
Design of Smart Nanodiamonds: Introducing pH Sensitivity to Improve Nucleic Acid Carrier Efficiency of Diamoplexes


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
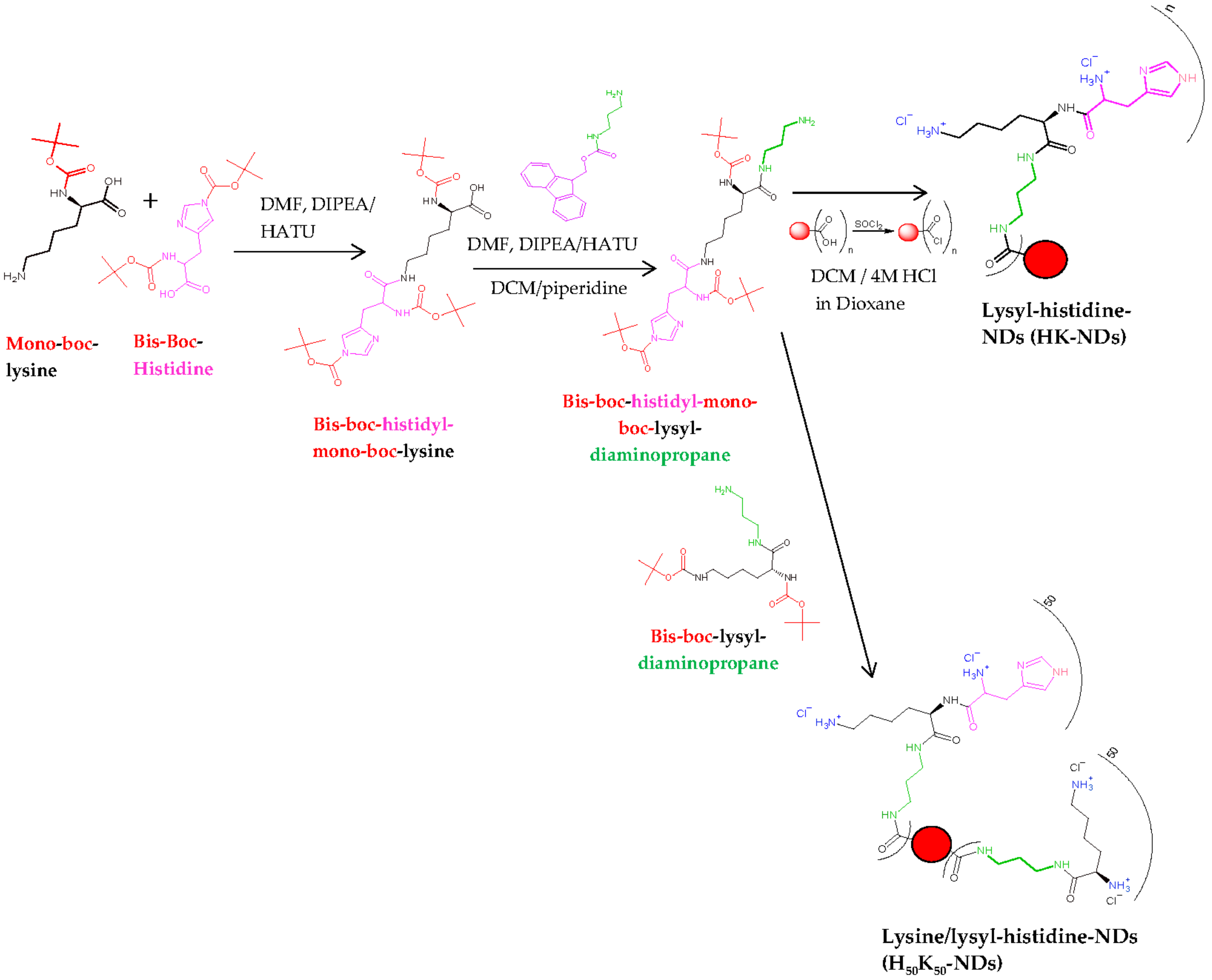
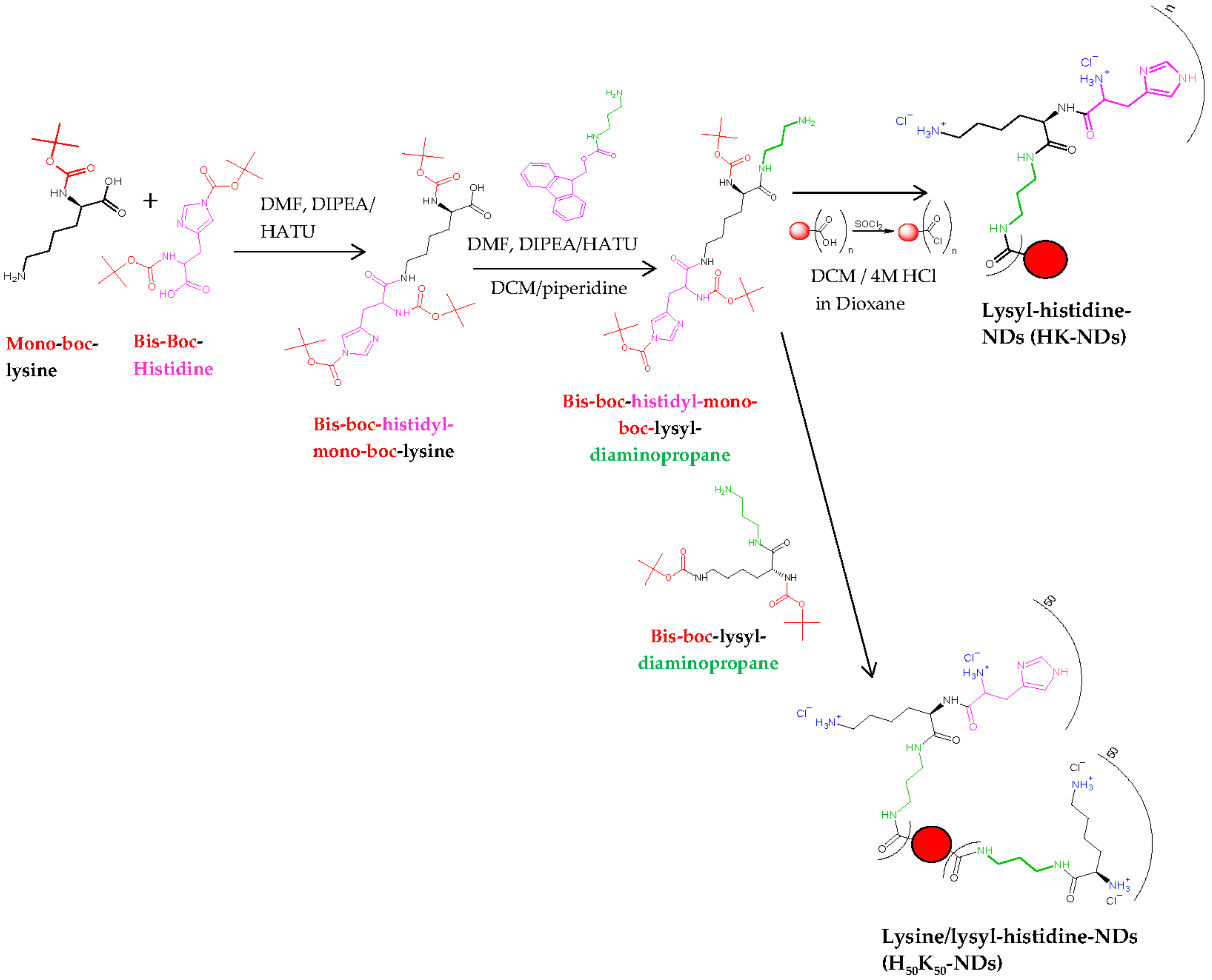
2.2. Synthesis of HK-NDs and H50K50-NDs
2.2.1. Synthesis of HK-NDs
2.2.2. Synthesis of H50K50-NDs
2.3. Size and Zeta Potential Measurements
2.4. Transmission Electron Microscopy (TEM)
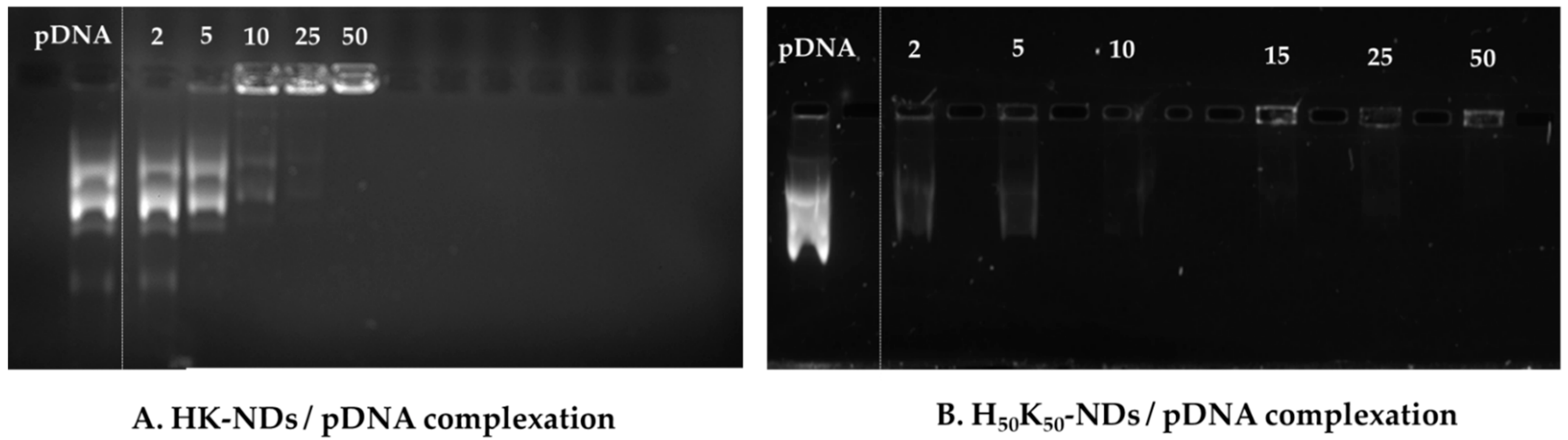
2.5. Gel Electrophoresis
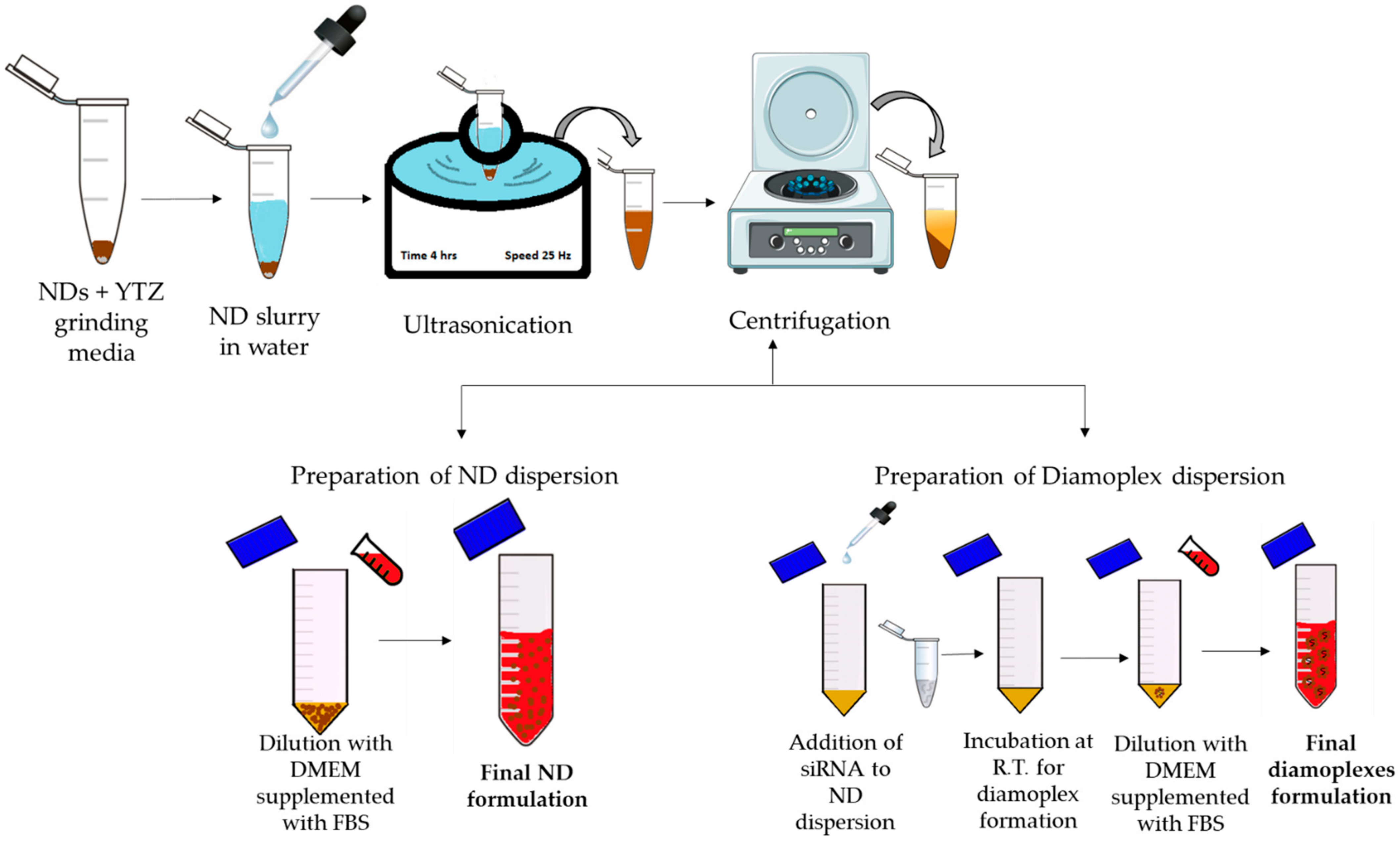
2.6. Formulation Development for In Vitro Assays
2.7. MTT-Cell Viability Assay
2.8. Flow Cytometry Analysis to Compare Cellular Entry and Exit Profile
2.9. Flow Cytometry Analysis of siRNA-Mediated Green Fluorescent Protein (GFP) Knockdown
2.10. Statistical Analysis
3. Results
3.1. Physicochemical Properties of HK-NDs and H50K50-NDs
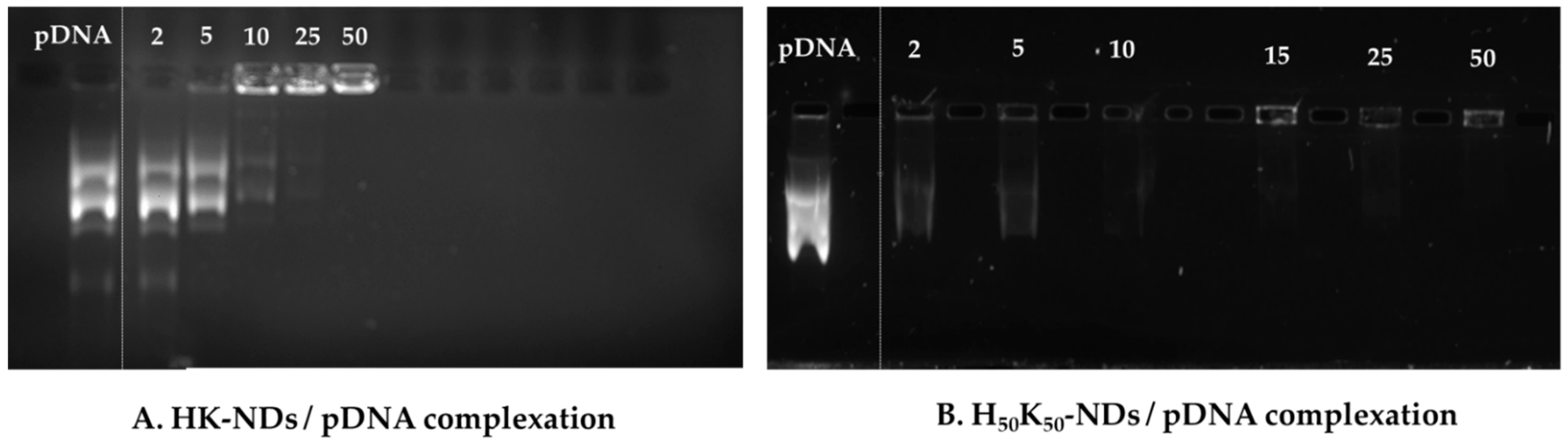
3.2. Nucleic Acid Binding Affinity of HK-NDs and H50K50-NDs
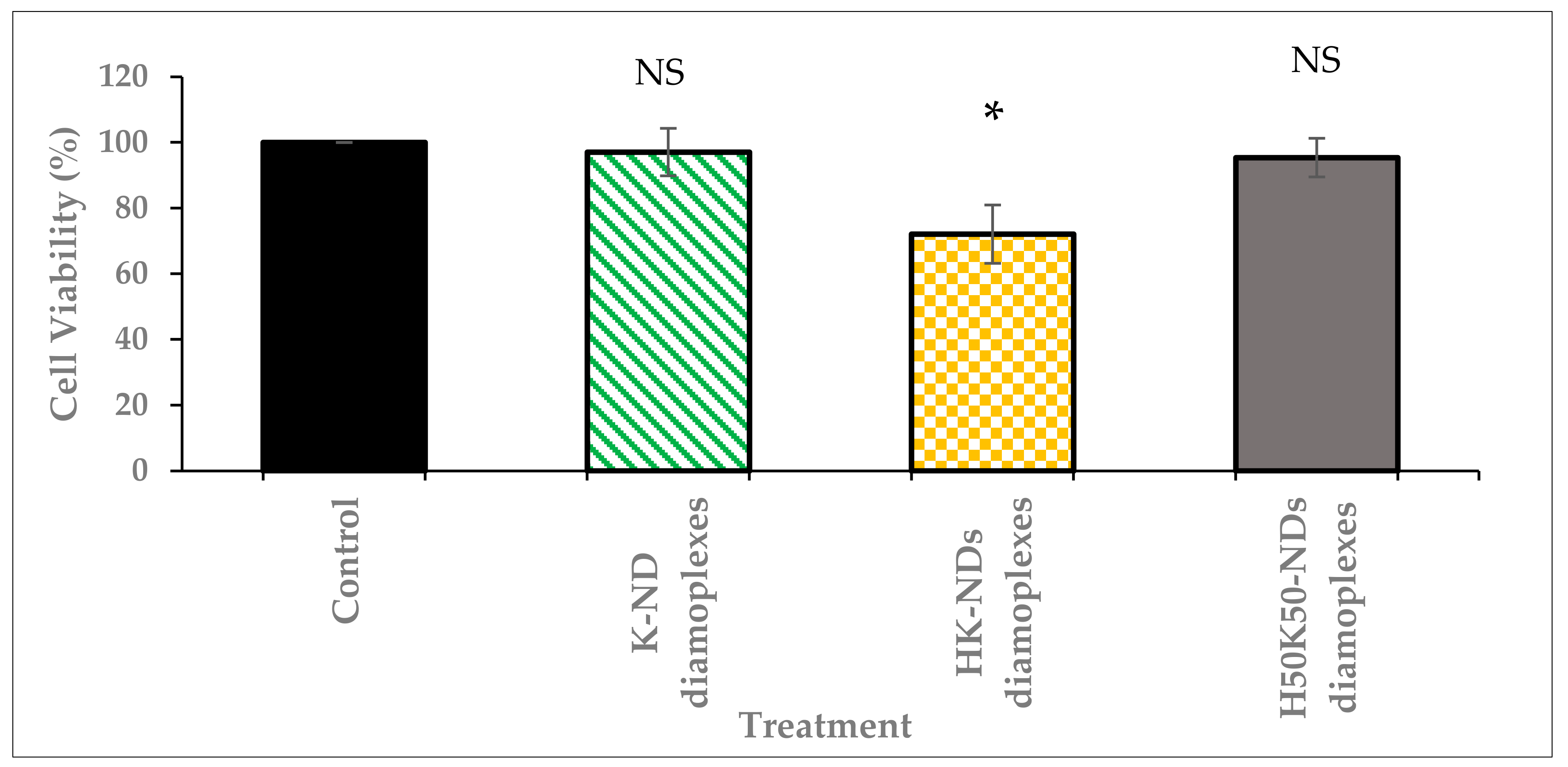
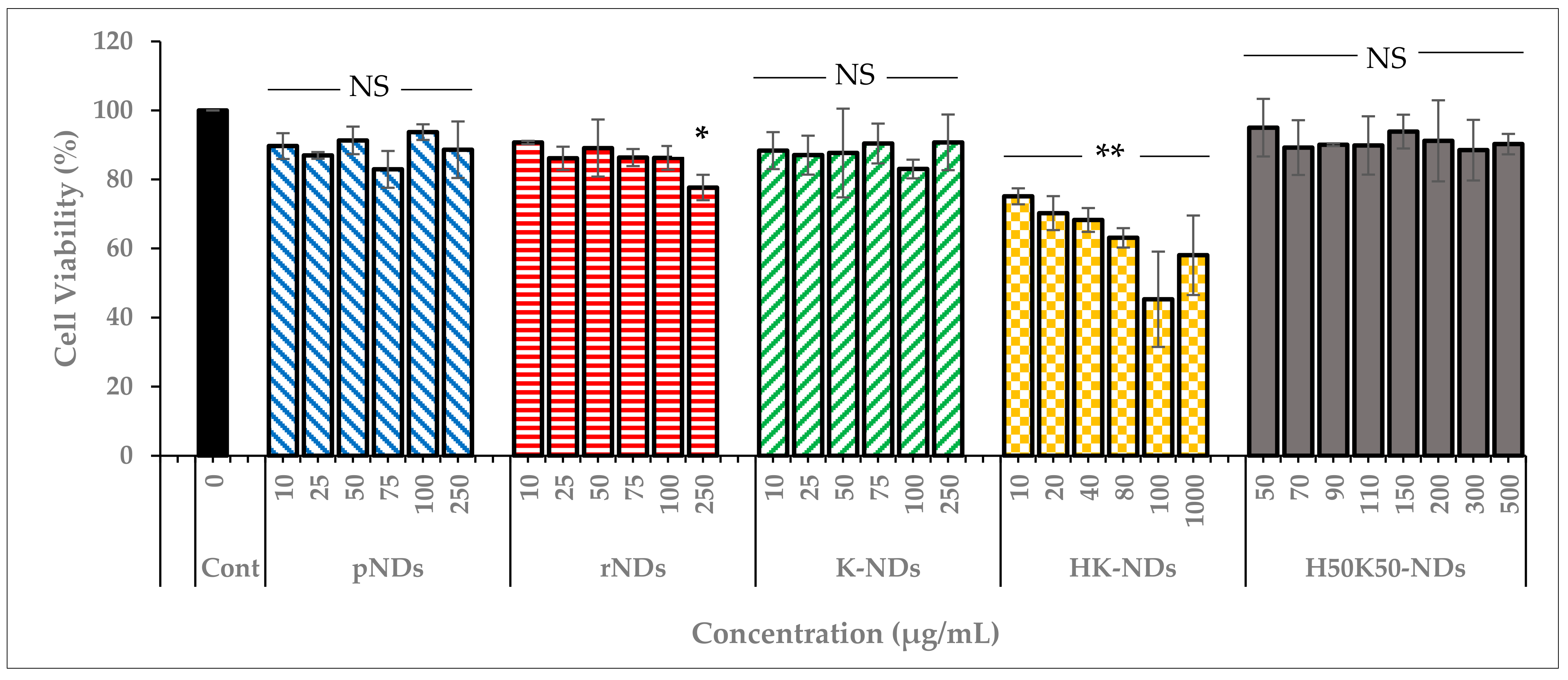
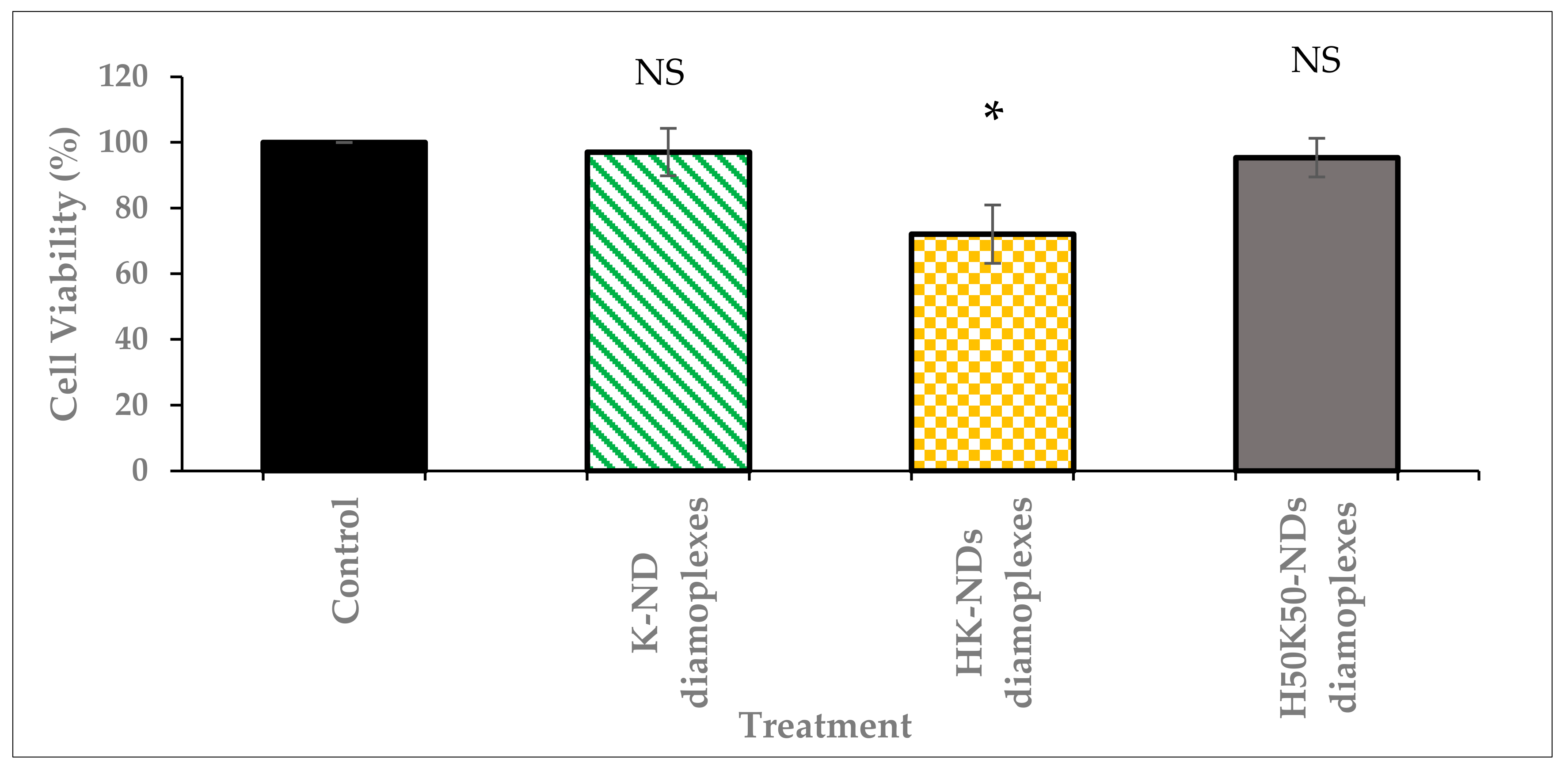
3.3. Concentration Dependent Biocompatibility of fNDs
3.4. Effect of Functionalization on Cellular Uptake and Exit of fNDs
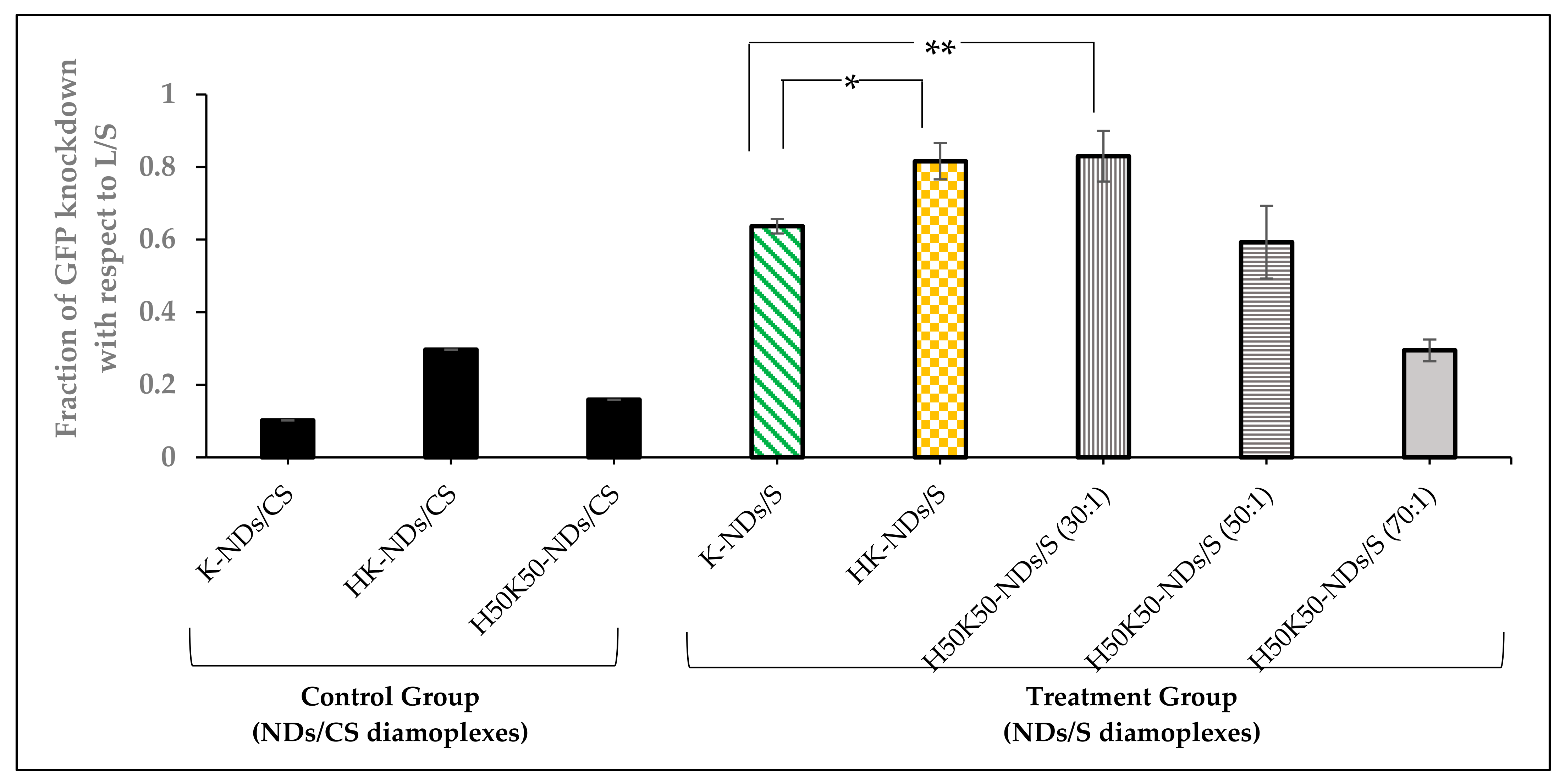
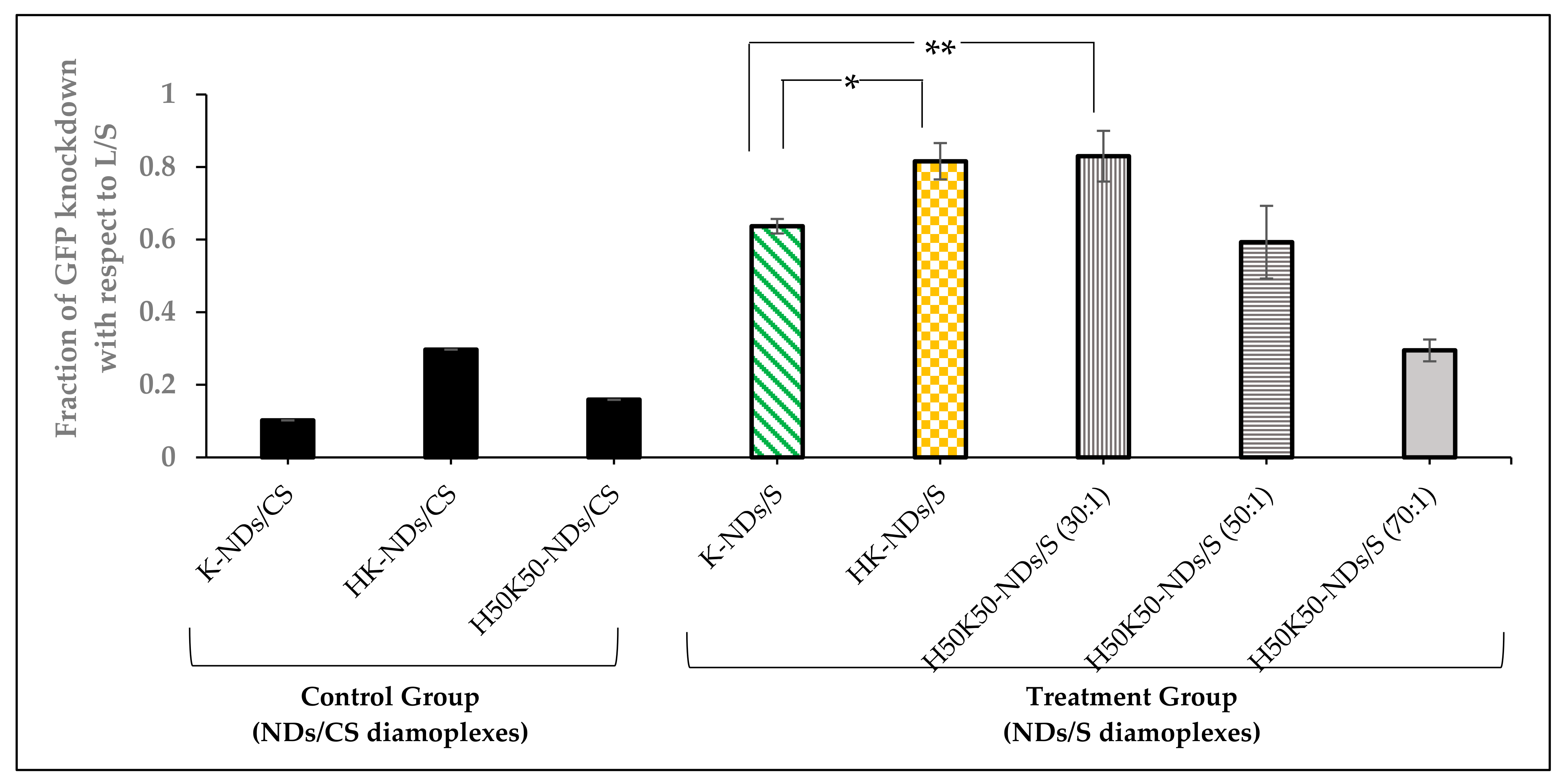
3.5. Transfection Efficiency—GFP Knockdown
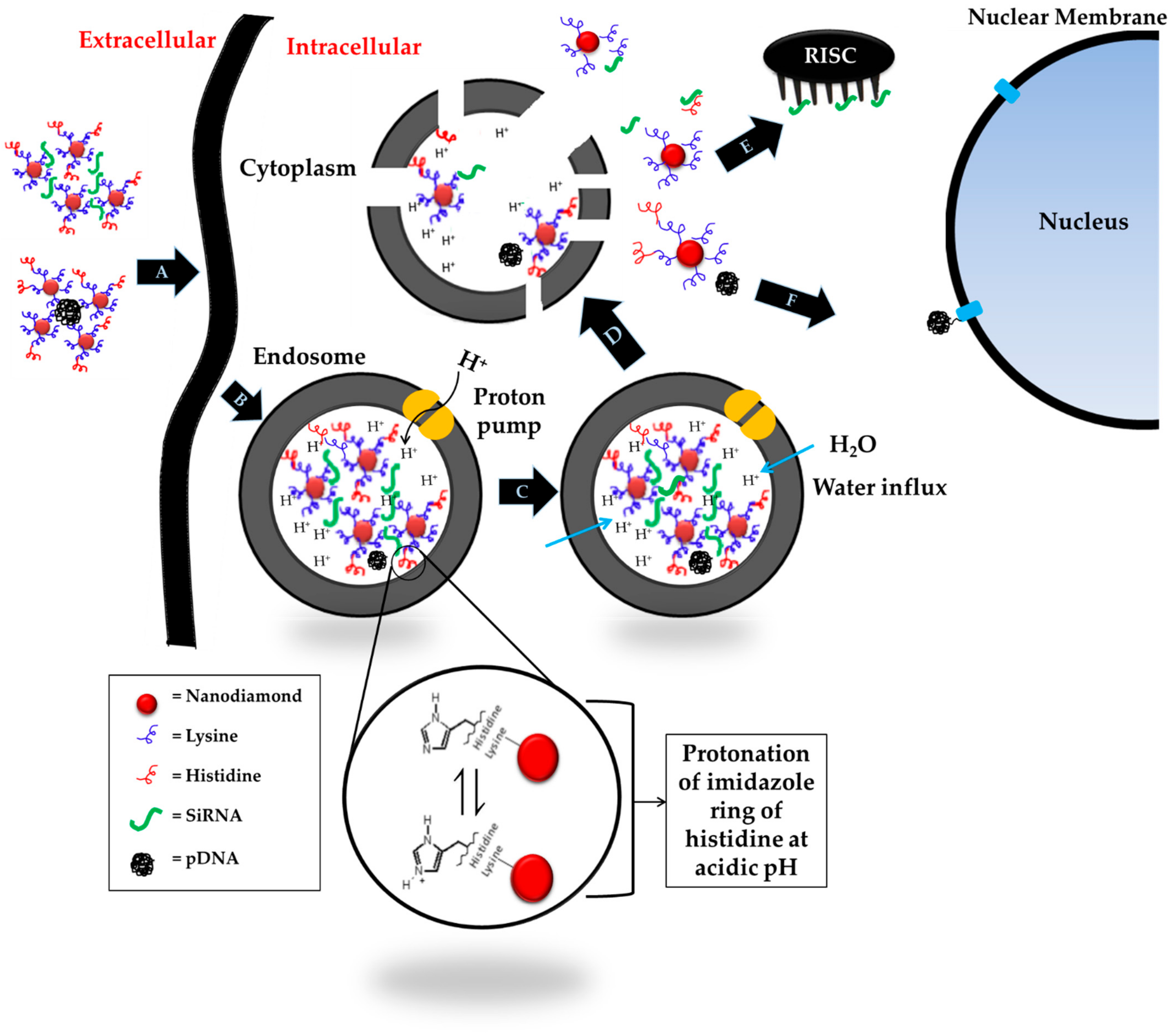
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Sun, W.; Shi, Q.; Zhang, H.; Yang, K.; Ke, Y.; Wang, Y.; Qiao, L. Advances in the techniques and methodologies of cancer gene therapy. Discov. Med. 2019, 27, 45–55. [Google Scholar] [PubMed]
- Banerjee, A.; Sharma, D.; Trivedi, R.; Singh, J. Treatment of insulin resistance in obesity-associated type 2 diabetes mellitus through adiponectin gene therapy. Int. J. Pharm. 2020, 583, 119357. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; McCray, P., Jr.; Engelhardt, J. Advances in gene therapy for cystic fibrosis lung disease. Hum. Mol. Genet. 2019, 28, R88–R94. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.; Kundu, S.; Hassan, R. Gene Therapy Approaches in an Autoimmune Demyelinating Disease: Multiple Sclerosis. Curr. Gene. Ther. 2020, 19, 376–385. [Google Scholar] [CrossRef]
- Lee, J.; Kumar, S.; Jhan, Y.; Bishop, C. Engineering DNA vaccines against infectious diseases. Acta Biomater. 2018, 80, 31–47. [Google Scholar] [CrossRef]
- Zhang, C.; Maruggi, C.; Shan, H.; Li, J. Advances in mRNA Vaccines for Infectious Diseases. Front. Immunol. 2019, 10, 594. [Google Scholar] [CrossRef]
- Baden, L.; El Sahly, H.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.; Rouphael, N.; Creech, B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef]
- Polack, F.; Thomas, S.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.; Marc, G.; Moreira, E.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA COVID-19 Vaccine. N. Engl. J. Med. 2020, 383, 2261–2603. [Google Scholar] [CrossRef]
- Smith, T.; Patel, A.; Ramos, S.; Elwood, D.; Zhu, X.; Yan, J.; Gary, E.; Walker, S.; Schultheis, K.; Purwar, M.; et al. Immunogenicity of a DNA vaccine candidate for COVID-19. Nat. Commun. 2020, 11, 2601. [Google Scholar] [CrossRef]
- Roma-Rodrigues, C.; Rivas-García, L.; Baptista, P.; Fernandes, A. Gene Therapy in Cancer Treatment: Why Go Nano? Pharmaceutics 2020, 12, 233. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Liu, L.; Wei, Y.-Q.; Gao, G.-P.; Wei, X.-W. Clinical Evaluations of Toxicity and Efficacy of Nanoparticle-Mediated Gene Therapy. Hum. Gene Ther. 2018, 29, 1227–1234. [Google Scholar] [CrossRef]
- Chen, J.; Guo, Z.; Tian, H.; Chen, X. Production and clinical development of nanoparticles for gene delivery. Mol. Ther. Methods Clin. Dev. 2016, 3, 16023. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Jiang, X.; Deng, Z.; Narain, R. Cationic glyco-functionalized single-walled carbon nanotubes as efficient gene delivery vehicles. Bioconj. Chem. 2009, 20, 2017–2022. [Google Scholar] [CrossRef] [PubMed]
- Jia, N.; Lian, Q.; Shen, H.; Wang, C.; Li, X.; Yang, Z. Intracellular delivery of quantum dots tagged antisense oligodeoxynucleotides by functionalized multiwalled carbon nanotubes. Nano Lett. 2007, 7, 2976–2980. [Google Scholar] [CrossRef] [PubMed]
- Alwani, S.; Kaur, R.; Michel, D.; Chitanda, M.; Verrall, R.; Karunakaran, C.; Badea, I. Lysine-functionalized nanodiamonds as gene carriers: Development of stable colloidal dispersion for in vitro cellular uptake studies and siRNA delivery application. Int. J. Nanomed. 2016, 11, 687–702. [Google Scholar]
- Claveau, S.; Nehlig, E.; Garcia-Argote, S.; Feuillastre, S.; Pieters, G.; Girard, H.; Arnault, J.-C.; Treussart, F.; Bertrand, J.-R. Delivery of siRNA to Ewing Sarcoma Tumor Xenografted on Mice, Using Hydrogenated Detonation Nanodiamonds: Treatment Efficacy and Tissue Distribution. Nanomaterials 2020, 10, 553. [Google Scholar] [CrossRef]
- Schrand, A.; Dai, L.; Schlager, J.; Hussain, S.; Osawa, E. Differential biocompatibility of carbon nanotubes and nanodiamonds. Diam. Relat. Mater. 2007, 16, 2118–2123. [Google Scholar] [CrossRef]
- Schrand, A.M.; Johnson, J.; Dai, L.; Hussain, S.M.; Schlager, J.J.; Zhu, L.; Hong, Y.; Ōsawa, E. Chapter 8 Cytotoxicity and Genetoxicity of Carbon Nanomaterials. In Safety of Nanoparticles: From Manufacturing to Medical Applications (Nanostructure Science and Technology); Springer: Berlin/Heidelberg, Germany, 2009; pp. 159–187. [Google Scholar]
- Yuan, Y.; Chen, Y.; Liu, J.; Wang, H.; Liu, Y. Biodistribution and Fate of Nanodiamonds In vivo. Diam. Relat. Mater. 2009, 18, 95–100. [Google Scholar] [CrossRef]
- Kruger, A.; Kataoka, F.; Ozawa, M.; Fujino, T.; Suzuki, Y.; Aleksenskii, A.E.; Vul, A.Y.; Osawa, E. Unusually tight aggregation in detonation nanodiamond: Identification and disintegration. Carbon 2005, 43, 1722–1730. [Google Scholar] [CrossRef]
- Norouzi, N.; Ong, Y.; Damle, V.; Najafi, M.; Schirhagl, R. Effect of medium and aggregation on antibacterial activity of nanodiamonds. Mater. Sci. Eng. C 2020, 112, 110930. [Google Scholar] [CrossRef]
- Edgington, R.; Spillane, K.; Papageorgiou, G.; Wray, W.; Ishiwata, H.; Labarca, M.; Leal-Ortiz, S.; Reid, G.; Webb, M.; Foord, J.; et al. Functionalisation of Detonation Nanodiamond for Monodispersed, Soluble DNA-Nanodiamond Conjugates Using Mixed Silane Bead-Assisted Sonication Disintegration. Sci. Rep. 2018, 8, 728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, R.; Chitanda, J.; Michel, D.; Maley, J.; Borondics, F.; Yang, P.; Verrall, E.R.; Badea, I. Lysine functionalized nano diamonds: Synthesis, physiochemical characterization and nucleic acid binding studies. Int. J. Nanomed. 2012, 7, 3851–3866. [Google Scholar]
- Krueger, A.; Boedeker, T. Deagglomeration and functionalization of detonation nanodiamond with long alkyl chains. Diam. Relat. Mater. 2008, 17, 1367–1370. [Google Scholar] [CrossRef]
- Alwani, S. Amino Acid Functionalized Nanodiamonds as Gene Delivery Vectors: Synthesis, Physicochemical Characterization and Cellular Interaction Studies. Master’s Thesis, University of Saskatchewan, Saskatoon, SK, Canada, 2015. [Google Scholar]
- Alwani, S.; Hua, Q.; Iftikhar, S.; Appathurai, N.; Michel, D.; Karunakaran, C.; Badea, I. Lysine functionalized nanodiamonds as gene carriers—Investigation of internalization pathways and intracellular trafficking. Diam. Relat. Mater. 2019, 98, 107477. [Google Scholar] [CrossRef]
- Li, J.; Chen, Y.-C.; Tseng, Y.-C.; Mozumdar, S.; Huang, L. Biodegradable calcium phosphate nanoparticle with lipid coating for systemic siRNA delivery. J. Control. Release 2010, 142, 416–421. [Google Scholar] [CrossRef]
- Pantarotto, D.; Singh, R.; McCarthy, D.; Erhardt, M.; Briand, J.-P.; Prato, M.; Kostarelos, K.; Bianco, A. Functionalized carbon nanotubes for plasmid DNA gene delivery. Agnew. Chem. Int. Ed. Engl. 2004, 43, 5242–5246. [Google Scholar] [CrossRef]
- Varkouhi, A.; Scholte, M.; Storm, G.; Haisma, H. Endosomal escape pathways for delivery of biologicals. J. Control. Release 2011, 151, 220–228. [Google Scholar] [CrossRef]
- Thuy, L.; Mallick, S.; Choi, J. Polyamidoamine (PAMAM) dendrimers modified with short oligopeptides for early endosomal escape and enhanced gene delivery. Int. J. Pharm. 2015, 492, 233–243. [Google Scholar] [CrossRef]
- Vicennati, P.; Giuliano, A.; Ortaggi, G.; Masotti, A. Polyethylenimine in medicinal chemistry. Curr. Med. Chem. 2008, 15, 2826–2839. [Google Scholar] [CrossRef]
- Akinc, A.; Thomas, M.; Klibanov, A.; Langer, R. Exploring polyethylenimine-mediated DNA transfection and the proton sponge hypothesis. J. Gene Med. 2005, 7, 657–963. [Google Scholar] [CrossRef]
- Liu, N.; Bechinger, B.; Suss, R. The histidine-rich peptide LAH4-L1 strongly promotes PAMAM-mediated transfection at low nitrogen to phosphorus ratios in the presence of serum. Sci. Rep. 2017, 7, 9585. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-L.; Nguyen, T.; Gillespie, D.; Jensen, R.; Lu, Z.-R. A multifunctional and reversibly polymerizable carrier for efficient siRNA delivery. Biomaterials 2008, 29, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Summerton, J. Endo-Porter: A novel reagent for safe, effective delivery of substances into cells. Ann. N. Y. Acad. Sci. 2005, 1058, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Midoux, P.; Kichler, A.; Boutin, V.; Maurizot, J.; Monsigny, M. Membrane permeabilization and efficient gene transfer by a peptide containing several histidines. Bioconjug. Chem. 1998, 9, 260–267. [Google Scholar] [CrossRef]
- Dominska, M.; Dykxhoorn, D. Breaking down the barriers: siRNA delivery and endosome escape. J. Cell Sci. 2010, 123, 1183–1189. [Google Scholar] [CrossRef]
- Medina-Kauwe, L.; Xie, J.; Hamm-Alvarez, S. Intracellular trafficking of nonviral vectors. Gene Therap. 2005, 12, 1734–1751. [Google Scholar] [CrossRef]
- Rai, R.; Alwani, S.; Khan, B.; Vishwas, R.; Vuong, S.; Krol, E.; Fonge, H.; Badea, I. Nanodiamonds as vectors for nucleic acid therapeutics: Biodistribution and positron emission tomography imaging. Nanomedicine 2022. (manuscript submitted). [Google Scholar]
- Sato, S.; Rancourt, A.; Sato, Y.; Satoh, M. Single-cell lineage tracking analysis reveals that an established cell line comprises putative cancer stem cells and their heterogeneous progeny. Sci. Rep. 2016, 6, 23328. [Google Scholar] [CrossRef]
- Bakke, A. The Principles of Flow Cytometry. Lab. Med. 2001, 32, 207–211. [Google Scholar] [CrossRef]
- Foroozandeh, P.; Abdul Aziz, A. Insight into Cellular Uptake and Intracellular Trafficking of Nanoparticles. Nanoscale Res. Lett. 2018, 13, 339. [Google Scholar] [CrossRef]
- Tang, M.; Dong, H.; Cai, X.; Zhu, H.; Ren, T.; Li, Y. Disulfide-Bridged Cleavable PEGylation of Poly-L-Lysine for SiRNA Delivery. Methods Mol. Biol. 2016, 1364, 49–61. [Google Scholar] [PubMed]
- Inoue, Y.; Kurihara, R.; Tsuchida, A.; Hasegawa, M.; Nagashima, T.; Mori, T.; Niidome, T.; Katayama, Y.; Okitsu, O. Efficient delivery of siRNA using dendritic poly(L-lysine) for loss-of-function analysis. J. Control. Release 2008, 126, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.; Son, H.; Nam, Y. Layer-by-layer siRNA/poly(L-lysine) Multilayers on Polydopamine-coated Surface for Efficient Cell Adhesion and Gene Silencing. Sci. Rep. 2018, 8, 7738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fröhlich, E. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int. J. Nanomed. 2012, 7, 5577–5591. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Liu, Y.; Reineke, T. General structure-activity relationship for poly(glycoamidoamine)s: The effect of amine density on cytotoxicity and DNA delivery efficiency. Bioconjug. Chem. 2008, 19, 428–440. [Google Scholar] [CrossRef]
- Zhang, Y.; Pan, H.; Zhang, P.; Gao, N.; Lin, Y.; Luo, Z.; Li, P.; Wang, C.; Liu, L.; Pang, D.; et al. Functionalized quantum dots induce proinflammatory responses in vitro: The role of terminal functional group-associated endocytic pathways. Nanoscale 2013, 5, 5919–5929. [Google Scholar] [CrossRef]
- Marina, O.; Sanders, C.; Mourant, J. Correlating light scattering with internal cellular structures. Biomed. Opt. Express 2012, 3, 296–312. [Google Scholar] [CrossRef]
- Smith, B.; Niebert, M.; Plakhotnik, T.; Zvyagin, A. Transfection and imaging of diamond nanocrystals as scattering optical labels. J. Lumin. 2007, 127, 260–263. [Google Scholar] [CrossRef]
- Hui, Y.Y.; Cheng, C.-L.; Chang, H.-C. Nanodiamonds for optical bioimaging. J. Phys. D Appl. Phys. 2010, 43, 374021. [Google Scholar] [CrossRef]
- Suzuki, H.; Toyooka, T.; Ibuki, Y. Simple and easy method to evaluate uptake potential of nanoparticles in mammalian cells using a flow cytometric light scatter analysis. Environ. Sci. Technol. 2007, 41, 3018–3024. [Google Scholar] [CrossRef]
- Manouchehri, S.; Zarrintaj, P.; Saeb, M.; Ramsey, J. Advanced Delivery Systems Based on Lysine or Lysine Polymers. Mol. Pharm. 2021, 18, 3652–3670. [Google Scholar] [CrossRef] [PubMed]
- Serda, R.E.; Mack, A.; van de Ven, A.L.; Ferrati, S.; Dunner, K., Jr.; Godin, B.; Chiappini, C.; Landry, M.; Brousseau, L.; Liu, X.; et al. Logic-embedded vectors for intracellular partitioning, endosomal escape, and exocytosis of nanoparticles. Small 2010, 6, 2691–2700. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Vaijayanthimala, V.; Cheng, C.; Yeh, S.; Chang, C.; Li, C.; Chang, H. The exocytosis of fluorescent nanodiamond and its use as a long-term cell tracker. Small 2011, 7, 3363–3370. [Google Scholar] [CrossRef]
- Chithrani, B.; Chan, W. Elucidating the mechanism of cellular uptake and removal of protein-coated gold nanoparticles of different sizes and shapes. Nano Lett. 2007, 7, 1542–1550. [Google Scholar] [CrossRef]
- Behzadi, S.; Serpooshan, V.; Tao, W.; Hamaly, M.; Alkawareek, M.; Dreaden, O.; Brown, D.; Alkilany, A.; Farokhzad, O.; Mahmoudi, M. Cellular Uptake of Nanoparticles: Journey Inside the Cell. Chem. Soc. Rev. 2017, 46, 4218–4244. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Han, T.; Lee, K.; Han, S.; Hwang, J.; Moon, D.; Kim, S.; Cho, Y. N-acetyl histidine-conjugated glycol chitosan self-assembled nanoparticles for intracytoplasmic delivery of drugs: Endocytosis, exocytosis and drug release. J. Control. Release 2006, 115, 37–45. [Google Scholar] [CrossRef]
- Wang, T.; Larcher, L.; Ma, L.; Veedu, R. Systematic Screening of Commonly Used Commercial Transfection Reagents towards Efficient Transfection of Single-Stranded Oligonucleotides. Molecules 2018, 23, 2564. [Google Scholar] [CrossRef]
- Kumar, V.; Pichon, C.; Refregiers, M.; Guerin, B.; Midoux, P.; Chaudhuri, A. Single histidine residue in head-group region is sufficient to impart remarkable gene transfection properties to cationic lipids: Evidence for histidine-mediated membrane fusion at acidic pH. Gene Ther. 2003, 10, 1206–1215. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Volume Particle Size Distribution | Average Particle Size (nm) | Polydispersity Index (PDI) | Zeta Potential ± Standard Deviation (mV) | |
|---|---|---|---|---|---|
| Size Range (nm) | Percent Distribution (%) | ||||
| HK-NDs (2 mg/mL) | 30–90 | 56 | 250 | 0.2 | 30 ± 3 |
| 100–200 | 31 | ||||
| 210–300 | 9 | ||||
| H50K50-NDs (2 mg/mL) | <200 | 0 | 1949 | 0.1 | 18 ± 4 |
| 210–500 | 1.7 | ||||
| 510–1000 | 0.6 | ||||
| >1000 | 97.5 | ||||
| H50K50-NDs (1.5 mg/mL) | <200 | 3.1 | 559 | 0.4 | 19 ± 6 |
| 210–500 | 11 | ||||
| 510–1000 | 38.1 | ||||
| >1000 | 48.8 | ||||
| H50K50-NDs (1 mg/mL) | <200 | 50.5 | 194 | 0.2 | 23 ± 6 |
| 210–500 | 30.8 | ||||
| 510–1000 | 10.7 | ||||
| >1000 | 8.2 | ||||
| Time-Point | Sample | Cells in QA3 (%) | Cells in QA1 (%) |
|---|---|---|---|
| 24 h post treatment | Untreated | 94.86 | 5.14 |
| K-NDs | 75.75 | 24.25 | |
| HK-NDs | 79.27 | 20.73 | |
| H50K50-NDs | 51.45 | 48.55 | |
| 48 h post-treatment | Untreated | 95.78 | 4.22 |
| K-NDs | 73.86 | 26.14 | |
| HK-NDs | 82.44 | 17.56 | |
| H50K50-NDs | 76.61 | 23.29 | |
| 120 h post treatment | Untreated | 98.79 | 1.21 |
| K-NDs | 90.69 | 9.31 | |
| HK-NDs | 91.64 | 8.36 | |
| H50K50-NDs | 89.03 | 10.97 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alwani, S.; Rai, R.; Zittlau, I.; Rekve, J.; Michel, D.; Badea, I. Design of Smart Nanodiamonds: Introducing pH Sensitivity to Improve Nucleic Acid Carrier Efficiency of Diamoplexes. Pharmaceutics 2022, 14, 1794. https://doi.org/10.3390/pharmaceutics14091794
Alwani S, Rai R, Zittlau I, Rekve J, Michel D, Badea I. Design of Smart Nanodiamonds: Introducing pH Sensitivity to Improve Nucleic Acid Carrier Efficiency of Diamoplexes. Pharmaceutics. 2022; 14(9):1794. https://doi.org/10.3390/pharmaceutics14091794
Chicago/Turabian StyleAlwani, Saniya, Raj Rai, Isabella Zittlau, Jonathan Rekve, Deborah Michel, and Ildiko Badea. 2022. "Design of Smart Nanodiamonds: Introducing pH Sensitivity to Improve Nucleic Acid Carrier Efficiency of Diamoplexes" Pharmaceutics 14, no. 9: 1794. https://doi.org/10.3390/pharmaceutics14091794
APA StyleAlwani, S., Rai, R., Zittlau, I., Rekve, J., Michel, D., & Badea, I. (2022). Design of Smart Nanodiamonds: Introducing pH Sensitivity to Improve Nucleic Acid Carrier Efficiency of Diamoplexes. Pharmaceutics, 14(9), 1794. https://doi.org/10.3390/pharmaceutics14091794