
Fine-tuning of BERT Model to Accurately Predict Drug–Target Interactions

 , , ,
, , ,  , and
, and
Abstract
:1. Introduction
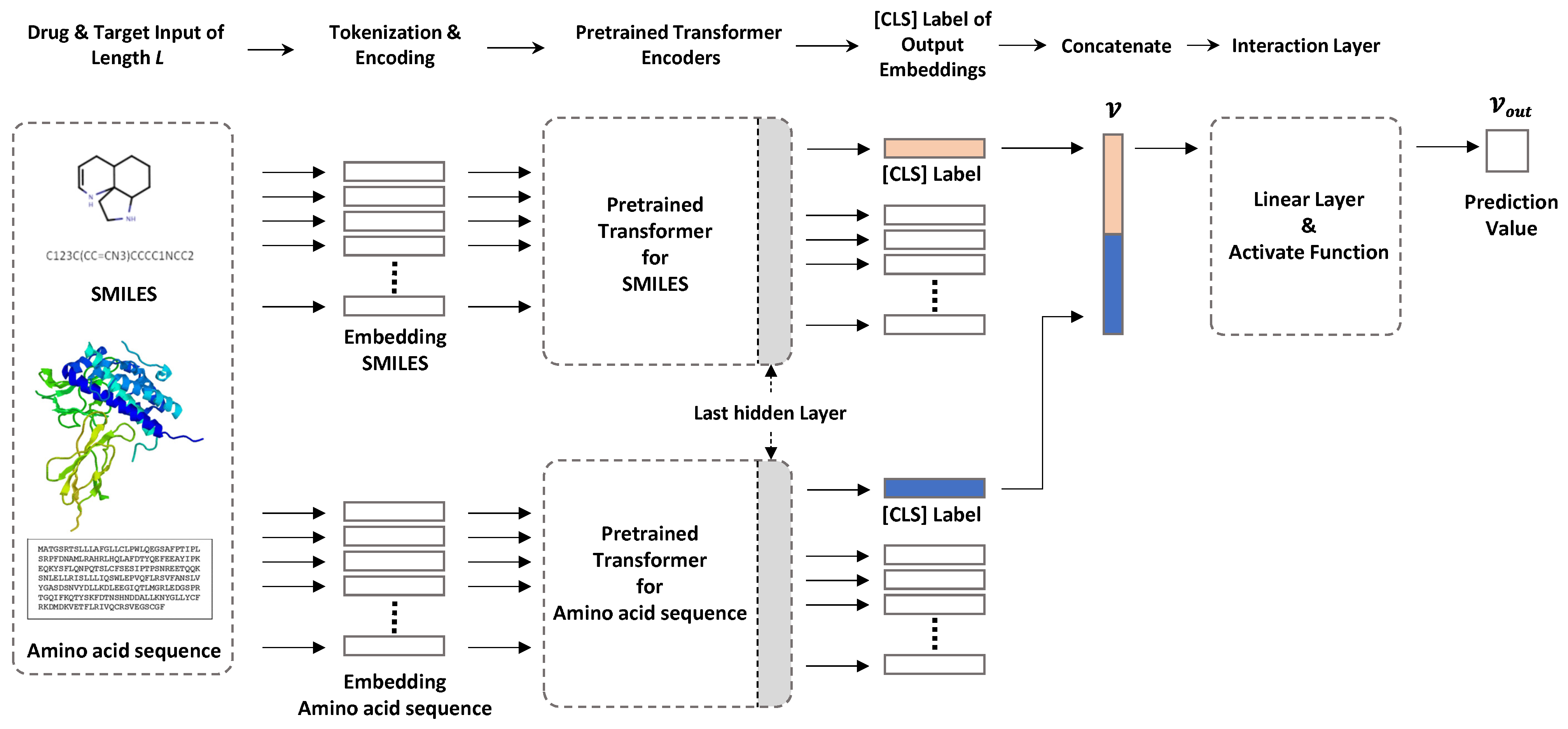
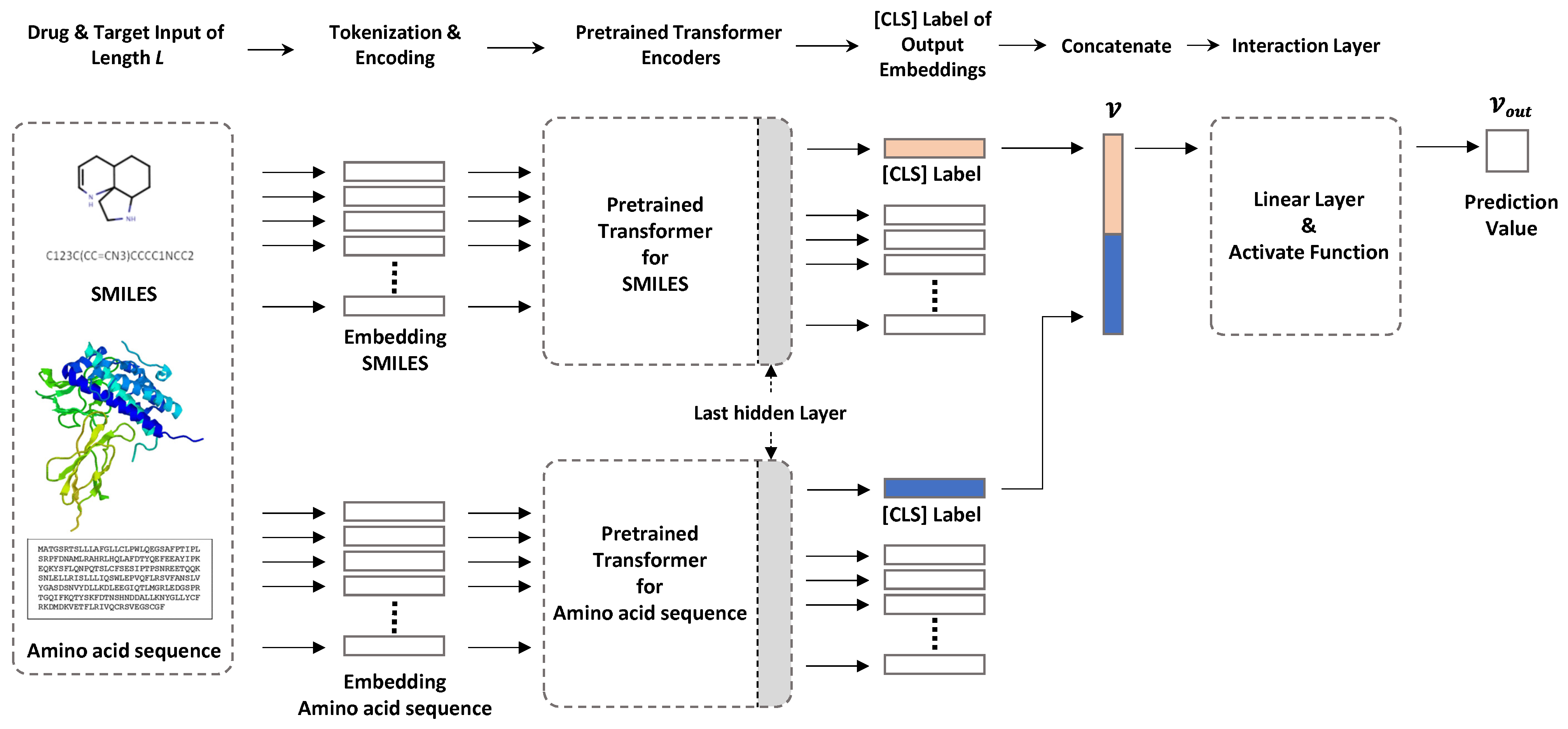
2. Datasets and Methods
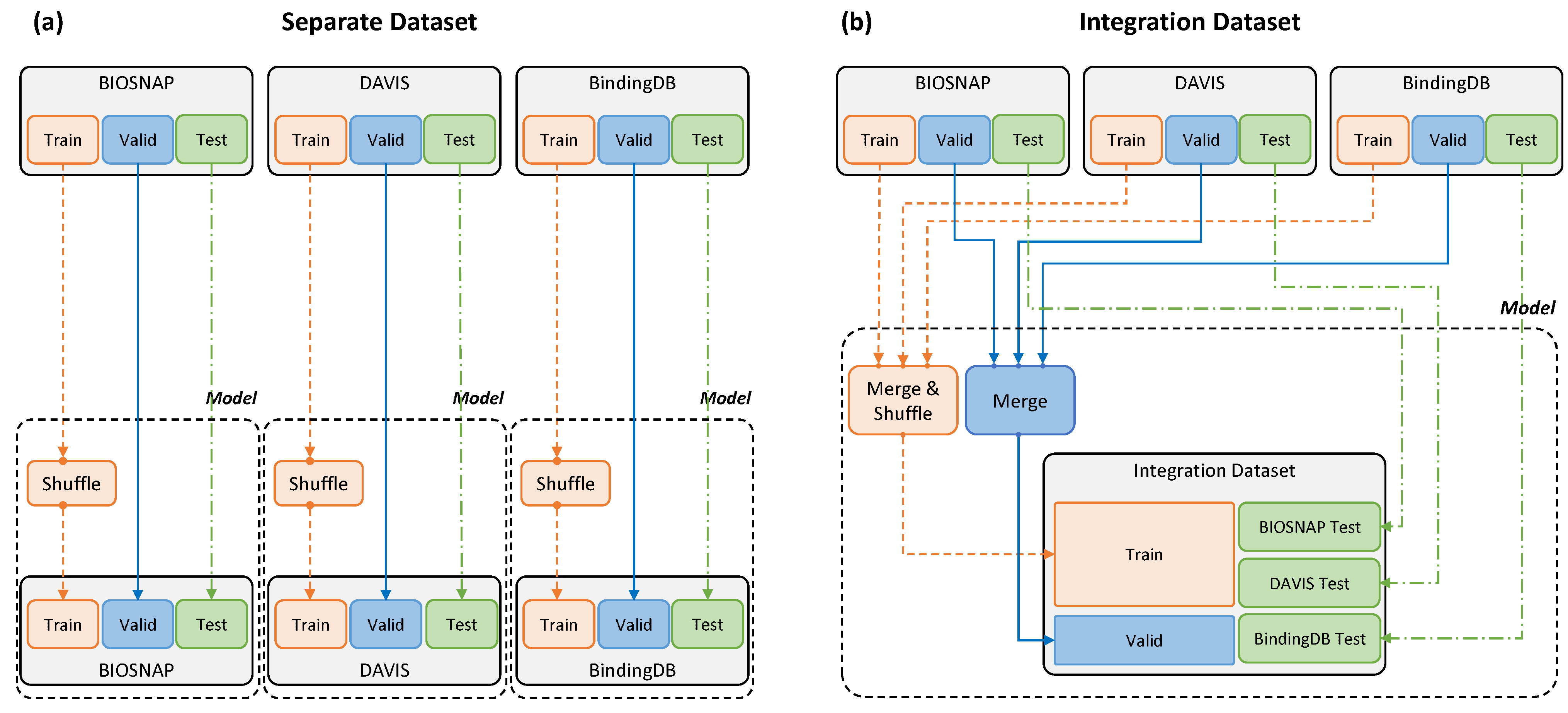
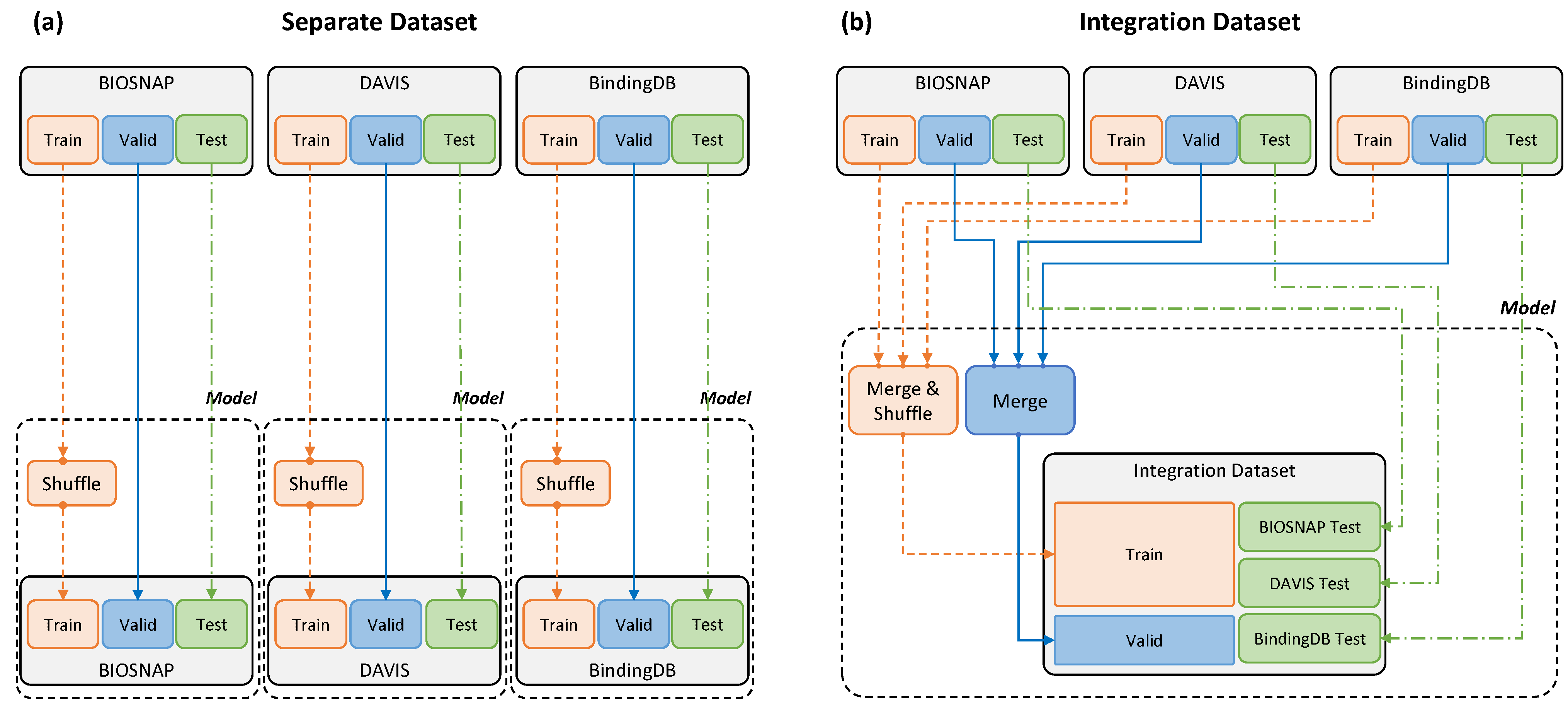
2.1. Dataset Configuration
2.2. Model Configuration
2.3. Implementation Settings
2.4. Model Performance Evaluation
2.5. Additional Experiments: Prediction Dissociation Constant
2.6. Model Evaluation Using an External Dataset
3. Results
3.1. Performance Evaluation
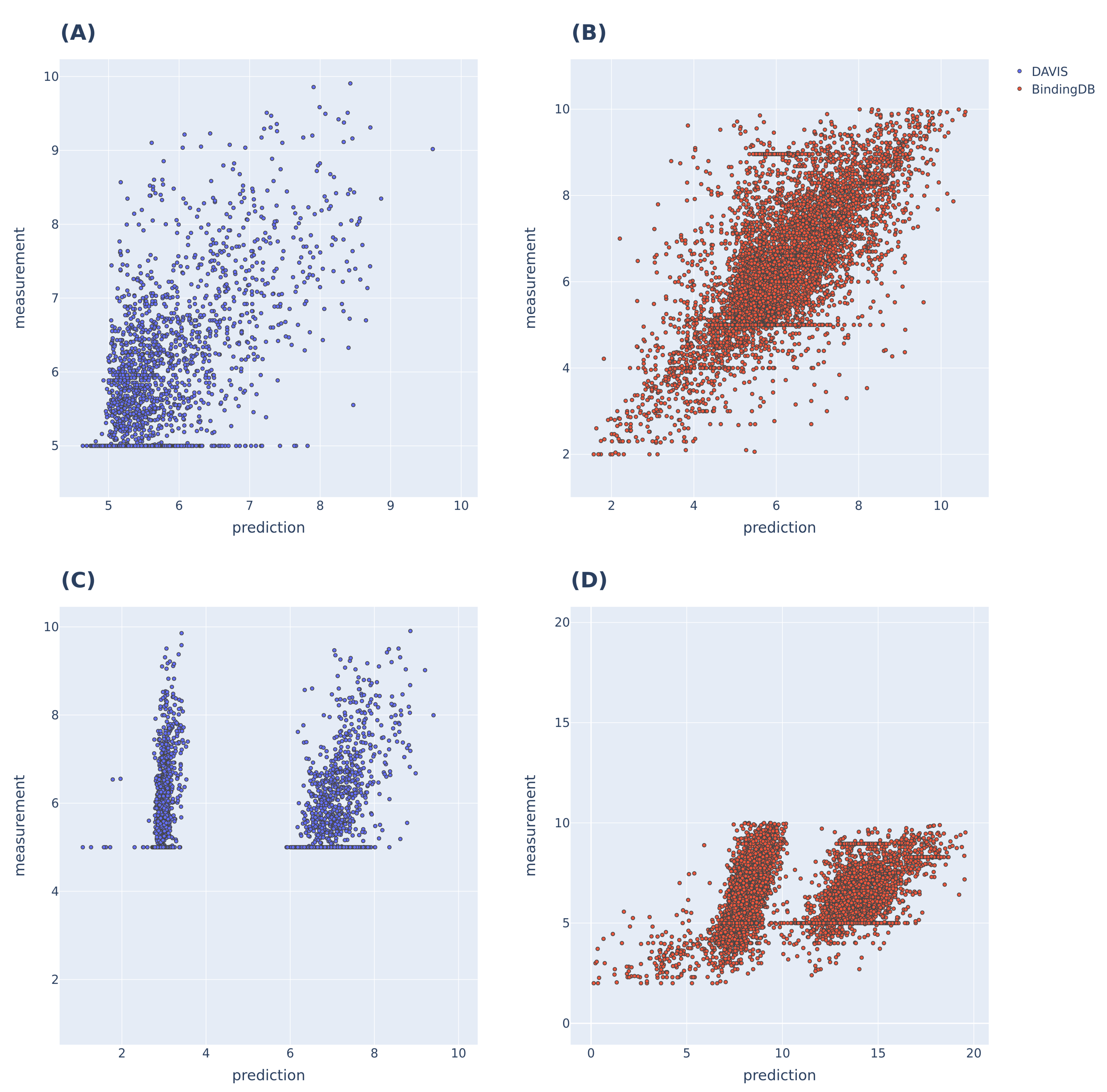
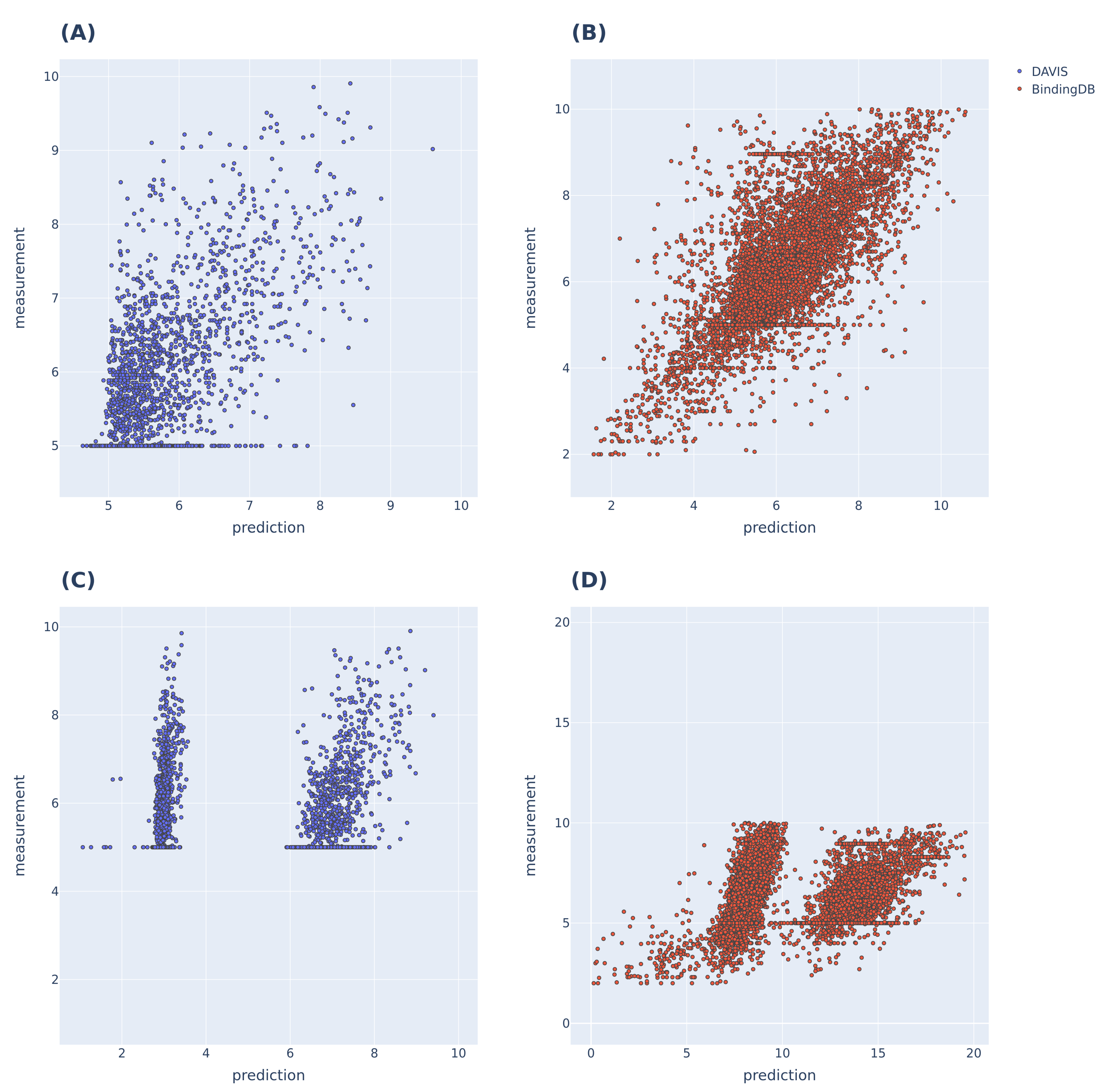
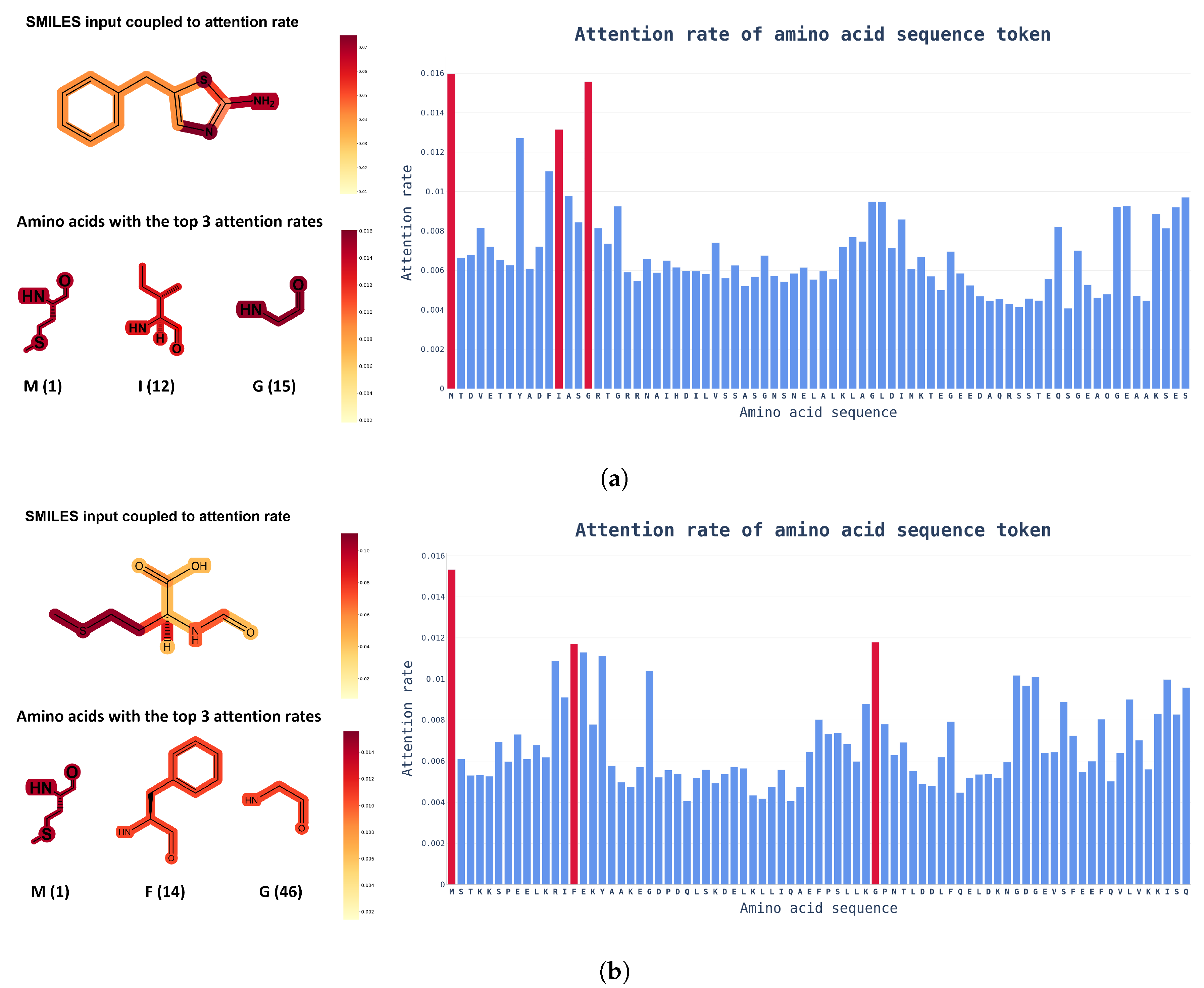
3.2. Additional Experimental Visualization of the Prediction Dissociation Constant
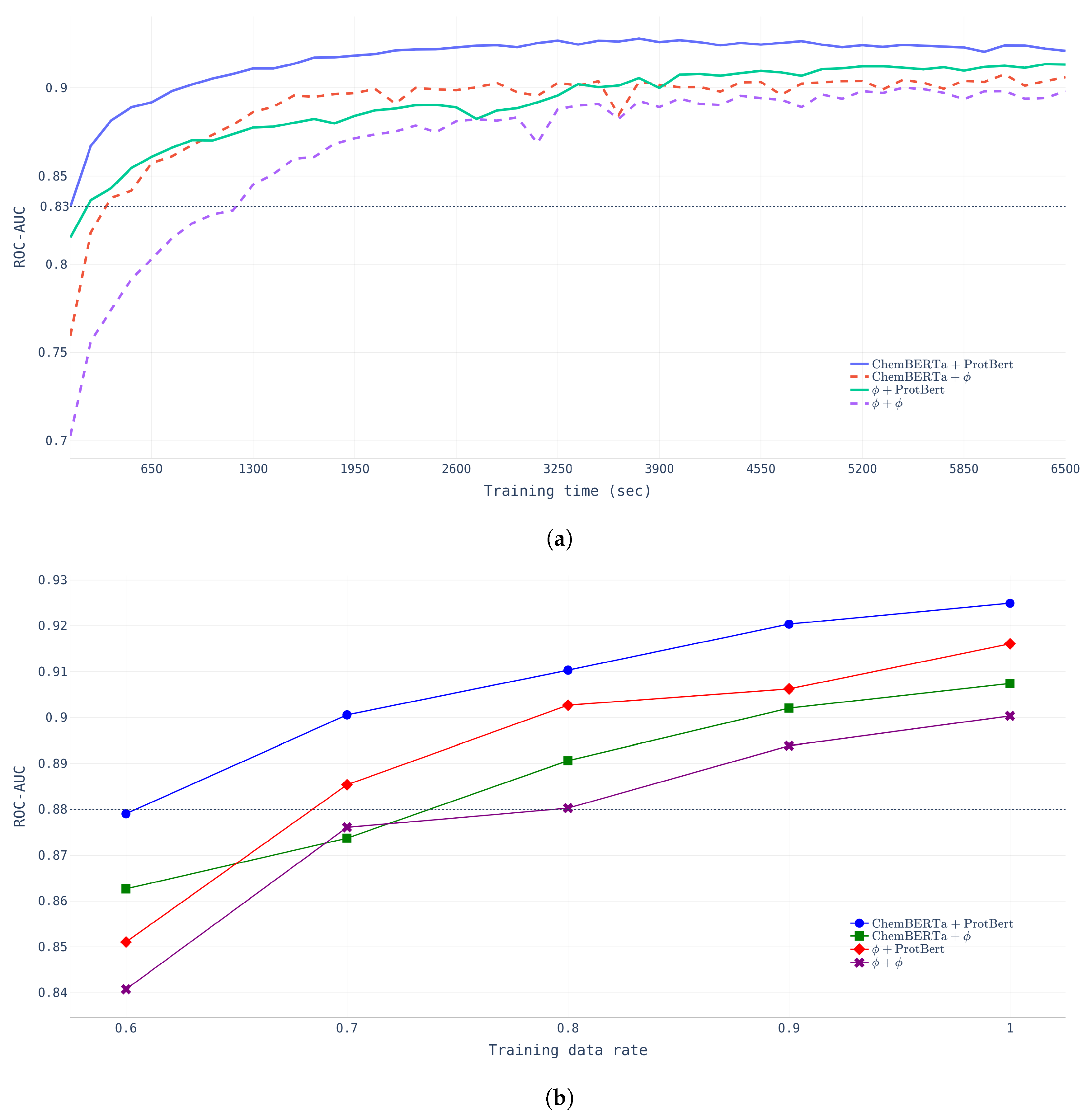
- Learning Time.Figure 4a shows that FP-Model-BDB converged much faster and performed better than NP-Model-BDB which did not undergo any pretraining. The NP-Model-BDB took nine epochs (1170 s) to reach a ROC-AUC of 0.8326, whereas the FP-Model-BDB achieved the same performance after only one epoch. The figure indicates that chemical compound encoding pretraining affects the initial training stage, but protein encoding pretraining had more powerful effects in determining model performance at the final stage.
- Data-Size.Figure 4b shows that FP-Model-BDB achieved the same performance with fewer data than the NP-Model-BDB. FP-Model-BDB only needed 60% of the total data, whereas the NP-Model-BDB required 80% of the total training data.

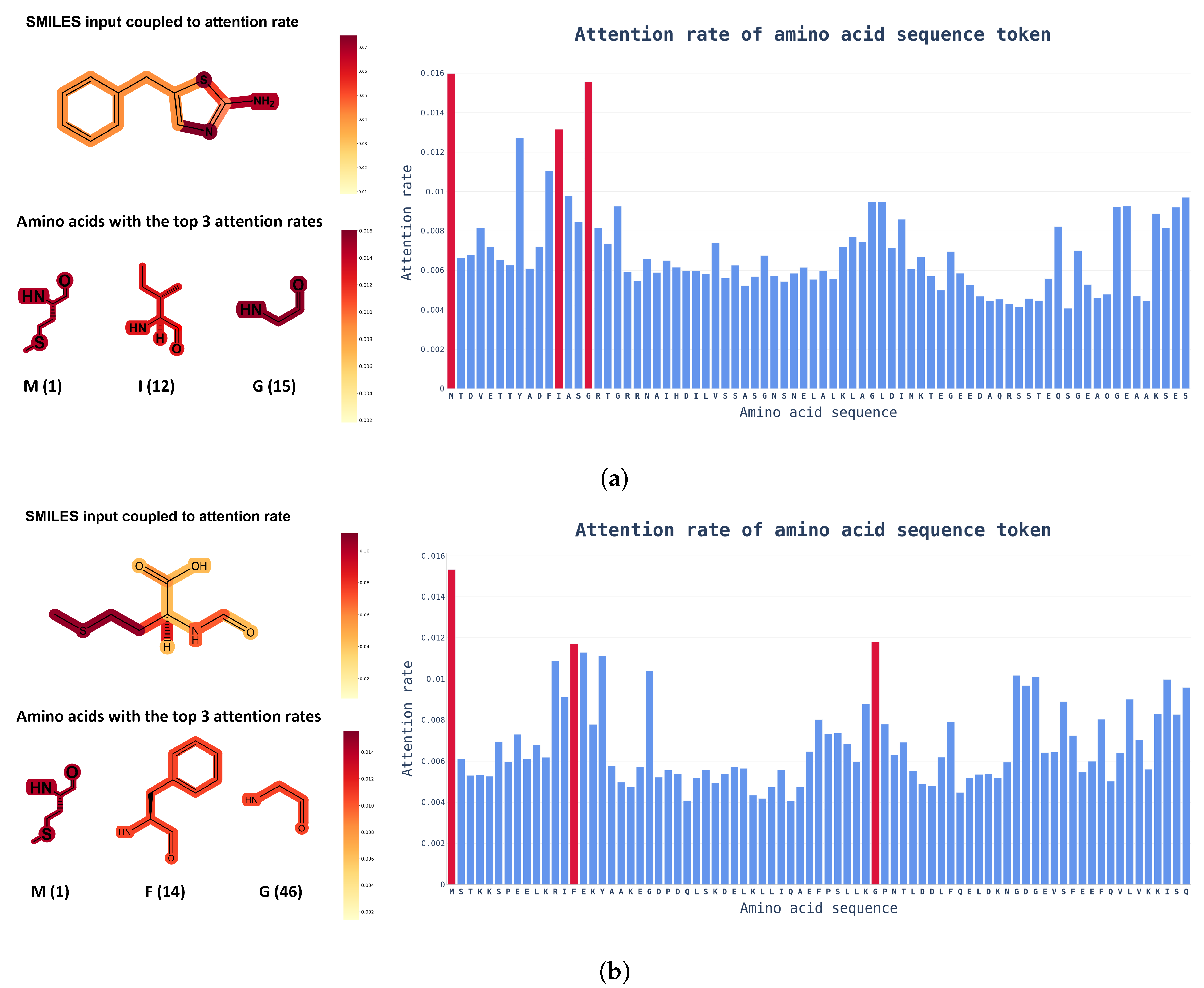
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DTI | drug–target interaction |
| DNN | deep neural network |
| DBN | deep belief network |
| CNN | convolution neural network |
| SMILES | simplified molecular-input line-entry system |
| BERT | Bidirectional Encoder Representations from Transformers |
| CLS | vector from a specially prepared token |
| FCN | fully connected layer |
| MSE | mean squared error |
References
- Unterthiner, T.; Mayr, A.; Klambauer, G.; Steijaert, M.; Wegner, J.K.; Ceulemans, H.; Hochreiter, S. Deep learning as an opportunity in virtual screening. In Proceedings of the Deep Learning Workshop at NIPS, Montreal, QC, Canada, 8–13 December 2014; Volume 27, pp. 1–9. [Google Scholar]
- Wen, M.; Zhang, Z.; Niu, S.; Sha, H.; Yang, R.; Yun, Y.; Lu, H. Deep-learning-based drug–target interaction prediction. J. Proteome Res. 2017, 16, 1401–1409. [Google Scholar] [CrossRef] [PubMed]
- Mayr, A.; Klambauer, G.; Unterthiner, T.; Steijaert, M.; Wegner, J.K.; Ceulemans, H.; Clevert, D.A.; Hochreiter, S. Large-scale comparison of machine learning methods for drug target prediction on ChEMBL. Chem. Sci. 2018, 9, 5441–5451. [Google Scholar] [CrossRef] [PubMed]
- Öztürk, H.; Özgür, A.; Ozkirimli, E. DeepDTA: Deep drug–target binding affinity prediction. Bioinformatics 2018, 34, i821–i829. [Google Scholar] [CrossRef] [PubMed]
- Öztürk, H.; Ozkirimli, E.; Özgür, A. WideDTA: Prediction of drug-target binding affinity. arXiv 2019, arXiv:1902.04166. [Google Scholar]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, Ł.; Polosukhin, I. Attention is all you need. In Proceedings of the Advances in Neural Information Processing Systems 30 (NIPS 2017), Long Beach, CA, USA, 4–9 December 2017; Volume 30. [Google Scholar]
- Wolf, T.; Debut, L.; Sanh, V.; Chaumond, J.; Delangue, C.; Moi, A.; Cistac, P.; Rault, T.; Louf, R.; Funtowicz, M.; et al. Transformers: State-of-the-Art Natural Language Processing. In Proceedings of the 2020 Conference on Empirical Methods in Natural Language Processing: System Demonstrations, Online, 16–20 November 2020; Association for Computational Linguistics: Stroudsburg, PA, USA, 2020; pp. 38–45. [Google Scholar] [CrossRef]
- Chithrananda, S.; Grand, G.; Ramsundar, B. Chemberta: Large-scale self-supervised pretraining for molecular property prediction. arXiv 2020, arXiv:2010.09885. [Google Scholar]
- Elnaggar, A.; Heinzinger, M.; Dallago, C.; Rehawi, G.; Wang, Y.; Jones, L.; Gibbs, T.; Feher, T.; Angerer, C.; Steinegger, M.; et al. ProtTrans: Towards Cracking the Language of Lifes Code Through Self-Supervised Deep Learning and High Performance Computing. IEEE Trans. Pattern Anal. Mach. Intell. 2021, 1. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Liu, X.; Jin, S.; Lin, J.; Liu, J. Machine learning for drug-target interaction prediction. Molecules 2018, 23, 2208. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- Zitnik, M.; Rok Sosič, S.M.; Leskovec, J. BioSNAP Datasets: Stanford Biomedical Network Dataset Collection. 2018. Available online: http://snap.stanford.edu/biodata (accessed on 22 July 2022).
- Davis, M.I.; Hunt, J.P.; Herrgard, S.; Ciceri, P.; Wodicka, L.M.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Lin, Y.; Wen, X.; Jorissen, R.N.; Gilson, M.K. BindingDB: A web-accessible database of experimentally determined protein–ligand binding affinities. Nucleic Acids Res. 2007, 35, D198–D201. [Google Scholar] [CrossRef] [PubMed]
- Devlin, J.; Chang, M.W.; Lee, K.; Toutanova, K. BERT: Pre-training of Deep Bidirectional Transformers for Language Understanding. In Proceedings of the 2019 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies, Volume 1 (Long and Short Papers), Minneapolis, MN, USA, 2–7 June 2019; Association for Computational Linguistics: Minneapolis, MN, USA, 2019; pp. 4171–4186. [Google Scholar] [CrossRef]
- Bepler, T.; Berger, B. Learning protein sequence embeddings using information from structure. arXiv 2019, arXiv:1902.08661. [Google Scholar]
- Liu, Y.; Ott, M.; Goyal, N.; Du, J.; Joshi, M.; Chen, D.; Levy, O.; Lewis, M.; Zettlemoyer, L.; Stoyanov, V. Roberta: A robustly optimized bert pretraining approach. arXiv 2019, arXiv:1907.11692. [Google Scholar]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem substance and compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Suzek, B.E.; Huang, H.; McGarvey, P.; Mazumder, R.; Wu, C.H. UniRef: Comprehensive and non-redundant UniProt reference clusters. Bioinformatics 2007, 23, 1282–1288. [Google Scholar] [CrossRef] [PubMed]
- Steinegger, M.; Söding, J. Clustering huge protein sequence sets in linear time. Nat. Commun. 2018, 9, 2542. [Google Scholar] [CrossRef] [PubMed]
- Girshick, R. Fast r-cnn. In Proceedings of the IEEE International Conference on Computer Vision, Washington, DC, USA, 7–13 December 2015; pp. 1440–1448. [Google Scholar]
- Huang, K.; Xiao, C.; Glass, L.M.; Sun, J. MolTrans: Molecular Interaction Transformer for drug–target interaction prediction. Bioinformatics 2021, 37, 830–836. [Google Scholar] [CrossRef]
- Gönen, M.; Heller, G. Concordance probability and discriminatory power in proportional hazards regression. Biometrika 2005, 92, 965–970. [Google Scholar] [CrossRef]
- Roy, K.; Chakraborty, P.; Mitra, I.; Ojha, P.K.; Kar, S.; Das, R.N. Some case studies on application of “rm2” metrics for judging quality of quantitative structure–activity relationship predictions: Emphasis on scaling of response data. J. Comput. Chem. 2013, 34, 1071–1082. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. In Vitro Drug INTERACTION studies—Cytochrome P450 Enzyme-and Transporter-Mediated Drug Interactions Guidance for Industry; Center for Drug Evaluation and Research, US Food and Drug Administration: Silver Spring, MD, USA, 2020. Available online: https://www.fda.gov/media/134582/download (accessed on 22 July 2022).
- Abnar, S.; Zuidema, W. Quantifying Attention Flow in Transformers. In Proceedings of the 58th Annual Meeting of the Association for Computational Linguistics, Online, 5–10 July 2020; Association for Computational Linguistics: Stroudsburg, PA, USA, 2020; pp. 4190–4197. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Drug | Proteins | Interactions |
|---|---|---|---|
| BIOSNAP | 4510 | 2181 | 27,482 (13,741/13,741) |
| DAVIS | 68 | 379 | 11,103 (1506/9597) |
| BindingDB | 10,665 | 1413 | 32,601 (9166/23,435) |
| Dataset | Training | Validation | Test |
|---|---|---|---|
| BIOSNAP | 19,238 | 2748 | 5496 |
| DAVIS | 2086 | 3006 | 6011 |
| BindingDB | 12,668 | 6644 | 13,289 |
| Integration | 33,992 | 12,398 | (5496/6011/13,289) |
| Dataset | BIOSNAP (BS) | DAVIS (DV) | BindingDB (BDB) | Integration (INT) |
|---|---|---|---|---|
| Non-pretrained (NP) ( + ) | NP-Model-BS | NP-Model-DV | NP-Model-BDB | NP-Model-INT |
| ChemBERTa-pretrained (CP) (ChemBERTa + ) | CP-Model-BS | CP-Model-DV | CP-Model-BDB | CP-Model-INT |
| ProtBERT-pretrained (PP) ( + ProtBERT) | PP-Model-BS | PP-Model-DV | PP-Model-BDB | PP-Model-INT |
| Full-pretrained (FP) (ChemBERTa + ProtBERT) | FP-Model-BS | FP-Model-DV | FP-Model-BDB | FP-Model-INT |
| Method | ROC-AUC | PR-AUC | Sensitivity | Specificity | CI | |
|---|---|---|---|---|---|---|
| Dataset 1. BIOSNAP | ||||||
| MolTrans | 0.895 ± 0.002 | 0.901 ± 0.004 | 0.775 ± 0.032 | 0.851 ± 0.014 | 0.889 | 0.449 |
| NP-Model-BS | 0.882 ± 0.004 | 0.871 ± 0.015 | 0.779 ± 0.020 | 0.850 ± 0.012 | 0.895 | 0.428 |
| CP-Model-BS | 0.881 ± 0.009 | 0.859 ± 0.017 | 0.811 ± 0.018 | 0.835 ± 0.008 | 0.891 | 0.406 |
| PP-Model-BS | 0.893 ± 0.003 | 0.874 ± 0.006 | 0.803 ± 0.033 | 0.851 ± 0.019 | 0.896 | 0.425 |
| FP-Model-BS | 0.914 ± 0.006 | 0.900 ± 0.007 | 0.862 ± 0.025 | 0.847 ± 0.007 | 0.913 | 0.467 |
| NP-Model-INT | 0.877 ± 0.007 | 0.860 ± 0.010 | 0.785 ± 0.007 | 0.842 ± 0.008 | 0.897 | 0.421 |
| CP-Model-INT | 0.875 ± 0.006 | 0.851 ± 0.009 | 0.775 ± 0.023 | 0.844 ± 0.016 | 0.885 | 0.401 |
| PP-Model-INT | 0.895 ± 0.003 | 0.880 ± 0.008 | 0.802 ± 0.018 | 0.852 ± 0.009 | 0.896 | 0.435 |
| FP-Model-INT | 0.910 ± 0.012 | 0.897 ± 0.014 | 0.830 ± 0.029 | 0.863 ± 0.011 | 0.911 | 0.447 |
| Dataset 2. DAVIS | ||||||
| MolTrans | 0.907 ± 0.002 | 0.404 ± 0.016 | 0.800 ± 0.022 | 0.876 ± 0.013 | 0.903 | 0.156 |
| NP-Model-DV | 0.870 ± 0.003 | 0.283 ± 0.005 | 0.738 ± 0.030 | 0.871 ± 0.026 | 0.875 | 0.118 |
| CP-Model-DV | 0.882 ± 0.006 | 0.250 ± 0.023 | 0.744 ± 0.021 | 0.888 ± 0.019 | 0.878 | 0.117 |
| PP-Model-DV | 0.866 ± 0.003 | 0.263 ± 0.007 | 0.747 ± 0.020 | 0.856 ± 0.012 | 0.864 | 0.115 |
| FP-Model-DV | 0.920 ± 0.002 | 0.395 ± 0.007 | 0.824 ± 0.026 | 0.889 ± 0.015 | 0.917 | 0.167 |
| NP-Model-INT | 0.899 ± 0.008 | 0.322 ± 0.030 | 0.814 ± 0.039 | 0.857 ± 0.028 | 0.892 | 0.141 |
| CP-Model-INT | 0.904 ± 0.011 | 0.351 ± 0.035 | 0.814 ± 0.030 | 0.859 ± 0.020 | 0.917 | 0.169 |
| PP-Model-INT | 0.923 ± 0.005 | 0.417 ± 0.028 | 0.844 ± 0.017 | 0.876 ± 0.021 | 0.916 | 0.162 |
| FP-Model-INT | 0.942 ± 0.005 | 0.517 ± 0.017 | 0.903 ± 0.017 | 0.866 ± 0.015 | 0.940 | 0.201 |
| Dataset 3. BindingDB | ||||||
| MolTrans | 0.914 ± 0.001 | 0.622 ± 0.007 | 0.797 ± 0.005 | 0.896 ± 0.007 | 0.899 | 0.267 |
| NP-Model-BDB | 0.891 ± 0.005 | 0.515 ± 0.014 | 0.774 ± 0.012 | 0.897 ± 0.013 | 0.899 | 0.309 |
| CP-Model-BDB | 0.914 ± 0.003 | 0.585 ± 0.021 | 0.803 ± 0.011 | 0.904 ± 0.010 | 0.907 | 0.320 |
| PP-Model-BDB | 0.897 ± 0.003 | 0.557 ± 0.013 | 0.775 ± 0.019 | 0.900 ± 0.009 | 0.913 | 0.324 |
| FP-Model-BDB | 0.922 ± 0.001 | 0.623 ± 0.010 | 0.814 ± 0.025 | 0.916 ± 0.016 | 0.927 | 0.365 |
| NP-Model-INT | 0.904 ± 0.001 | 0.574 ± 0.008 | 0.766 ± 0.015 | 0.910 ± 0.015 | 0.907 | 0.315 |
| CP-Model-INT | 0.909 ± 0.005 | 0.600 ± 0.019 | 0.787 ± 0.008 | 0.907 ± 0.008 | 0.918 | 0.330 |
| PP-Model-INT | 0.918 ± 0.001 | 0.607 ± 0.012 | 0.787 ± 0.014 | 0.920 ± 0.010 | 0.916 | 0.344 |
| FP-Model-INT | 0.926 ± 0.001 | 0.639 ± 0.018 | 0.802 ± 0.022 | 0.928 ± 0.013 | 0.926 | 0.362 |
| CYP Subtype (Targets) | Drugs | MolTrans | NP-Model-BDB | FP-Model-BDB | NP-Model-INT | FP-Model-INT |
|---|---|---|---|---|---|---|
| 1A2 | Phenacetin | 0.391 | 0.745 | 0.774 | 0.767 | 0.764 |
| 1A2 | 7-Ethoxyresorufin | 0.255 | 0.747 | 0.786 | 0.762 | 0.784 |
| 2B6 | Efavirenz | 0.755 | 0.728 | 0.755 | 0.687 | 0.749 |
| 2B6 | Bupropion | 0.524 | 0.727 | 0.757 | 0.712 | 0.733 |
| 3A4 | Midazolam | 0.352 | 0.707 | 0.694 | 0.719 | 0.721 |
| 3A4 | Testosterone | 0.125 | 0.741 | 0.699 | 0.714 | 0.662 |
| 2C8 | Paclitaxel | 0.823 | 0.698 | 0.676 | 0.722 | 0.732 |
| 2C8 | Amodiaquine | 0.631 | 0.694 | 0.712 | 0.748 | 0.759 |
| 2C9 | S-Warfarin | 0.546 | 0.725 | 0.655 | 0.701 | 0.705 |
| 2C9 | Diclofenac | 0.556 | 0.645 | 0.758 | 0.726 | 0.711 |
| 2C19 | S-Mephenytoin | 0.313 | 0.738 | 0.742 | 0.740 | 0.744 |
| 2D6 | Bufuralol | 0.475 | 0.355 | 0.749 | 0.756 | 0.699 |
| 2D6 | Dextromethorphan | - | 0.490 | 0.740 | 0.750 | 0.740 |
| Average BP | 0.479 | 0.687 | 0.730 | 0.731 | 0.731 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, H.; Goo, S.; Lee, H.; Chae, J.-w.; Yun, H.-y.; Jung, S. Fine-tuning of BERT Model to Accurately Predict Drug–Target Interactions. Pharmaceutics 2022, 14, 1710. https://doi.org/10.3390/pharmaceutics14081710
Kang H, Goo S, Lee H, Chae J-w, Yun H-y, Jung S. Fine-tuning of BERT Model to Accurately Predict Drug–Target Interactions. Pharmaceutics. 2022; 14(8):1710. https://doi.org/10.3390/pharmaceutics14081710
Chicago/Turabian StyleKang, Hyeunseok, Sungwoo Goo, Hyunjung Lee, Jung-woo Chae, Hwi-yeol Yun, and Sangkeun Jung. 2022. "Fine-tuning of BERT Model to Accurately Predict Drug–Target Interactions" Pharmaceutics 14, no. 8: 1710. https://doi.org/10.3390/pharmaceutics14081710
APA StyleKang, H., Goo, S., Lee, H., Chae, J.-w., Yun, H.-y., & Jung, S. (2022). Fine-tuning of BERT Model to Accurately Predict Drug–Target Interactions. Pharmaceutics, 14(8), 1710. https://doi.org/10.3390/pharmaceutics14081710