhanced antidiabetic efficacy and safety of compound K⁄β-cyclodextrin inclusion complex in zebrafish. J. Ginseng Res. 2017, 41, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, D.H.; Engelke, A.; Wenz, G. Solubilizing steroidal drugs by β-cyclodextrin derivatives. Int. J. Pharm. 2017, 531, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Weiss-Errico, M.J.; O’Shea, K.E. Detailed NMR investigation of cyclodextrin-perfluorinated surfactant interactions in aqueous media. J. Hazard. Mater. 2017, 329, 57–65. [Google Scholar] [CrossRef]
- Igami, K.; Ozawa, M.; Inoue, S.; Iohara, D.; Miyazaki, T.; Shinoda, M.; Anraku, M.; Hirayama, F.; Uekama, K. The formation of an inclusion complex between a metabolite of ginsenoside, compound K and γ-cyclodextrin and its dissolution characteristics. J. Pharm. Pharmacol. 2016, 68, 646–654. [Google Scholar] [CrossRef]
- Uekama, K.; Irie, T.; Sunada, M.; Otagiri, M.; Iwasaki, K.; Okano, Y.; Miyata, T.; Kasé, Y. Effects of cyclodextrins on chlorpromazine-induced haemolysis and central nervous system responses. J. Pharm. Pharmacol. 1981, 33, 707–710. [Google Scholar] [CrossRef] [PubMed]
- Irie, T.; Uekama, K. Protection against the photosensitized skin irritancy of chlorpromazine by cyclodextrin complexation. J. Pharmacobiodyn. 1985, 8, 788–791. [Google Scholar] [CrossRef]
- Irie, T.; Kuwahara, S.; Otagiri, M.; Uekama, K.; Iwamasa, T. Reduction in the local tissue toxicity of chlorpromazine by beta-cyclodextrin complexation. J. Pharmacobiodyn. 1983, 6, 790–792. [Google Scholar] [CrossRef]
- Svendsen, O. β-Cyclodextrin and Local Muscle Toxicity of Intramuscular Drug Formulations. In The Target Organ and the Toxic Process; Chambers, P.L., Chambers, C.M., Dirheimer, G., Eds.; Archives of Toxicology; Springer: Berlin/Heidelberg, Germany, 1988; Volume 12. [Google Scholar] [CrossRef]
- Wang, Z.; Landy, D.; Sizun, C.; Cézard, C.; Solgadi, A.; Przybylski, C.; de Chaisemartin, L.; Herfindal, L.; Barratt, G.; Legrand, F.-X. Cyclodextrin complexation studies as the first step for repurposing of chlorpromazine. Int. J. Pharmaceut. 2020, 584, 119391. [Google Scholar] [CrossRef] [PubMed]
- Adan, A.; Kiraz, Y.; Baran, Y. Cell proliferation and cytotoxicity assays. Curr. Pharmaceut. Biotechnol. 2016, 17, 1213–1221. [Google Scholar] [CrossRef]
- Kocyigit, A.; Guler, E.M.; Karatas, E.; Caglar, H.; Bulut, H. Dose-dependent proliferative and cytotoxic effects of melatonin on human epidermoid carcinoma and normal skin fibroblast cells. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2018, 829–830, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Csepregi, R.; Temesfői, V.; Poór, M.; Faust, Z.; Kőszegi, T. Green fluorescent protein-based viability assay in a multiparametric configuration. Molecules 2018, 23, 1575. [Google Scholar] [CrossRef] [PubMed]
- Csenki, Z.; Garai, E.; Faisal, Z.; Csepregi, R.; Garai, K.; Kánainé Sipos, D.; Szabó, I.; Kőszegi, T.; Czéh, Á.; Czömpöly, T.; et al. The individual and combined effects of ochratoxin A with citrinin and their metabolites (ochratoxin B, ochratoxin C, and dihydrocitrinone) on 2D/3D cell cultures, and zebrafish embryo models. Food Chem. Toxicol. 2021, 158, 112674. [Google Scholar] [CrossRef] [PubMed]
- Sali, N.; Nagy, S.; Poór, M.; Kőszegi, T. Multiparametric luminescent cell viability assay in toxicology models: A critical evaluation. J. Pharmacol. Toxicol. Methods 2016, 79, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Bruce, R.D. An up-and-down procedure for acute toxicity testing. Fundam. Appl. Toxicol. 1985, 5, 151–157. [Google Scholar] [CrossRef]
- Dixon, W.J. The Up-and-Down Method for Small Samples. J. Am. Stat. Assoc. 1965, 60, 967–978. [Google Scholar] [CrossRef]
- Kovacs, K.; Ancha, M.; Jane, M.; Lee, S.; Angalakurthi, S.; Negrito, M.; Rasheed, S.; Nwaneri, A.; Petrikovics, I. Identification, solubility enhancement and in vivo testing of a cyanide antidote candidate. Eur. J. Pharmaceut. Sci. 2013, 49, 352–358. [Google Scholar] [CrossRef]
- Kovacs, K.; Duke, A.C.; Shifflet, M.; Winner, B.; Lee, S.A.; Rockwood, G.A.; Petrikovics, I. Parenteral dosage form development and testing of dimethyl trisulfide, as an antidote candidate to combat cyanide intoxication. Pharm. Dev. Technol. 2017, 22, 958–963. [Google Scholar] [CrossRef]
- Campbell, D.E.S.; Richter, W. An Observational Method Estimating Toxicity and Drug. Actions in Mice applied to 68 Reference Drugs. Acta Pharmacol. Toxicol. 1967, 25, 345–363. [Google Scholar] [CrossRef]
- Dandiya, P.C.; Johnson, G.; Sellers, E.A. Influence of variation in environmental temperature on the acute toxicity of reserpine and chlorpromazine in mice. Can. J. Biochem. Physiol. 1960, 38, 591. [Google Scholar] [CrossRef]
- Hoshino, T.; Ishida, K.; Irie, T.; Uekama, K.; Ono, T. An attempt to reduce the photosensitizing potential of chlorpromazine with the simultaneous use of beta- and dimethyl-beta-cyclodextrins in guinea pigs. Arch. Dermatol. Res. 1989, 281, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Roubein, I.F.; Samuelly, M.; Keup, W. The toxicity of chlorpromazine and mescaline on mouse cerebellum and fibroblast cells in culture. Acta Pharmacol. Toxicol. 1973, 33, 326–329. [Google Scholar] [CrossRef]
- Shenoy, M.A.; Biaglow, J.E.; Varnes, M.E.; Daniel, J.W. A biochemical basis for the radiosensitizing and cytotoxic effects of chlorpromazine hydrochloride in vitro and in vivo. Int. J. Radiat. Oncol. Biol. Phys. 1982, 8, 725–728. [Google Scholar] [CrossRef]
- Shin, S.Y.; Lee, K.S.; Choi, Y.-K.; Lim, H.J.; Lee, H.G.; Lim, Y.; Lee, Y.H. The antipsychotic agent chlorpromazine induces autophagic cell death by inhibiting the Akt/mTOR pathway in human U-87MG glioma cells. Carcinogenesis 2013, 34, 2080–2089. [Google Scholar] [CrossRef]
- Okimoto, K.; Ohike, A.; Ibuki, R.; Aoki, O.; Ohnishi, N.; Irie, T.; Uekama, K.; Rajewski, R.A.; Stella, V.J. Design and evaluation of an osmotic pump tablet (OPT) for chlorpromazine using (SBE)7m-beta-CD. Pharm. Res. 1999, 16, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Szmeja, S.; Gubica, T.; Ostrowski, A.; Zalewska, A.; Szeleszczuk, Ł.; Zawada, K.; Zielińska-Pisklak, M.; Skowronek, K.; Wiweger, M. Caffeine-Cyclodextrin Complexes as Solids: Synthesis, Biological and Physicochemical Characterization. Int. J. Mol. Sci. 2021, 22, 4191. [Google Scholar] [CrossRef]
- Prabu, S.; Swaminathan, M.; Sivakumar, K.; Rajamohan, R. Preparation, characterization and molecular modeling studies of the inclusion complex of Caffeine with Beta-cyclodextrin. J. Mol. Struct. 2015, 1099, 616–624. [Google Scholar] [CrossRef]
- Fliszár-Nyúl, E.; Lemli, B.; Kunsági-Máté, S.; Szente, L.; Poór, M. Interactions of Mycotoxin Alternariol with Cyclodextrins and its Removal from Aqueous Solution by Beta-Cyclodextrin Bead Polymer. Biomolecules 2019, 9, 428. [Google Scholar] [CrossRef]
- Mottram, A.R.; Bryant, S.M.; Aks, S.E. Effect of Cyclodextrin Infusion in a Rat Model of Verapamil Toxicity. Am. J. Ther. 2011, 18, 371–374. [Google Scholar] [CrossRef]
- Mottram, A.R.; Bryant, S.M.; Aks, S.E. Dose-Dependent Response to Cyclodextrin Infusion in a Rat Model of Verapamil Toxicity. West. J. Emerg. Med. 2012, 13, 63–67. [Google Scholar] [CrossRef][Green Version]
- Ozbilgin, S.; Ozbilgin, M.; Kucukoztas, B.; Kamaci, G.; Unek, T.; Yurtlu, B.S.; Güneli, M.E.; Hanci, V.; Gunerli, A. Evaluation of the effectiveness of sugammadex for verapamil intoxication. Basic Clin. Pharmacol. Toxicol. 2013, 113, 280–285. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CPZ (Control) | CPZ + 500 mg/kg SBECD | CPZ + 2000 mg/kg SBECD | CPZ + 500 mg/kg SGD | CPZ + 2000 mg/kg SGD | |
|---|---|---|---|---|---|
| LD50 (mg/kg, based on 24 h mortality) | 194.0 | 206.2 | 232.8 | 200.0 | 171.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fliszár-Nyúl, E.; Csepregi, R.; Benkovics, G.; Szente, L.; Poór, M. Testing the Protective Effects of Sulfobutylether-Βeta-Cyclodextrin (SBECD) and Sugammadex against Chlorpromazine-Induced Acute Toxicity in SH-SY5Y Cell Line and in NMRI Mice. Pharmaceutics 2022, 14, 1888. https://doi.org/10.3390/pharmaceutics14091888
Fliszár-Nyúl E, Csepregi R, Benkovics G, Szente L, Poór M. Testing the Protective Effects of Sulfobutylether-Βeta-Cyclodextrin (SBECD) and Sugammadex against Chlorpromazine-Induced Acute Toxicity in SH-SY5Y Cell Line and in NMRI Mice. Pharmaceutics. 2022; 14(9):1888. https://doi.org/10.3390/pharmaceutics14091888
Chicago/Turabian StyleFliszár-Nyúl, Eszter, Rita Csepregi, Gábor Benkovics, Lajos Szente, and Miklós Poór. 2022. "Testing the Protective Effects of Sulfobutylether-Βeta-Cyclodextrin (SBECD) and Sugammadex against Chlorpromazine-Induced Acute Toxicity in SH-SY5Y Cell Line and in NMRI Mice" Pharmaceutics 14, no. 9: 1888. https://doi.org/10.3390/pharmaceutics14091888
APA StyleFliszár-Nyúl, E., Csepregi, R., Benkovics, G., Szente, L., & Poór, M. (2022). Testing the Protective Effects of Sulfobutylether-Βeta-Cyclodextrin (SBECD) and Sugammadex against Chlorpromazine-Induced Acute Toxicity in SH-SY5Y Cell Line and in NMRI Mice. Pharmaceutics, 14(9), 1888. https://doi.org/10.3390/pharmaceutics14091888