
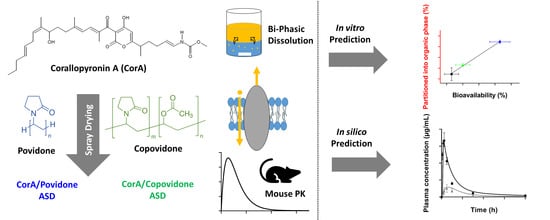
In Vitro–In Vivo Relationship in Mini-Scale—Enabling Formulations of Corallopyronin A

 , , , , , ,
, , , , , ,  ,
,  ,
, 

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Materials
2.2. Manufacture of the Spray-Dried CorA-Amorphous Solid Dispersion Formulations
2.3. Solubility Determination in Biorelevant Medium
2.4. Biorelevant Dissolution
2.4.1. Monophasic Dissolution
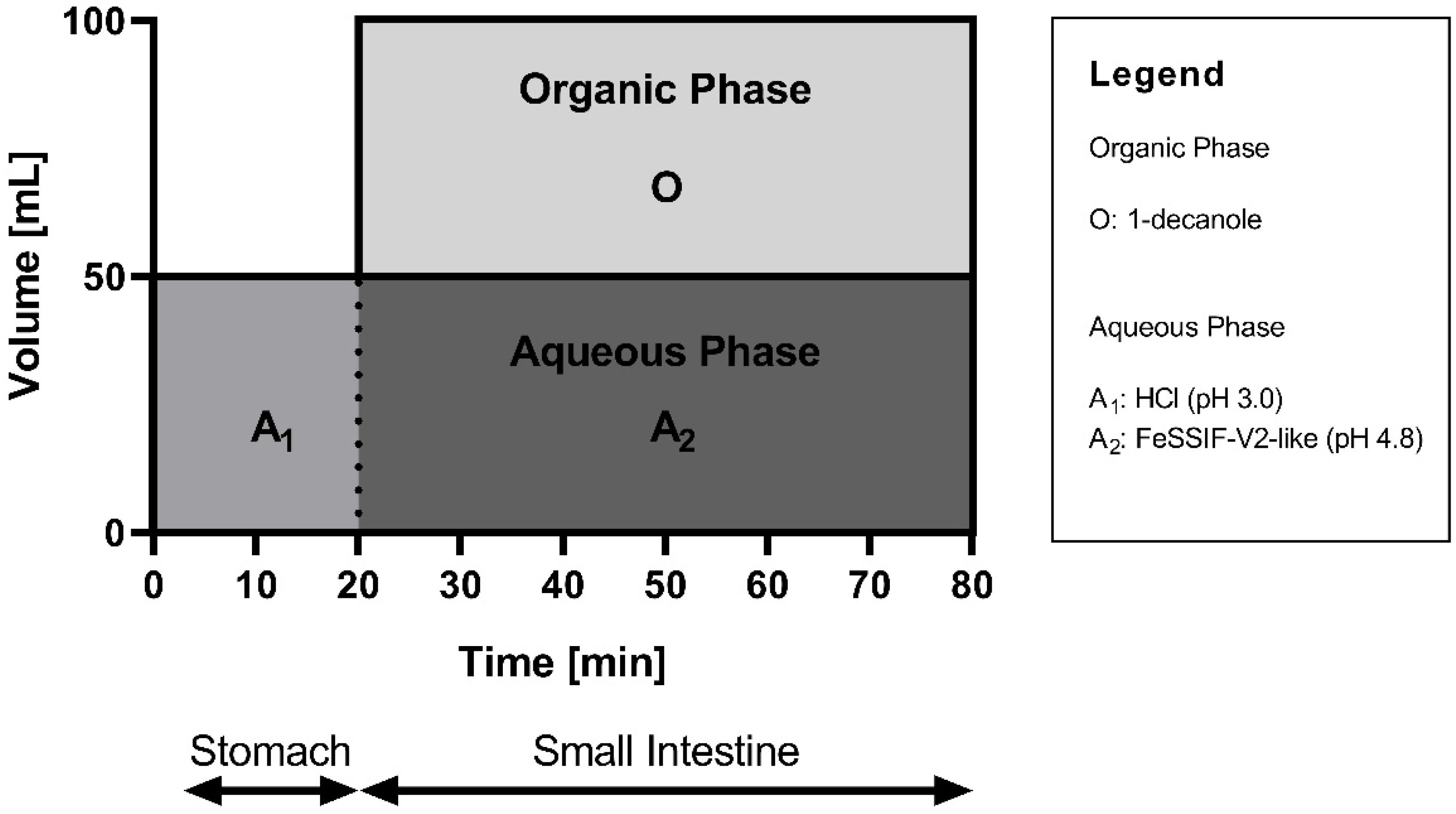
2.4.2. Biphasic Dissolution
2.5. Pharmacokinetic Study Setup
2.6. Bioanalysis of Corallopyronin A
2.7. Pharmacokinetic Analysis
2.8. Cell Culture Experiments
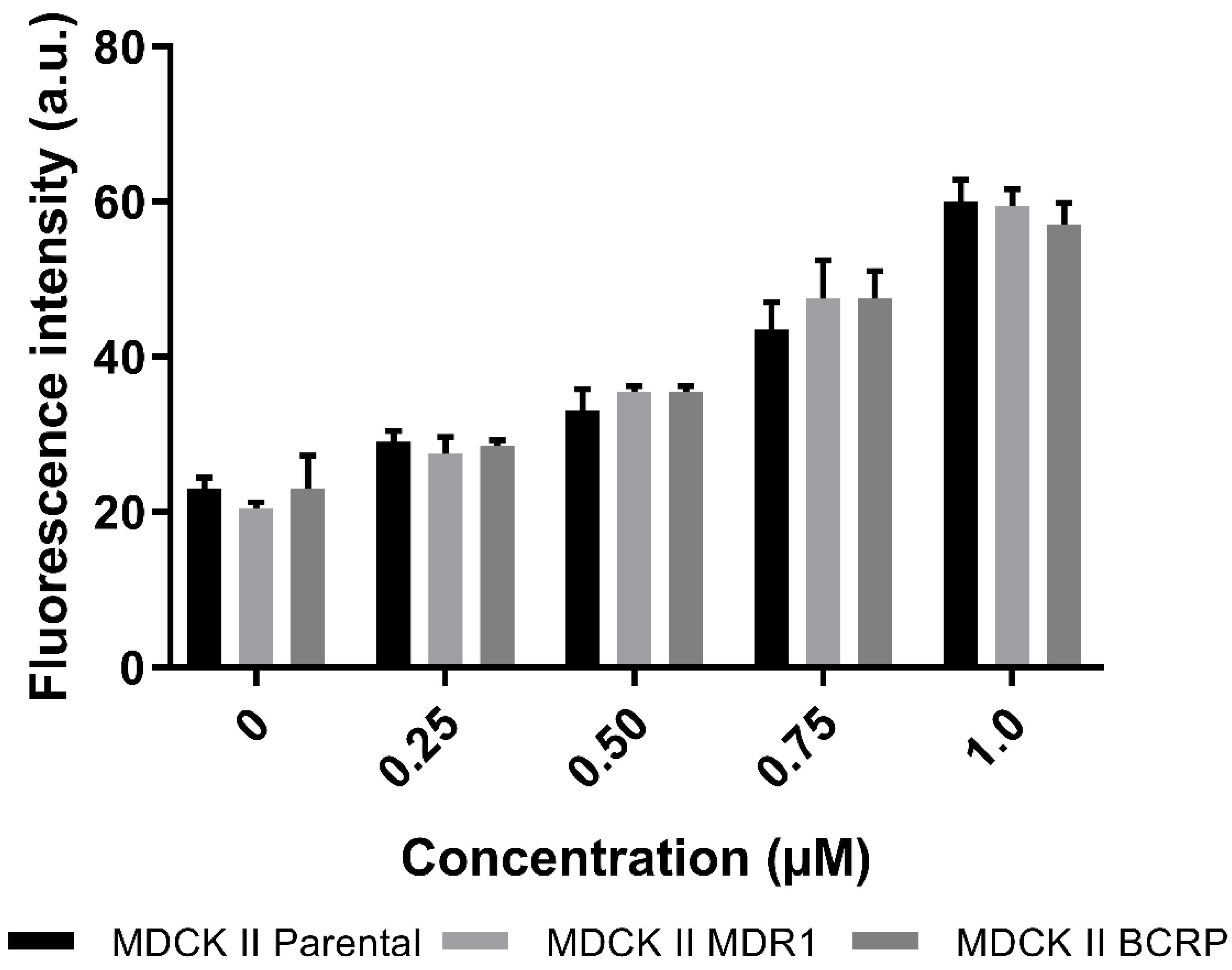
2.8.1. Investigation of Active Transport via P-gp and BCRP
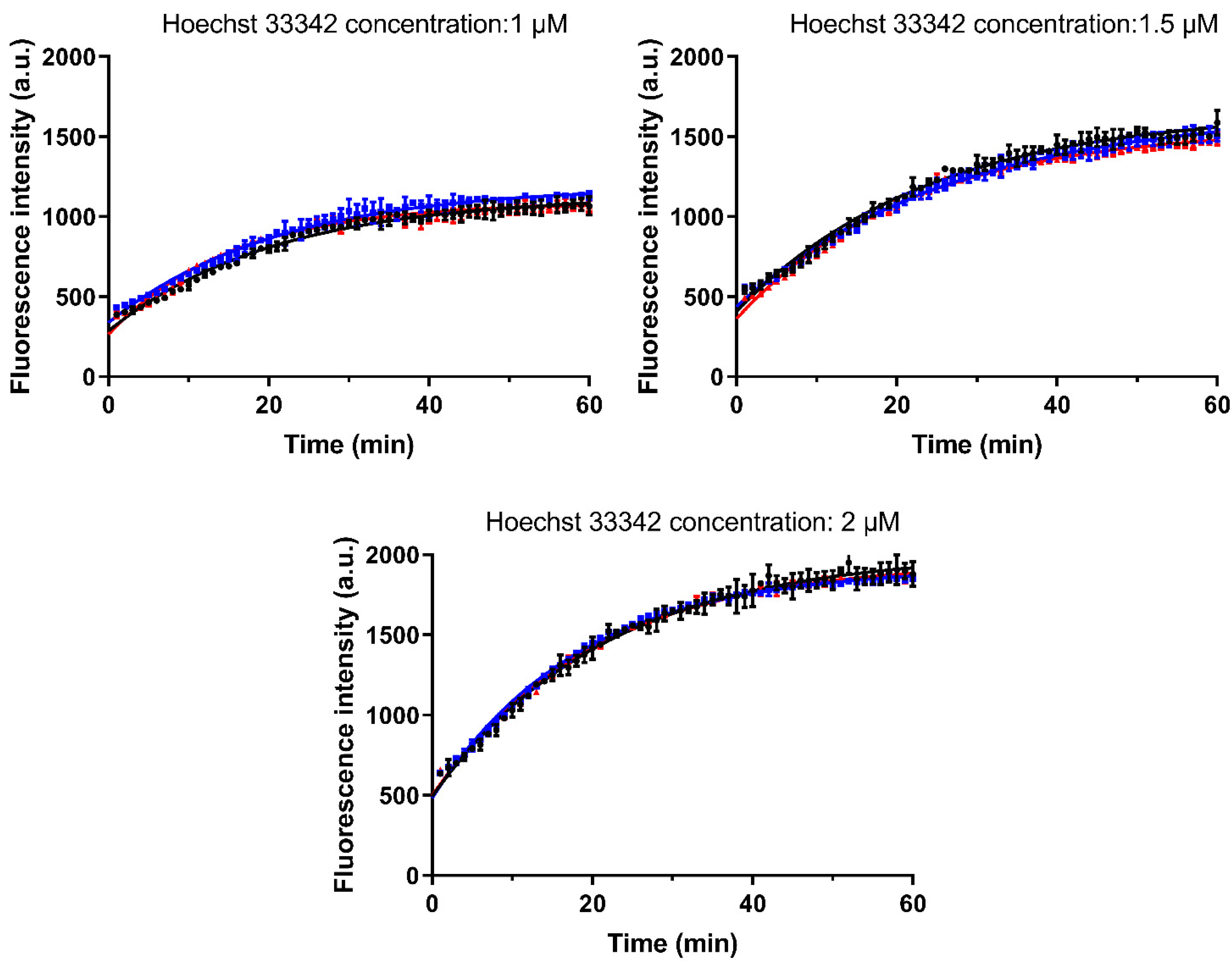
2.8.2. Investigation of the Interaction with Hoechst 33342
2.9. PBPK Modeling
3. Results
3.1. Solubility Determination in Biorelevant Medium
3.2. Biorelevant Dissolution
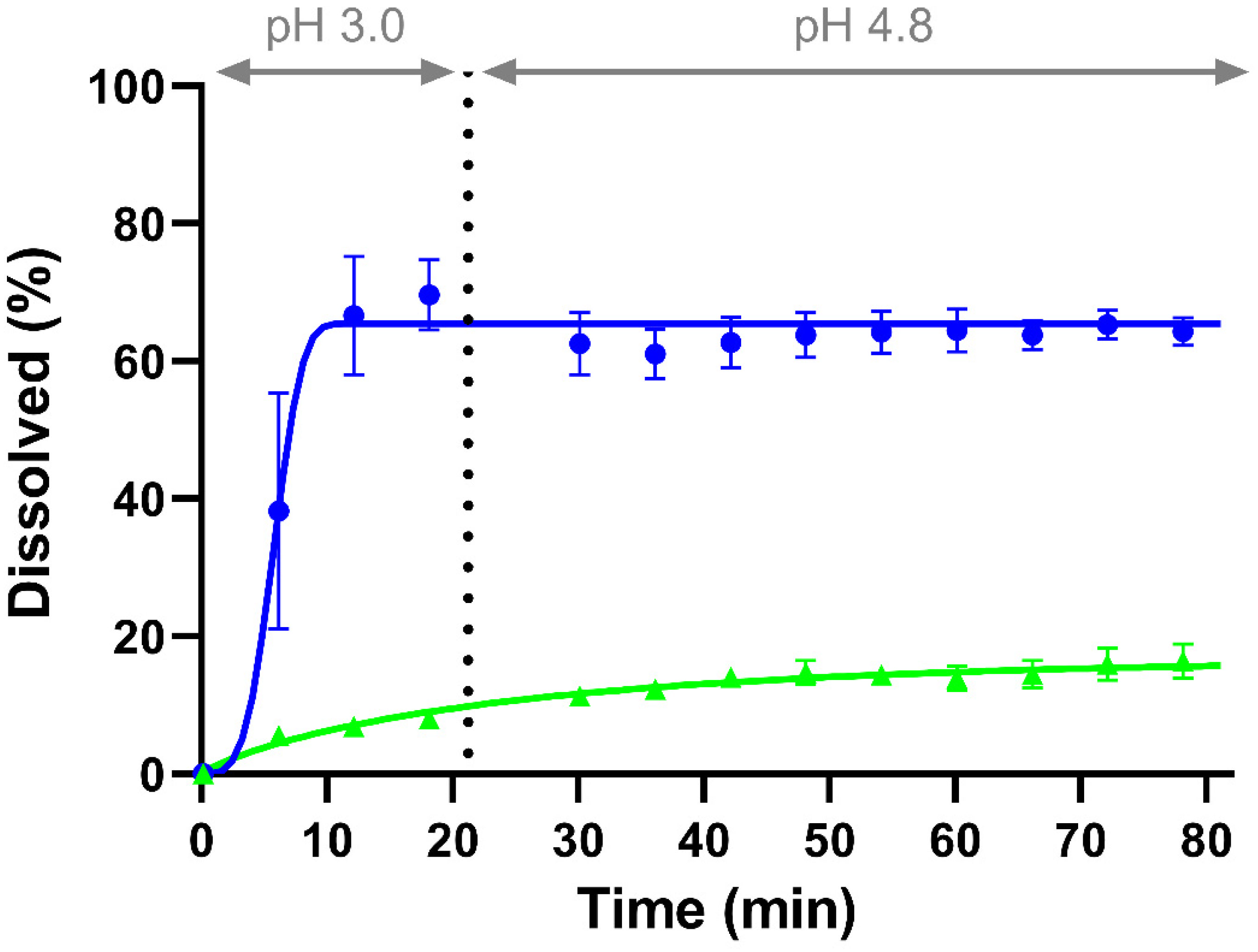
3.2.1. Monophasic Dissolution
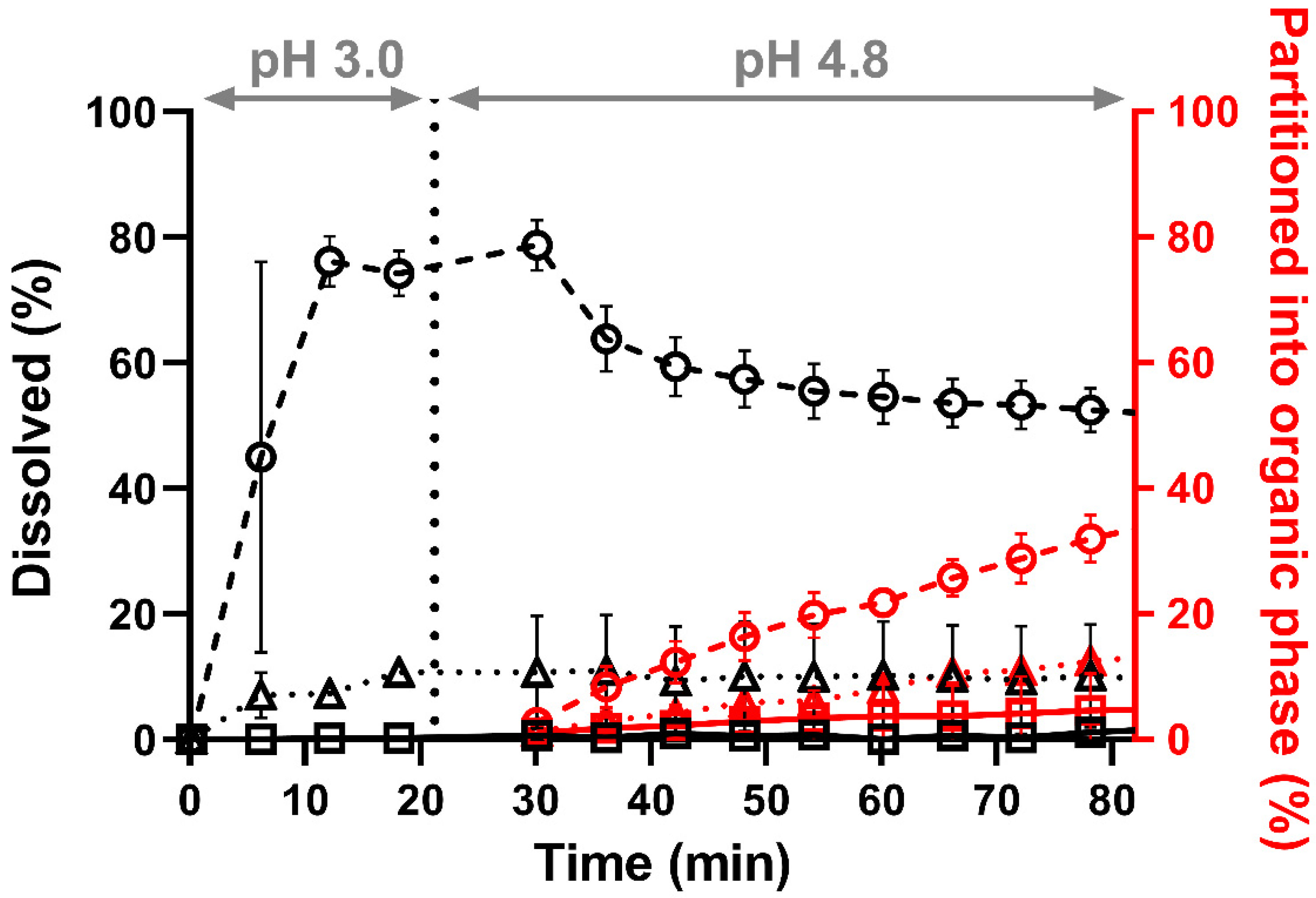
3.2.2. Biphasic Dissolution
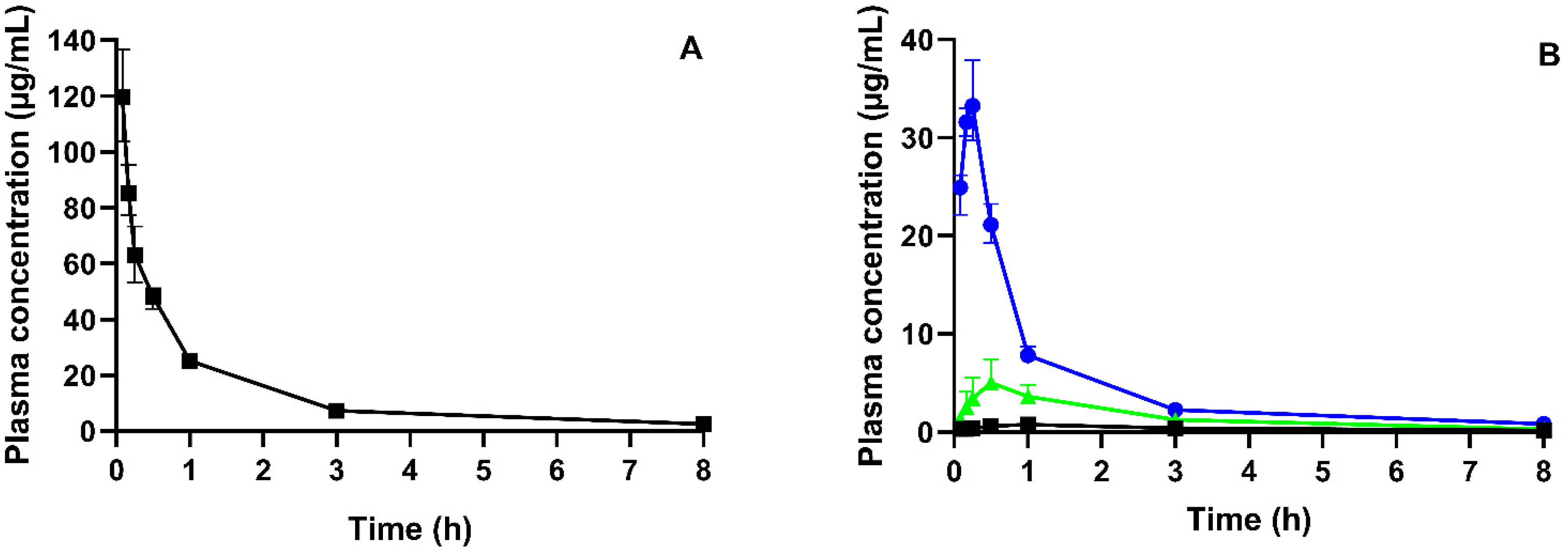
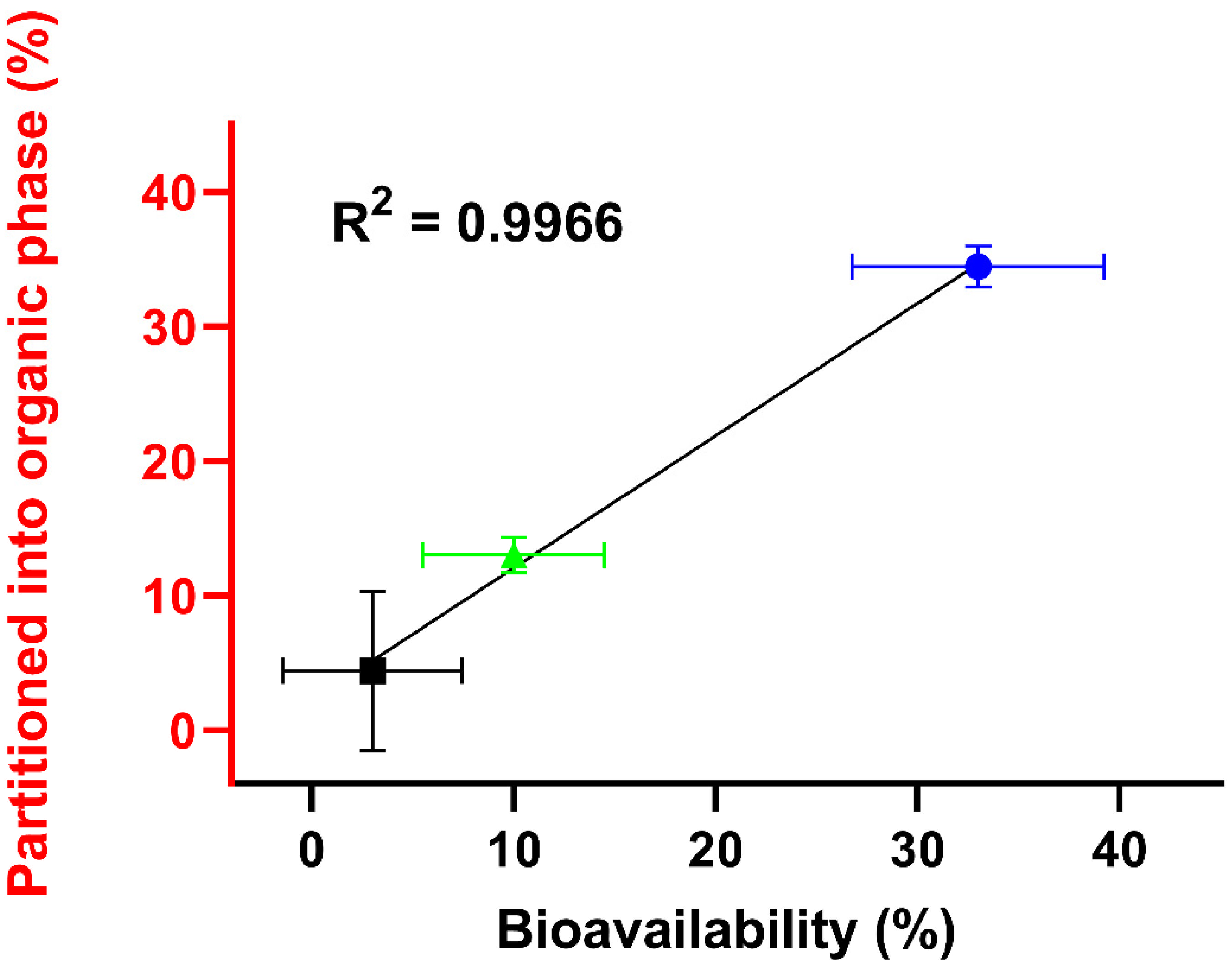
3.3. Pharmacokinetic Study
3.4. Cell Culture Experiments
3.4.1. Active Transport of CorA via P-gp and BCRP
3.4.2. Investigation of the Interaction with Hoechst 33342
3.5. PBPK Modeling
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Maas, J.; Kamm, W.; Hauck, G. An Integrated Early Formulation Strategy—From Hit Evaluation to Preclinical Candidate Profiling. Eur. J. Pharm. Biopharm. 2007, 66, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Balbach, S.; Korn, C. Pharmaceutical Evaluation of Early Development Candidates “the 100 Mg-Approach”. Int. J. Pharm. 2004, 275, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Saka, R.; Chella, N. Nanotechnology for Delivery of Natural Therapeutic Substances: A Review. Environ. Chem. Lett. 2021, 19, 1097–1106. [Google Scholar] [CrossRef]
- Theuretzbacher, U.; Outterson, K.; Engel, A.; Karlén, A. The Global Preclinical Antibacterial Pipeline. Nat. Rev. Microbiol. 2020, 18, 275–285. [Google Scholar] [CrossRef] [Green Version]
- Shahiwala, A. Formulation Approaches in Enhancement of Patient Compliance to Oral Drug Therapy. Expert Opin. Drug Deliv. 2011, 8, 1521–1529. [Google Scholar] [CrossRef]
- Li, H.K.; Agweyu, A.; English, M.; Bejon, P. An Unsupported Preference for Intravenous Antibiotics. PLoS Med. 2015, 12, e1001825. [Google Scholar] [CrossRef] [Green Version]
- Schäberle, T.F.; Schiefer, A.; Schmitz, A.; König, G.M.; Hoerauf, A.; Pfarr, K. Corallopyronin A—A Promising Antibiotic for Treatment of Filariasis. Int. J. Med. Microbiol. 2014, 304, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Schäberle, T.F.; Schmitz, A.; Zocher, G.; Schiefer, A.; Kehraus, S.; Neu, E.; Roth, M.; Vassylyev, D.G.; Stehle, T.; Bierbaum, G.; et al. Insights into Structure—Activity Relationships of Bacterial RNA Polymerase Inhibiting Corallopyronin Derivatives. J. Nat. Prod. 2015, 78, 2505–2509. [Google Scholar] [CrossRef]
- Schiefer, A.; Schmitz, A.; Schäberle, T.F.; Specht, S.; Lämmer, C.; Johnston, K.L.; Vassylyev, D.G.; König, G.M.; Hoerauf, A.; Pfarr, K. Corallopyronin A Specifically Targets and Depletes Essential Obligate Wolbachia Endobacteria From Filarial Nematodes In Vivo. J. Infect. Dis. 2012, 206, 249–257. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, A.; Kehraus, S.; Schab, T.F.; Ko, G.M. Corallorazines from the Myxobacterium Corallococcus Coralloides. J. Nat. Prod. 2014, 77, 163–195. [Google Scholar] [CrossRef]
- Rupp, J. Elaborations on Corallopyronin A as a Novel Treatment Strategy Against Genital Chlamydial Infections. Front. Microbiol. 2019, 10, 943. [Google Scholar]
- Schiefer, A.; Hübner, M.P.; Krome, A.; Lämmer, C.; Ehrens, A.; Aden, T.; Koschel, M.; Neufeld, H.; Chaverra-Muñoz, L.; Jansen, R.; et al. Corallopyronin A for Short-Course Anti-Wolbachial, Macrofilaricidal Treatment of Filarial Infections. PLoS Negl. Trop. Dis. 2020, 14, e0008930. [Google Scholar] [CrossRef] [PubMed]
- Krome, A.K.; Becker, T.; Kehraus, S.; Schiefer, A.; Gütschow, M.; Chaverra-Muñoz, L.; Hüttel, S.; Jansen, R.; Stadler, M.; Ehrens, A.; et al. Corallopyronin A: Antimicrobial Discovery to Preclinical Development. Nat. Prod. Rep. 2022. [Google Scholar] [CrossRef]
- Krome, A.K.; Becker, T.; Kehraus, S.; Schiefer, A.; Steinebach, C.; Aden, T.; Frohberger, S.J.; Mármol, Á.L.; Kapote, D.; Jansen, R.; et al. Solubility and Stability Enhanced Oral Formulations for the Anti-Infective Corallopyronin A. Pharmaceutics 2020, 12, 1105. [Google Scholar] [CrossRef]
- Zhao, M.; Lepak, A.J.; Andes, D.R. Animal Models in the Pharmacokinetic/Pharmacodynamic Evaluation of Antimicrobial Agents. Bioorganic Med. Chem. 2016, 24, 6390–6400. [Google Scholar] [CrossRef] [PubMed]
- Takimoto, C.H. Preclinical Drug Development. Princ. Clin. Pharmacol. 2010, 67, 578–585. [Google Scholar]
- Clark, J.M. The 3Rs in Research: A Contemporary Approach to Replacement, Reduction and Refinement. Br. J. Nutr. 2018, 120, S1–S7. [Google Scholar] [CrossRef]
- Zhuang, X.; Lu, C. PBPK Modeling and Simulation in Drug Research and Development. Acta Pharm. Sin. B 2016, 6, 430–440. [Google Scholar] [CrossRef] [Green Version]
- Grignard, E.; Taylor, R.; McAllister, M.; Box, K.; Fotaki, N. Considerations for the Development of in Vitro Dissolution Tests to Reduce or Replace Preclinical Oral Absorption Studies. Eur. J. Pharm. Sci. 2017, 99, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Bergström, C.A.S.; Holm, R.; Jørgensen, S.A.; Andersson, S.B.E.; Artursson, P.; Beato, S.; Borde, A.; Box, K.; Brewster, M.; Dressman, J.; et al. Early Pharmaceutical Profiling to Predict Oral Drug Absorption: Current Status and Unmet Needs. Eur. J. Pharm. Sci. 2014, 57, 173–199. [Google Scholar] [CrossRef]
- Hatton, G.B.; Yadav, V.; Basit, A.W.; Merchant, H.A. Animal Farm: Considerations in Animal Gastrointestinal Physiology and Relevance to Drug Delivery in Humans. J. Pharm. Sci. 2015, 104, 2747–2776. [Google Scholar] [CrossRef] [PubMed]
- Henze, L.J.; Koehl, N.J.; Jansen, R.; Holm, R.; Vertzoni, M.; Whitfield, P.D.; Griffin, B.T. Development and Evaluation of a Biorelevant Medium Simulating Porcine Gastrointestinal Fluids. Eur. J. Pharm. Biopharm. 2020, 154, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Markopoulos, C.; Andreas, C.J.; Vertzoni, M.; Dressman, J.; Reppas, C. In-Vitro Simulation of Luminal Conditions for Evaluation of Performance of Oral Drug Products: Choosing the Appropriate Test Media. Eur. J. Pharm. Biopharm. 2015, 93, 173–182. [Google Scholar] [CrossRef] [PubMed]
- McConnell, E.L.; Basit, A.W.; Murdan, S. Measurements of Rat and Mouse Gastrointestinal PH, Fluid and Lymphoid Tissue, and Implications for in-Vivo Experiments. J. Pharm. Pharmacol. 2008, 60, 63–70. [Google Scholar] [CrossRef]
- Koziolek, M.; Grimm, M.; Becker, D.; Iordanov, V.; Zou, H.; Shimizu, J.; Wanke, C.; Garbacz, G.; Weitschies, W. Investigation of PH and Temperature Profiles in the GI Tract of Fasted Human Subjects Using the Intellicap® System. J. Pharm. Sci. 2015, 104, 2855–2863. [Google Scholar] [CrossRef]
- Woting, A.; Blaut, M. Small Intestinal Permeability and Gut-Transit Time Determined with Low and High Molecular Weight Fluorescein Isothiocyanate-Dextrans in C3H Mice. Nutrients 2018, 10, 685. [Google Scholar] [CrossRef] [Green Version]
- DeSesso, J.M.; Williams, A.L. Contrasting the Gastrointestinal Tracts of Mammals: Factors That Influence Absorption. In Annual Reports in Medicinal Chemistry; Elsevier: Amsterdam, The Netherlands, 2008; Volume 43, pp. 353–371. ISBN 978-0-12-374344-2. [Google Scholar]
- Christfort, J.F.; Strindberg, S.; Plum, J.; Hall-Andersen, J.; Janfelt, C.; Nielsen, L.H.; Müllertz, A. Developing a Predictive in Vitro Dissolution Model Based on Gastrointestinal Fluid Characterisation in Rats. Eur. J. Pharm. Biopharm. 2019, 142, 307–314. [Google Scholar] [CrossRef]
- McIlvaine, T.C. A Buffer Solution for Colorimetric Comparison. J. Biol. Chem. 1921, 49, 183–186. [Google Scholar] [CrossRef]
- Jang, S.-F.; Goins, B.A.; Phillips, W.T.; Santoyo, C.; Rice-Ficht, A.; McConville, J.T. Size Discrimination in Rat and Mouse Gastric Emptying: Size Discrimination in Gastric Emptying. Biopharm. Drug Dispos. 2013, 34, 107–124. [Google Scholar] [CrossRef]
- Denninger, A.; Westedt, U.; Rosenberg, J.; Wagner, K.G. A Rational Design of a Biphasic DissolutionSetup—Modelling of Biorelevant Kinetics for a Ritonavir Hot-Melt Extruded Amorphous Solid Dispersion. Pharmaceutics 2020, 12, 237. [Google Scholar] [CrossRef] [Green Version]
- Denninger, A.; Westedt, U.; Wagner, K.G. Shared IVIVR for Five Commercial Enabling Formulations Using the BiPHa + Biphasic Dissolution Assay. Pharmaceutics 2021, 13, 285. [Google Scholar] [CrossRef] [PubMed]
- Krapf, M.K.; Gallus, J.; Vahdati, S.; Wiese, M. New Inhibitors of Breast Cancer Resistance Protein (ABCG2) Containing a 2,4-Disubstituted Pyridopyrimidine Scaffold. J. Med. Chem. 2018, 61, 3389–3408. [Google Scholar] [CrossRef] [PubMed]
- Langenbucher, F. Letters to the Editor: Linearization of Dissolution Rate Curves by the Weibull Distribution. J. Pharm. Pharmacol. 2011, 24, 979–981. [Google Scholar] [CrossRef]
- Guideline on the Pharmacokinetic and Clinical Evaluation of Modified Release Dosage Forms. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-pharmacokinetic-clinical-evaluation-modified-release-dosage-forms_en.pdf (accessed on 15 May 2022).
- Emami, J. In vitro—in vivo Correlation: From Theory to Applications. J. Pharm. Pharm. Sci. 2006, 9, 169–189. [Google Scholar] [PubMed]
- Gajdács, M. The Concept of an Ideal Antibiotic: Implications for Drug Design. Molecules 2019, 24, 892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Science, P. Intravenous to Oral Antibiotic Switch Therapy. Drugs Today 2001, 37, 311–319. [Google Scholar]
- Kararli, T.T. Comparison of the Gastrointestinal Anatomy, Physiology, and Biochemistry of Humans and Commonly Used Laboratory Animals. Biopharm. Drug Dispos. 1995, 16, 351–380. [Google Scholar] [CrossRef]
- Dahlgren, D.; Venczel, M.; Ridoux, J.-P.; Skjöld, C.; Müllertz, A.; Holm, R.; Augustijns, P.; Hellström, P.M.; Lennernäs, H. Fasted and Fed State Human Duodenal Fluids: Characterization, Drug Solubility, and Comparison to Simulated Fluids and with Human Bioavailability. Eur. J. Pharm. Biopharm. 2021, 163, 240–251. [Google Scholar] [CrossRef]
- López Mármol, Á.; Denninger, A.; Touzet, A.; Dauer, K.; Becker, T.; Pöstges, F.; Pellequer, Y.; Lamprecht, A.; Wagner, K.G. The Relevance of Supersaturation and Solubilization in the Gastrointestinal Tract for Oral Bioavailability: An in vitro vs. in vivo Approach. Int. J. Pharm. 2021, 603, 120648. [Google Scholar] [CrossRef]
- Jambhekar, S.S.; Breen, P.J. Drug Dissolution: Significance of Physicochemical Properties and Physiological Conditions. Drug Discov. Today 2013, 18, 1173–1184. [Google Scholar] [CrossRef]
- Jamei, M.; Abrahamsson, B.; Brown, J.; Bevernage, J.; Bolger, M.B.; Heimbach, T.; Karlsson, E.; Kotzagiorgis, E.; Lindahl, A.; McAllister, M.; et al. Current Status and Future Opportunities for Incorporation of Dissolution Data in PBPK Modeling for Pharmaceutical Development and Regulatory Applications: OrBiTo Consortium Commentary. Eur. J. Pharm. Biopharm. 2020, 155, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Tsume, Y.; Langguth, P.; Garcia-Arieta, A.; Amidon, G.L. In Silico Prediction of Drug Dissolution and Absorption with Variation in Intestinal PH for BCS Class II Weak Acid Drugs: Ibuprofen and Ketoprofen: In silico Drug Dissolution and Absorption for BCS Class ii Weak Acids. Biopharm. Drug Dispos. 2012, 33, 366–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varum, F.J.O.; Merchant, H.A.; Basit, A.W. Oral Modified-Release Formulations in Motion: The Relationship between Gastrointestinal Transit and Drug Absorption. Int. J. Pharm. 2010, 395, 26–36. [Google Scholar] [CrossRef]
- Estudante, M.; Morais, J.G.; Soveral, G.; Benet, L.Z. Intestinal Drug Transporters: An Overview. Adv. Drug Deliv. Rev. 2013, 65, 1340–1356. [Google Scholar] [CrossRef]
- Pathak, S.M.; Schaefer, K.J.; Jamei, M.; Turner, D.B. Biopharmaceutic IVIVE—Mechanistic Modeling of Single- and Two-Phase In Vitro Experiments to Obtain Drug-Specific Parameters for Incorporation Into PBPK Models. J. Pharm. Sci. 2019, 108, 1604–1618. [Google Scholar] [CrossRef] [Green Version]
- O’Shea, J.P.; Augustijns, P.; Brandl, M.; Brayden, D.J.; Brouwers, J.; Griffin, B.T.; Holm, R.; Jacobsen, A.-C.; Lennernäs, H.; Vinarov, Z.; et al. Best Practices in Current Models Mimicking Drug Permeability in the Gastrointestinal Tract—An UNGAP Review. Eur. J. Pharm. Sci. 2022, 170, 106098. [Google Scholar] [CrossRef]
- Kostewicz, E.S.; Aarons, L.; Bergstrand, M.; Bolger, M.B.; Galetin, A.; Hatley, O.; Jamei, M.; Lloyd, R.; Pepin, X.; Rostami-Hodjegan, A.; et al. PBPK Models for the Prediction of in Vivo Performance of Oral Dosage Forms. Eur. J. Pharm. Sci. 2014, 57, 300–321. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FaSSIF-V2- Human | FeSSIF-V2- Human | FaSSIF-V2- Mouse | FeSSIF-V2- Mouse | |
|---|---|---|---|---|
| Lecithin (mM) | 3.0 | 10 | 3.0 | 10 |
| Sodium taurocholate (mM) | 0.2 | 2 | 0.2 | 2 |
| Glyceryl monooleate (mM) | 5 | 5 | ||
| Sodium monooleate (mM) | 0.8 | 0.8 | ||
| Sodium chloride (mM) | 68.6 | 125.5 | 68.6 | 125.5 |
| Sodium hydroxide (mM) | 34.8 | 102.4 | 34.8 | 102.4 |
| Maleic acid (mM) | 19.1 | 71.9 | 19.1 | 71.9 |
| Sodium hydroxide 1 N | q.s.; pH 6.5 | q.s.; pH 5.8 | ||
| Hydrochloric acid 1 N | q.s.; pH 5.2 | q.s.; pH 4.8 | ||
| Buffer capacity (mM/∆pH) | 10 | 25 | 10 | 25 |
| Osmolality (mOsm/kg) | 180 | 390 | 180 | 390 |
| pH | 6.5 | 5.8 | 5.2 | 4.8 |
| Reagents | FeSSIF-V2-like |
|---|---|
| Lecithin (mM) | 2 |
| Sodium taurocholate (mM) | 10 |
| Glyceryl monooleate (mM) | 5 |
| Sodium monooleate (mM) | 0.8 |
| Sodium chloride (mM) | 25.0 |
| Potassium citrate (mM) | 10 |
| Potassium phosphate (mM) | 4.3 |
| Sodium hydroxide (mM) | 10 |
| Physicochemical characteristics | |
| Buffer capacity (mM/∆pH) | 25.2 |
| Osmolality (mOsm/kg) | 389 |
| pH | 4.8 |
| (A) FaSSIF-V2 pH 6.5 (Human) | Solubility after 2 min (mg/mL) | Solubility after 4 h (mg/mL) |
| Neat CorA | 0.00 (±0.00) | 0.25 (±0.06) |
| CorA-povidone | 1.09 (±0.18) | 1.24 (±0.04) |
| CorA-copovidone | 0.96 (±0.28) | 1.40 (±0.14) |
| (B) FeSSIF-V2 pH 5.8 (Human) | Solubility after 2 min (mg/mL) | Solubility after 4 h (mg/mL) |
| Neat CorA | 0.00 (±0.00) | 0.26 (±0.01) |
| CorA-povidone | 1.06 (±0.02) | 0.94 (±0.07) |
| CorA-copovidone | 0.67 (±0.09) | 0.93 (±0.05) |
| (C) FaSSIF-V2-Mouse pH 5.2 | Solubility after 2 min (mg/mL) | Solubility after 1 h (mg/mL) |
| Neat CorA | 0.00 (±0.00) | 0.01 (±0.01) |
| CorA-povidone | 0.12 (±0.01) | 0.07 (±0.01) |
| CorA-copovidone | 0.09 (±0.01) | 0.19 (±0.01) |
| (D) FeSSIF-V2-Mouse pH 4.8 | Solubility after 2 min (mg/mL) | Solubility after 1 h (mg/mL) |
| Neat CorA | 0.00 (±0.00) | 0.02 (±0.01) |
| CorA-povidone | 0.66 (±0.05) | 0.54 (±0.01) |
| CorA-copovidone | 0.45 (±0.09) | 0.44 (±0.08) |
| Weibull Parameter | CorA-Povidone | CorA-Copovidone |
|---|---|---|
| Maximum of released API (%) | 65.43 | 16.78 |
| Time lag (h) | 0 | 0 |
| Shape factor | 3.73 | 0.85 |
| Time scale (hb) | 2 × 10−4 | 0.47 |
| R2 | 0.92 | 0.92 |
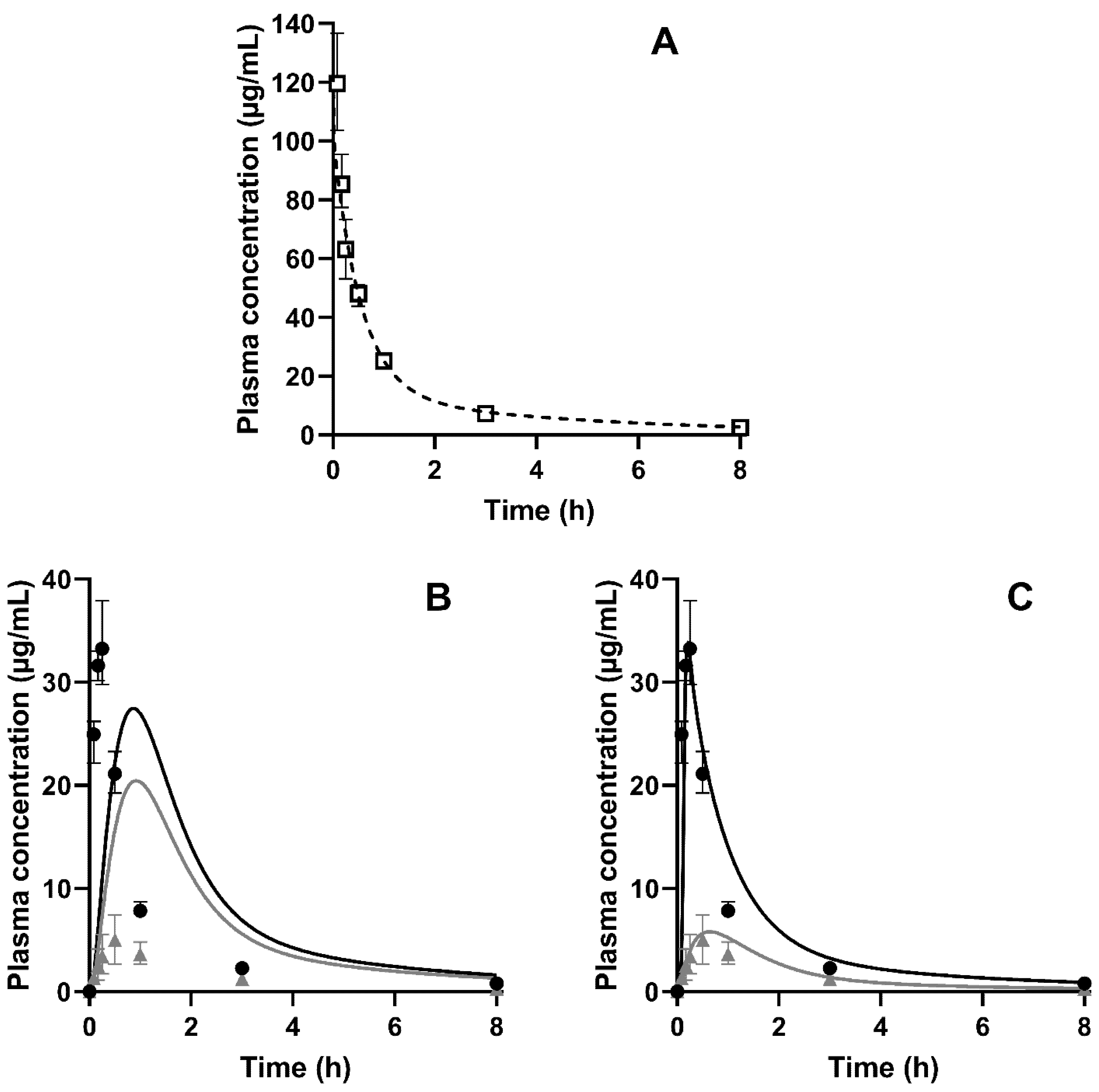
| AUC0–inf (µg·h/mL) | Cmax (µg/mL) | Tmax (min) | BA (%) | |
|---|---|---|---|---|
| Reference of CorA–IV # | 127.7 (110.2–149.0) | 119.6 (103.7–136.7) | 5 * | 100 ** |
| Neat CorA | 4.5 (3.8–7.4) | 0.9 (0.6–1.1) | 60 (53–90) | 3 |
| CorA-povidone | 41.9 (39.6–46.5) | 33.2 (30.3–37.9) | 15 (10–15) | 33 |
| CorA-copovidone | 13.4 (11.4–17.4) | 5.0 (3.0–7.4) | 30 (30–38) | 10 |
| Parameters | CorA-Povidone | ||||
| Observed | Predicted (w/o Dissolution) | PE (%) (w/o Dissolution) | Predicted (w/ Dissolution) | PE (%) (w/ Dissolution) | |
| Cmax (µg/mL) | 33.24 | 27.43 | 21.18 | 33.80 | −1.68 |
| AUC0–inf (µg·h/mL) | 41.83 | 64.63 | −54.51 | 46.44 | −11.02 |
| Parameters | CorA-Copovidone | ||||
| Observed | Predicted (w/o Dissolution) | PE (%) (w/o Dissolution) | Predicted (w/ Dissolution) | PE (%) (w/ Dissolution) | |
| Cmax (µg/mL) | 5.00 | 20.43 | −308.60 | 5.80 | −16.00 |
| AUC0–inf (µg·h/mL) | 13.35 | 56.60 | −323.97 | 15.00 | −12.36 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Becker, T.; Krome, A.K.; Vahdati, S.; Schiefer, A.; Pfarr, K.; Ehrens, A.; Aden, T.; Grosse, M.; Jansen, R.; Alt, S.; et al. In Vitro–In Vivo Relationship in Mini-Scale—Enabling Formulations of Corallopyronin A. Pharmaceutics 2022, 14, 1657. https://doi.org/10.3390/pharmaceutics14081657
Becker T, Krome AK, Vahdati S, Schiefer A, Pfarr K, Ehrens A, Aden T, Grosse M, Jansen R, Alt S, et al. In Vitro–In Vivo Relationship in Mini-Scale—Enabling Formulations of Corallopyronin A. Pharmaceutics. 2022; 14(8):1657. https://doi.org/10.3390/pharmaceutics14081657
Chicago/Turabian StyleBecker, Tim, Anna K. Krome, Sahel Vahdati, Andrea Schiefer, Kenneth Pfarr, Alexandra Ehrens, Tilman Aden, Miriam Grosse, Rolf Jansen, Silke Alt, and et al. 2022. "In Vitro–In Vivo Relationship in Mini-Scale—Enabling Formulations of Corallopyronin A" Pharmaceutics 14, no. 8: 1657. https://doi.org/10.3390/pharmaceutics14081657
APA StyleBecker, T., Krome, A. K., Vahdati, S., Schiefer, A., Pfarr, K., Ehrens, A., Aden, T., Grosse, M., Jansen, R., Alt, S., Hesterkamp, T., Stadler, M., Hübner, M. P., Kehraus, S., König, G. M., Hoerauf, A., & Wagner, K. G. (2022). In Vitro–In Vivo Relationship in Mini-Scale—Enabling Formulations of Corallopyronin A. Pharmaceutics, 14(8), 1657. https://doi.org/10.3390/pharmaceutics14081657