
Selective Targeting and Eradication of Various Human Non-Small Cell Lung Cancer Cell Lines Using Self-Assembled Aptamer-Decorated Nanoparticles



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Cultures
2.3. Methods
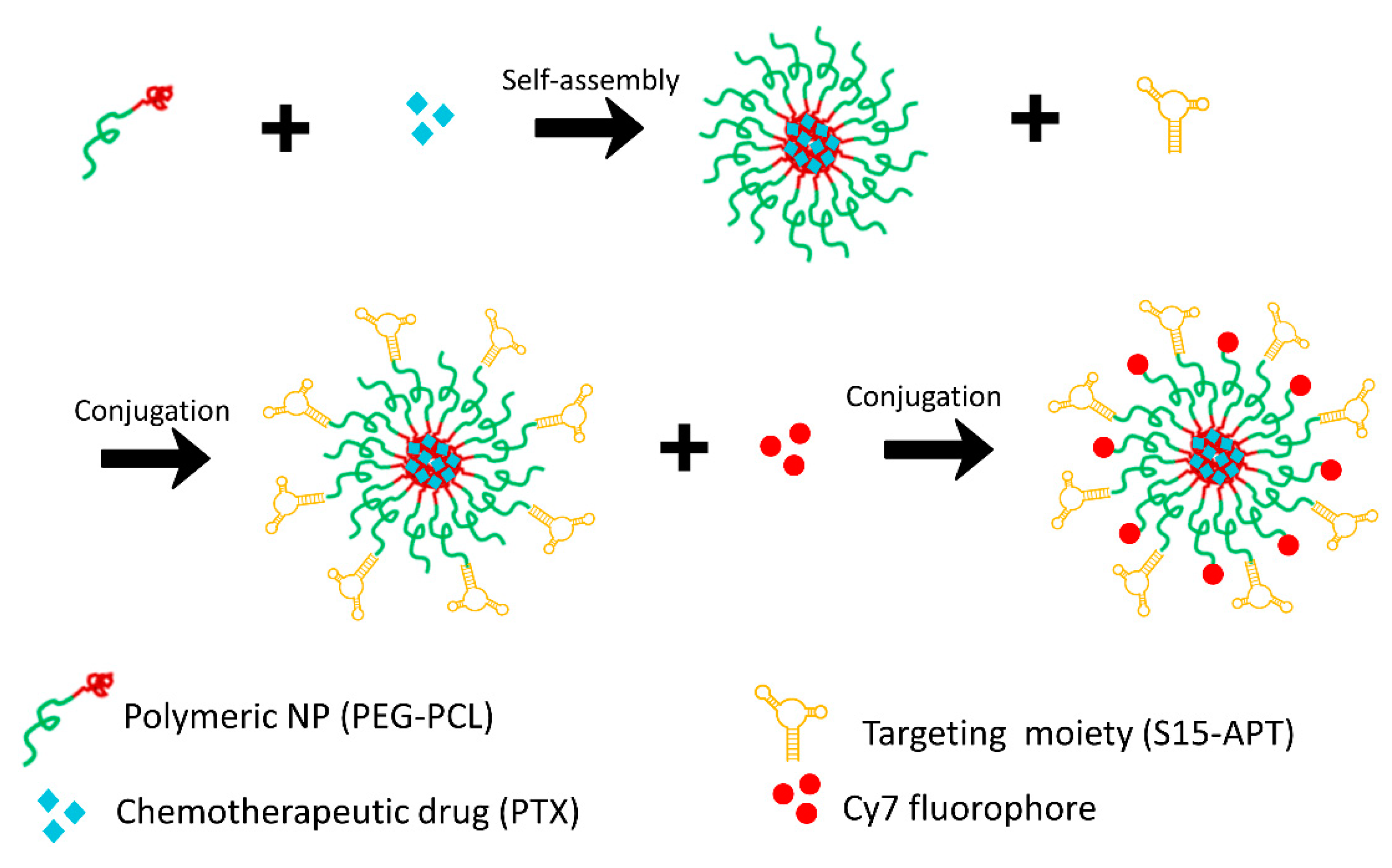
2.3.1. Preparation of Cy7-Labeled APT-NPs
2.3.2. Particle Size Distribution Analysis and Cryo-TEM Imaging
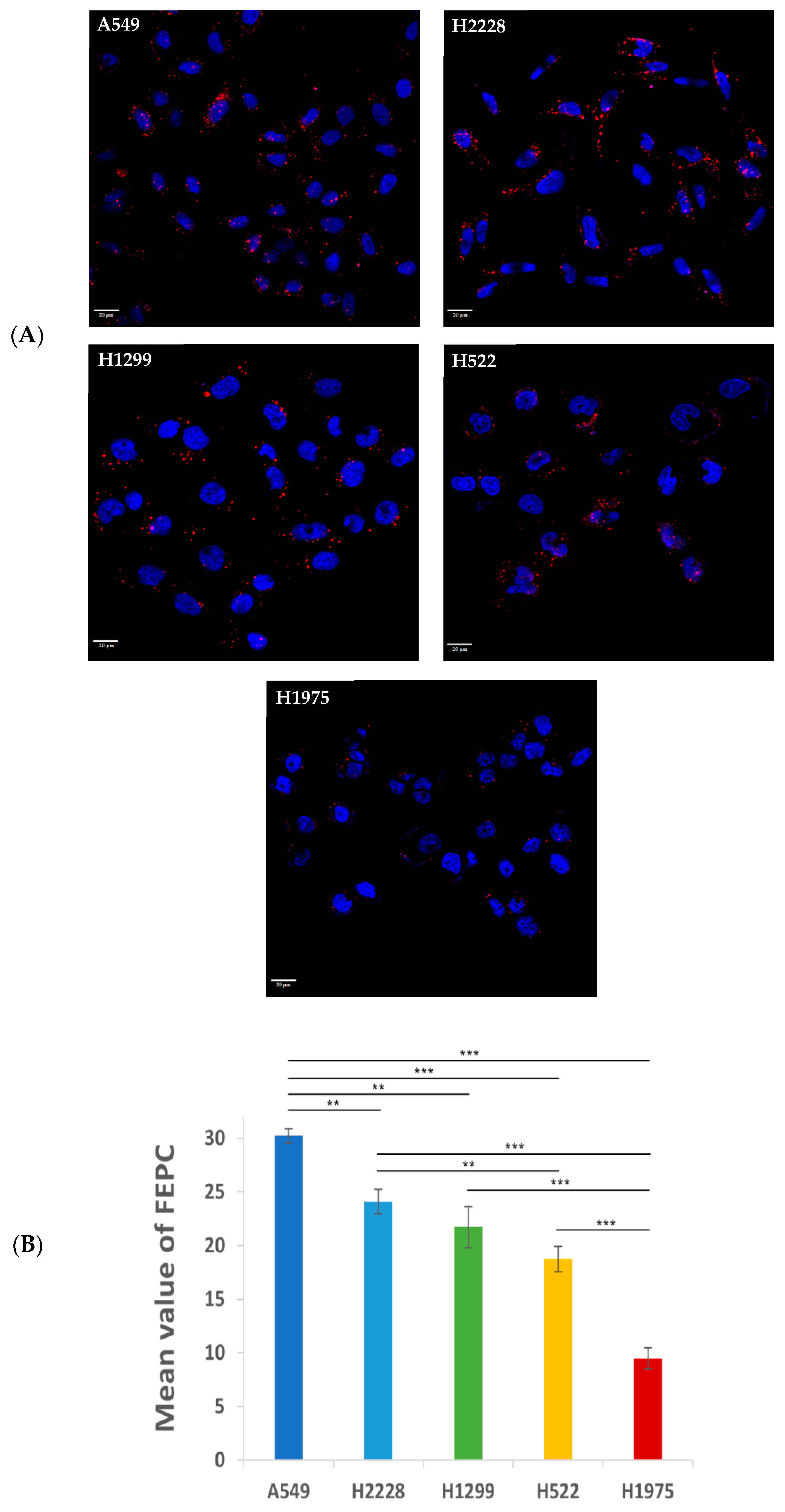
2.3.3. Characterization and Comparison of the Specificity of APT-NPs to Various NSCLC Cell Lines Using Confocal Laser Microscopy
2.3.4. Image Analysis
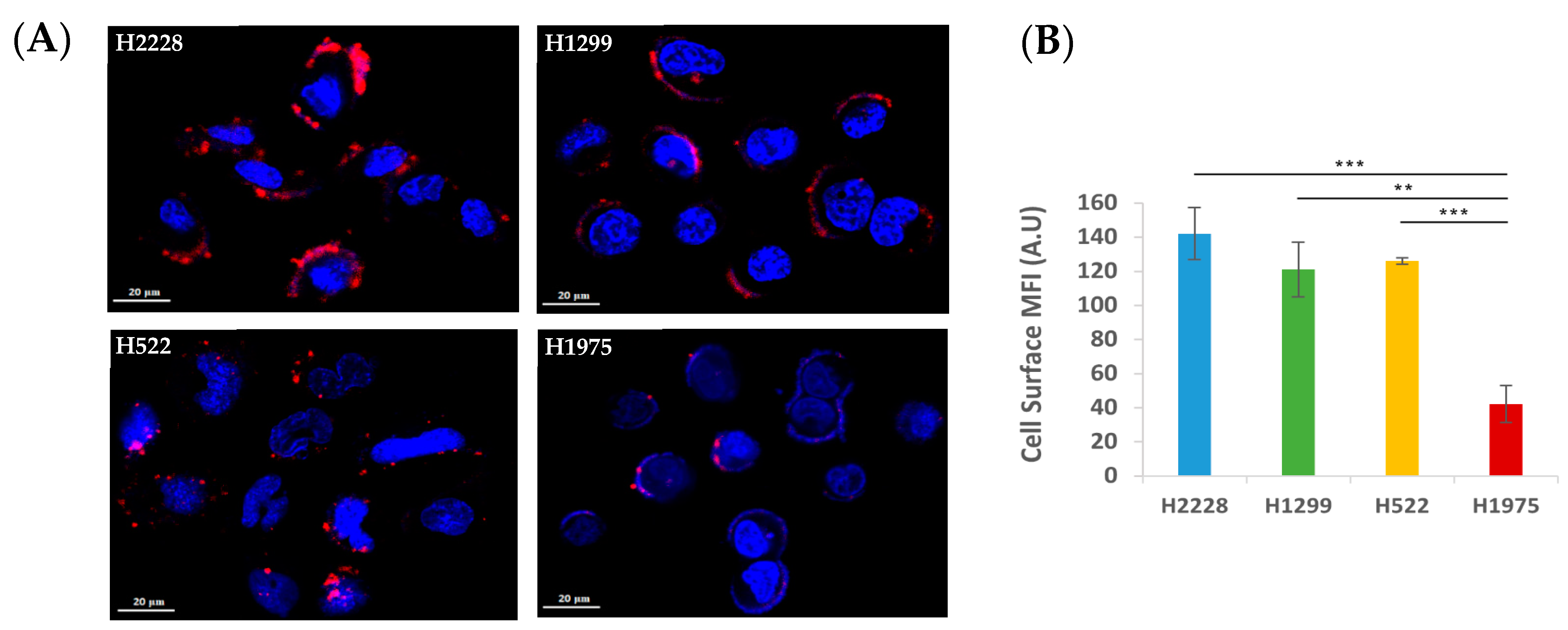
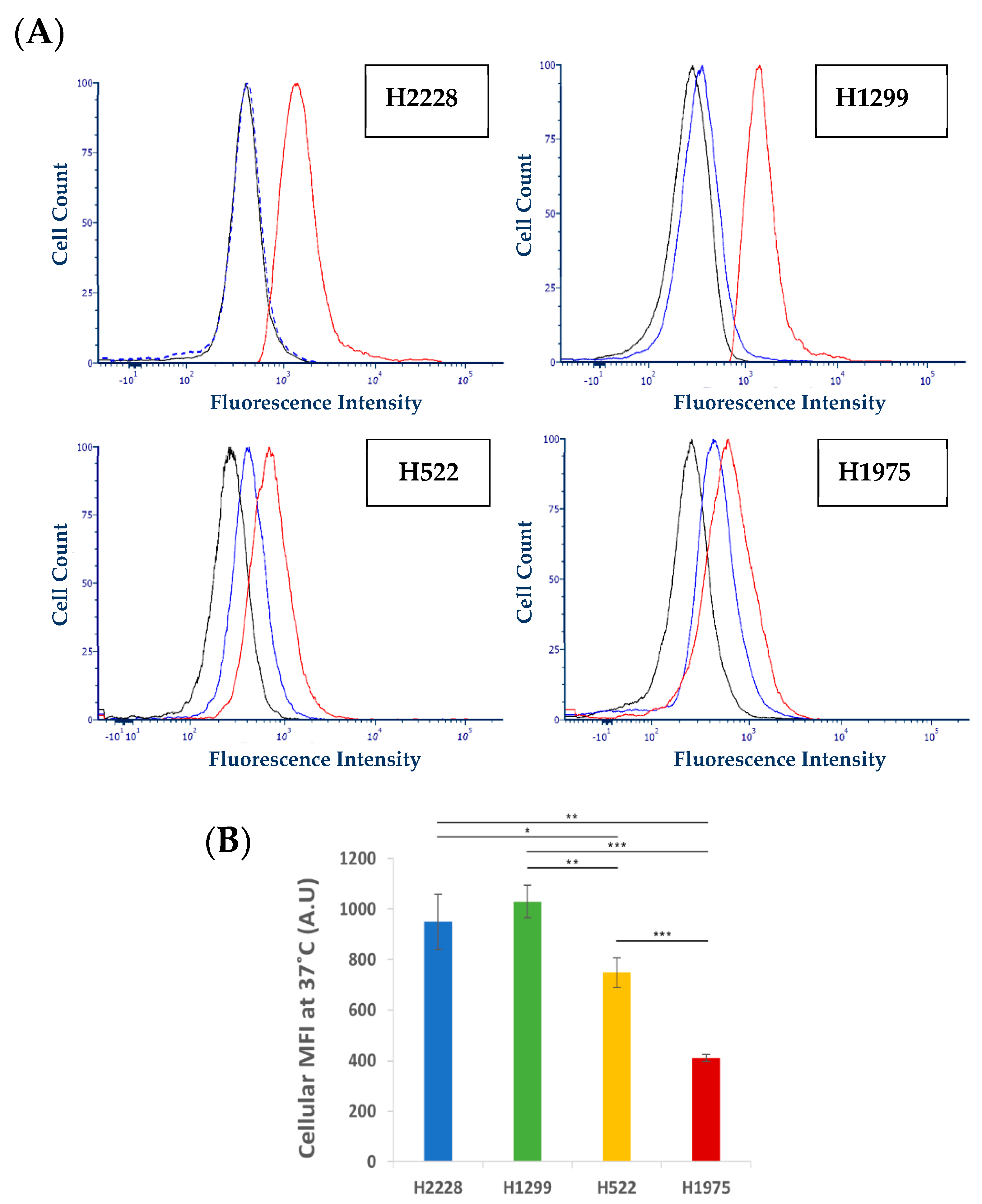
2.3.5. Characterization of Active S15 APT-NPs Internalization by NSCLC Cell Lines
Conjugation of QDs
Flow Cytometric Analysis
2.3.6. Cytotoxicity Assays
Preparation of Aptamer-Decorated Nanoparticles Harboring a Cytotoxic Drug
Cytotoxicity Assay
3. Results and Discussion
3.1. Size Distribution of NPs and Cryogenic Transmission Electron Microscopy (Cryo-TEM) Analysis
3.2. Specificity of Binding and Internalization of APT-NPs in Various Human NSCLC Cell Lines
3.3. Further Exploration of the Selective Binding and Internalization of the S15-Decorated NPs
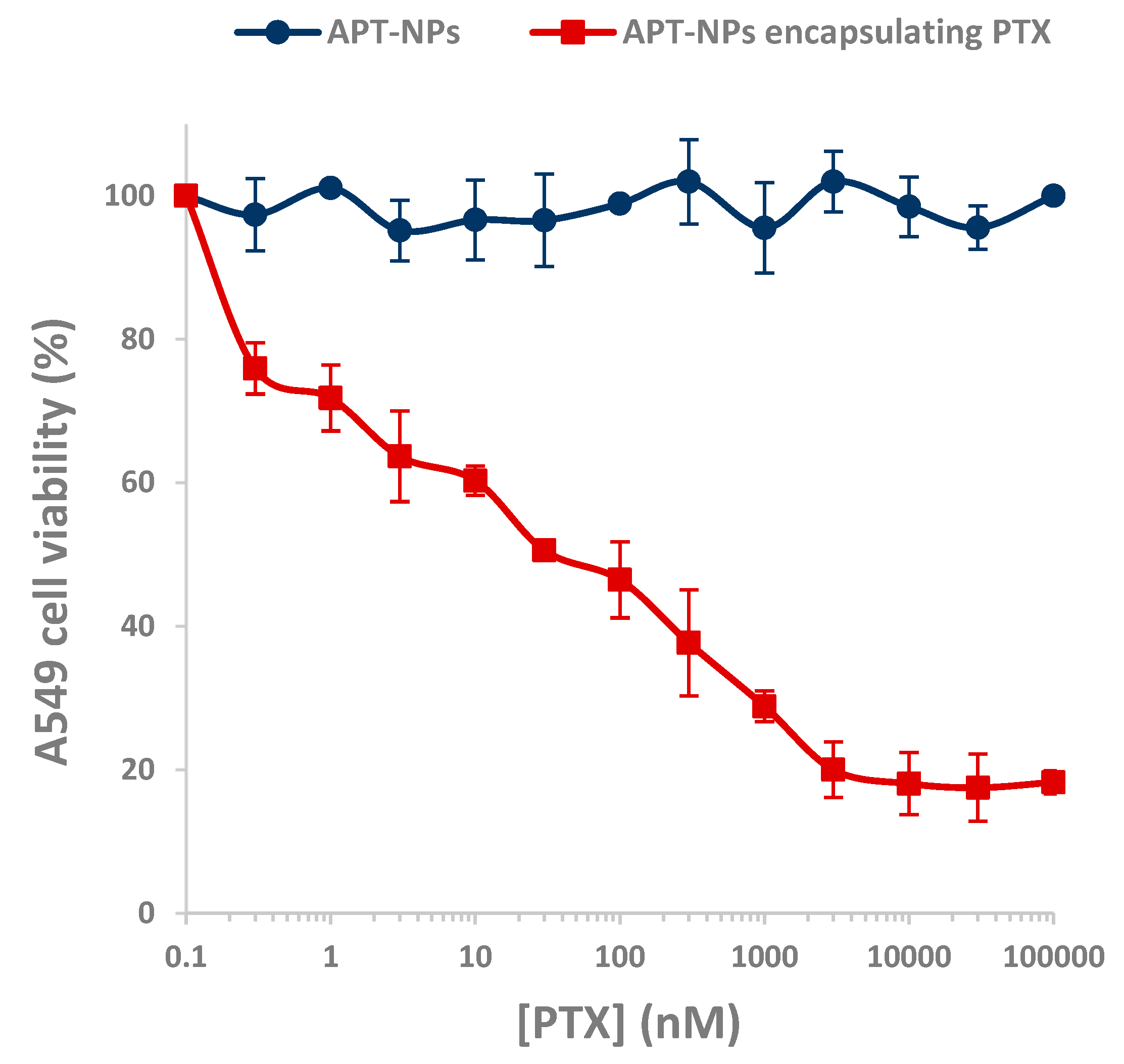
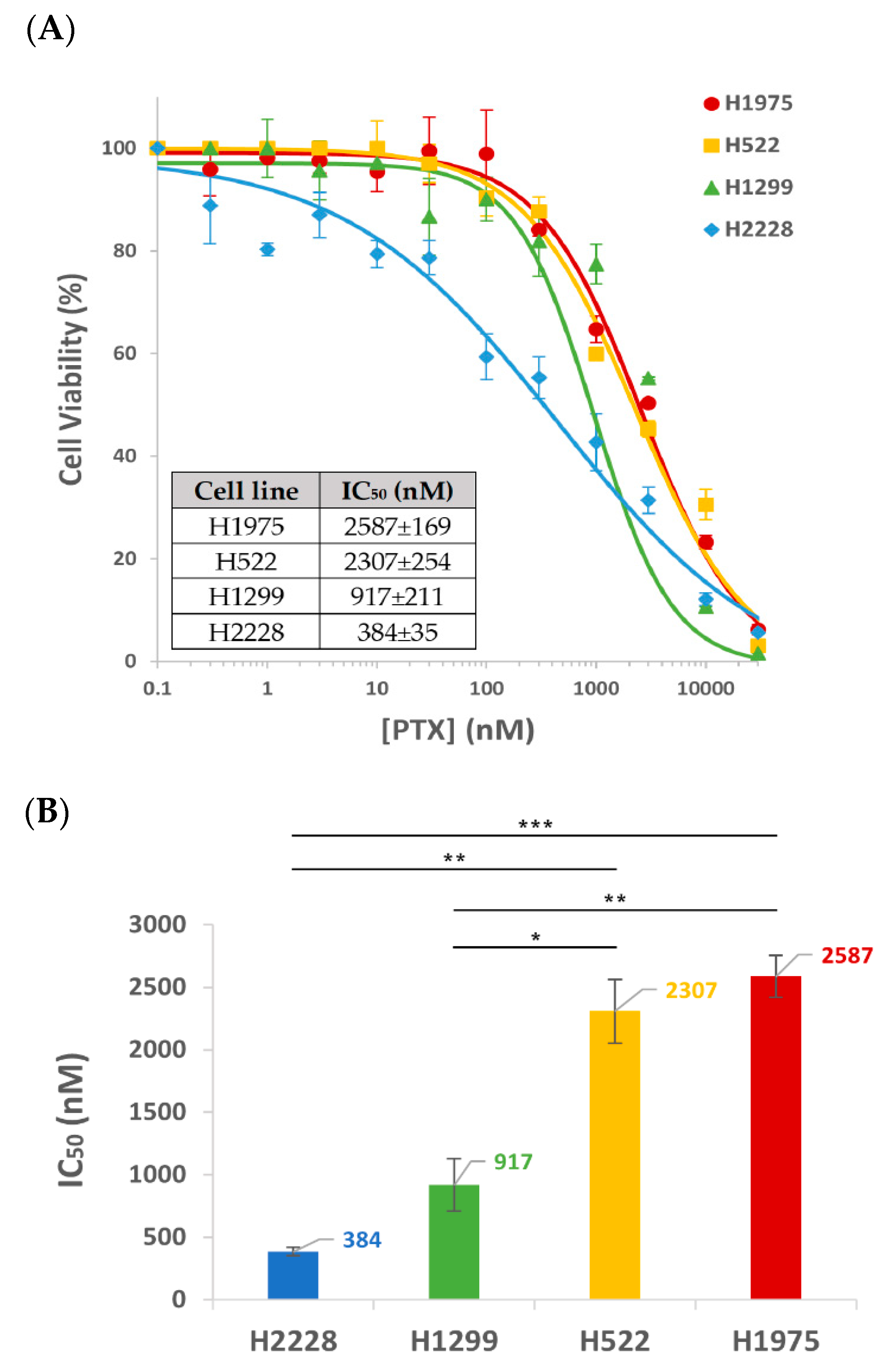
3.4. Cytotoxicity of APT-NPs to NSCLC Cell Lines
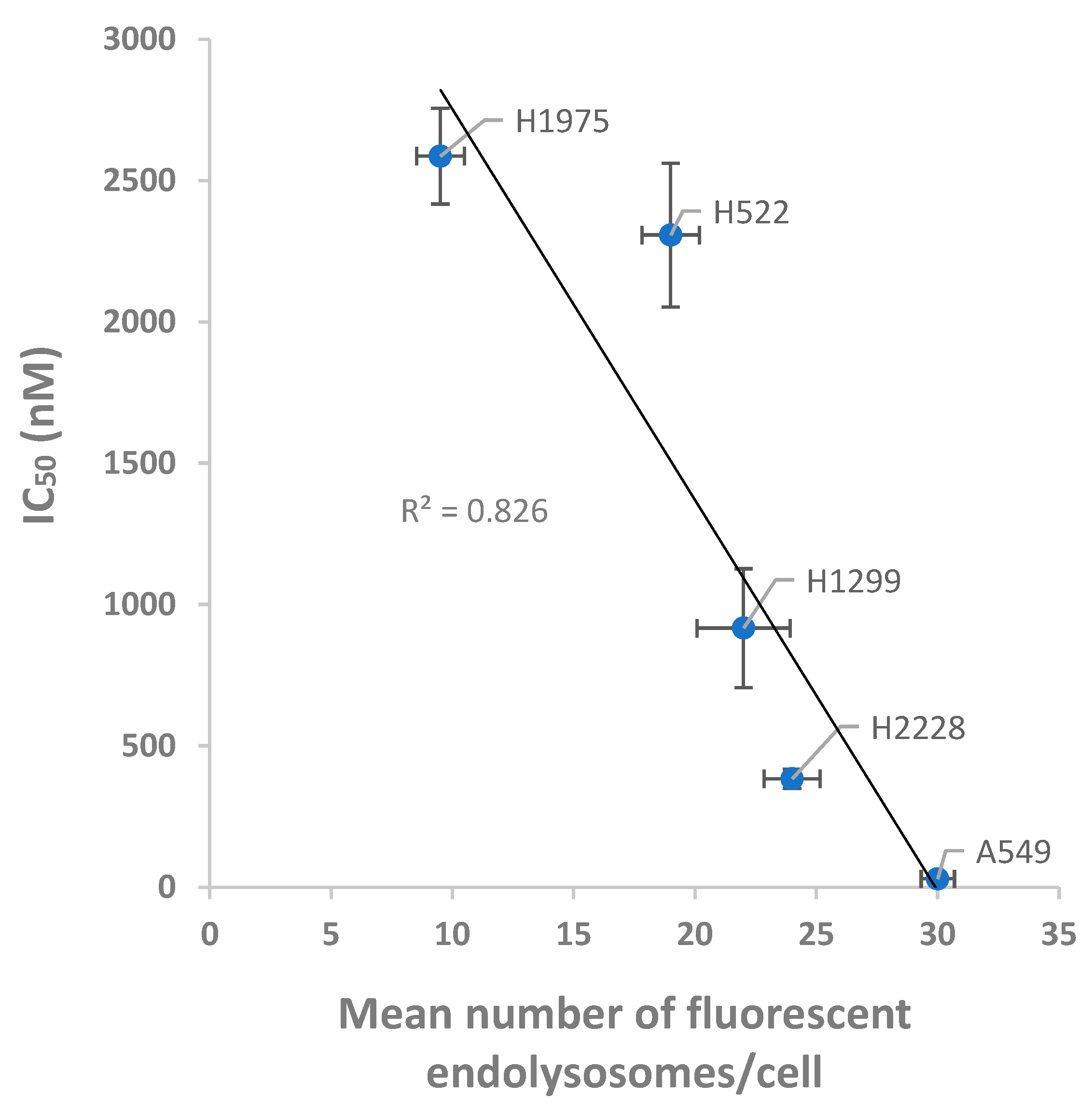
3.5. Relation between the Internalization and Cytotoxic Effect of PTX-Loaded NPs on NSCLC Cells
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schabath, M.B.; Cote, M.L. Cancer Progress and Priorities: Lung Cancer. Cancer Epidemiol. Biomarkers Prev. 2019, 28, 1563–1579. [Google Scholar] [CrossRef] [PubMed]
- Zappa, C.; Mousa, S.A. Non-Small Cell Lung Cancer: Current Treatment and Future Advances. Transl. Lung Cancer Res. 2016, 5, 288–300. [Google Scholar] [CrossRef] [PubMed]
- de Groot, P.M.; Wu, C.C.; Carter, B.W.; Munden, R.F. The Epidemiology of Lung Cancer. Transl. Lung Cancer Res. 2018, 7, 220–233. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Friedlaender, A.; Addeo, A.; Russo, A.; Gregorc, V.; Cortinovis, D.; Rolfo, C.D. Targeted Therapies in Early Stage NSCLC: Hype or Hope? Int. J. Mol. Sci. 2020, 21, 6329. [Google Scholar] [CrossRef]
- Chen, F.; Zhang, Y.; Parra, E.; Rodriguez, J.; Behrens, C.; Akbani, R.; Lu, Y.; Kurie, J.M.; Gibbons, D.L.; Mills, G.B.; et al. Multiplatform-Based Molecular Subtypes of Non-Small-Cell Lung Cancer. Oncogene 2017, 36, 1384–1393. [Google Scholar] [CrossRef] [PubMed]
- Hyun, S.H.; Ahn, M.S.; Koh, Y.W.; Lee, S.J. A Machine-Learning Approach Using PET-Based Radiomics to Predict the Histological Subtypes of Lung Cancer. Clin. Nucl. Med. 2019, 44, 956–960. [Google Scholar] [CrossRef]
- Islas-Muñoz, B.; Volkow-Fernández, P.; Ibanes-Gutiérrez, C.; Villamar-Ramírez, A.; Vilar-Compte, D.; Cornejo-Juárez, P. Bloodstream Infections in Cancer Patients. Risk Factors Associated with Mortality. Int. J. Infect. Dis. 2018, 71, 59–64. [Google Scholar] [CrossRef]
- Bernabeu, E.; Cagel, M.; Lagomarsino, E.; Moretton, M.; Chiappetta, D.A. Paclitaxel: What Has Been Done and the Challenges Remain Ahead. Int. J. Pharm. 2017, 526, 474–495. [Google Scholar] [CrossRef]
- Mosca, L.; Ilari, A.; Fazi, F.; Assaraf, Y.G.; Colotti, G. Taxanes in Cancer Treatment: Activity, Chemoresistance and Its Overcoming. Drug Resist. Updates 2021, 54, 100742. [Google Scholar] [CrossRef]
- Das, T.; Anand, U.; Pandey, S.K.; Ashby, C.R.; Assaraf, Y.G.; Chen, Z.S.; Dey, A. Therapeutic Strategies to Overcome Taxane Resistance in Cancer. Drug Resist. Updates 2021, 55, 100754. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.; Li, S.; Li, W.; Xie, M.; Wei, Z.; Guo, H.; Wei, W.; Zhang, S. Disruption of Protein Neddylation with MLN4924 Attenuates Paclitaxel-Induced Apoptosis and Microtubule Polymerization in Ovarian Cancer Cells. Biochem. Biophys. Res. Commun. 2019, 508, 986–990. [Google Scholar] [CrossRef] [PubMed]
- Vermeer, C.J.C.; Hiensch, A.E.; Cleenewerk, L.; May, A.M.; Eijkelkamp, N. Neuro-Immune Interactions in Paclitaxel-Induced Peripheral Neuropathy. Acta Oncol. 2021, 60, 1369–1382. [Google Scholar] [CrossRef] [PubMed]
- Babu, A.; Templeton, A.K.; Munshi, A.; Ramesh, R. Nanoparticle-Based Drug Delivery for Therapy of Lung Cancer: Progress and Challenges. J. Nanomater. 2013, 2013, 14. [Google Scholar] [CrossRef]
- Jadhav, S.R.; Bryant, G.; Mata, J.P.; Eldridge, D.S.; Palombo, E.A.; Harding, I.H.; Shah, R.M. Structural Aspects of a Self-Emulsifying Multifunctional Amphiphilic Excipient: Part II. The Case of Cremophor EL. J. Mol. Liq. 2021, 344, 117881. [Google Scholar] [CrossRef]
- Zeng, L.; Xin, X.; Zhang, Y. Development and Characterization of Promising Cremophor EL-Stabilized o/w Nanoemulsions Containing Short-Chain Alcohols as a Cosurfactant. RSC Adv. 2017, 7, 19815–19827. [Google Scholar] [CrossRef]
- Calzoni, E.; Cesaretti, A.; Polchi, A.; Di Michele, A.; Tancini, B.; Emiliani, C. Biocompatible Polymer Nanoparticles for Drug Delivery Applications in Cancer and Neurodegenerative Disorder Therapies. J. Funct. Biomater. 2019, 10, 4. [Google Scholar] [CrossRef]
- Su, Z.; Dong, S.; Zhao, S.C.; Liu, K.; Tan, Y.; Jiang, X.; Assaraf, Y.G.; Qin, B.; Chen, Z.S.; Zou, C. Novel Nanomedicines to Overcome Cancer Multidrug Resistance. Drug Resist. Updates 2021, 58, 100777. [Google Scholar] [CrossRef]
- Zhao, C.Y.; Cheng, R.; Yang, Z.; Tian, Z.M. Nanotechnology for Cancer Therapy Based on Chemotherapy. Molecules 2018, 23, 826. [Google Scholar] [CrossRef]
- Bar-Zeev, M.; Livney, Y.D.; Assaraf, Y.G. Targeted Nanomedicine for Cancer Therapeutics: Towards Precision Medicine Overcoming Drug Resistance. Drug Resist. Updates 2017, 31, 15–30. [Google Scholar] [CrossRef]
- Cohen, L.; Livney, Y.D.; Assaraf, Y.G. Targeted Nanomedicine Modalities for Prostate Cancer Treatment. Drug Resist. Updates 2021, 56, 100762. [Google Scholar] [CrossRef] [PubMed]
- Stylianopoulos, T. EPR-Effect: Utilizing Size-Dependent Nanoparticle Delivery to Solid Tumors. Ther. Deliv. 2013, 4, 421–423. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Rho, S.; Stiles, W.R.; Hu, S.; Baek, Y.; Hwang, D.W.; Kashiwagi, S.; Kim, M.S.; Choi, H.S. Size-Dependent EPR Effect of Polymeric Nanoparticles on Tumor Targeting. Adv. Healthc. Mater. 2020, 9, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Caro, C.; Pozo, D. Polysaccharide Colloids as Smart Vehicles in Cancer Therapy. Curr. Pharm. Des. 2015, 21, 4822–4836. [Google Scholar] [CrossRef]
- Wilczewska, A.Z.; Niemirowicz, K.; Markiewicz, K.H.; Car2, H. Nanoparticles as Drug Delivery Systems. Pharmacol. Rep. 2012, 64, 1020–1037. [Google Scholar] [CrossRef]
- Livney, Y.D.; Assaraf, Y.G. Rationally Designed Nanovehicles to Overcome Cancer Chemoresistance. Adv. Drug Deliv. Rev. 2013, 65, 1716–1730. [Google Scholar] [CrossRef]
- El-Say, K.M.; El-Sawy, H.S. Polymeric Nanoparticles: Promising Platform for Drug Delivery. Int. J. Pharm. 2017, 528, 675–691. [Google Scholar] [CrossRef]
- Liu, T.I.; Yang, Y.C.; Chiang, W.H.; Hung, C.K.; Tsai, Y.C.; Chiang, C.S.; Lo, C.L.; Chiu, H.C. Radiotherapy-Controllable Chemotherapy from Reactive Oxygen Species-Responsive Polymeric Nanoparticles for Effective Local Dual Modality Treatment of Malignant Tumors. Biomacromolecules 2018, 19, 3825–3839. [Google Scholar] [CrossRef]
- Rios-Doria, J.; Carie, A.; Costich, T.; Burke, B.; Skaff, H.; Panicucci, R.; Sill, K. A Versatile Polymer Micelle Drug Delivery System for Encapsulation and In Vivo Stabilization of Hydrophobic Anticancer Drugs. J. Drug Deliv. 2012, 2012, 951741. [Google Scholar] [CrossRef]
- Begines, B.; Ortiz, T.; Pérez-Aranda, M.; Martínez, G.; Merinero, M.; Argüelles-Arias, F.; Alcudia, A. Polymeric Nanoparticles for Drug Delivery: Recent Developments and Future Prospects. Nanomaterials 2020, 10, 1403. [Google Scholar] [CrossRef]
- Aravind, A.; Jeyamohan, P.; Nair, R.; Veeranarayanan, S.; Nagaoka, Y.; Yoshida, Y.; Maekawa, T.; Kumar, D.S. AS1411 Aptamer Tagged PLGA-Lecithin-PEG Nanoparticles for Tumor Cell Targeting and Drug Delivery. Biotechnol. Bioeng. 2012, 109, 2920–2931. [Google Scholar] [CrossRef] [PubMed]
- Hori, S.I.; Herrera, A.; Rossi, J.J.; Zhou, J. Current Advances in Aptamers for Cancer Diagnosis and Therapy. Cancers 2018, 10, 9. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Chen, X. Aptamer-Based Targeted Therapy. Adv. Drug Deliv. Rev. 2018, 134, 65–78. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, W.; Chen, S.; Zhuang, Z.; Zhang, Y.; Jiang, L.; Lin, J.S. SELEX Tool: A Novel and Convenient Gel-Based Diffusion Method for Monitoring of Aptamer-Target Binding. J. Biol. Eng. 2020, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Engelberg, S.; Lin, Y.; Assaraf, Y.G.; Livney, Y.D. Targeted Nanoparticles Harboring Jasmine-Oil-Entrapped Paclitaxel for Elimination of Lung Cancer Cells. Int. J. Mol. Sci. 2021, 22, 1019. [Google Scholar] [CrossRef] [PubMed]
- Ni, S.; Yao, H.; Wang, L.; Lu, J.; Jiang, F.; Lu, A.; Zhang, G. Chemical Modifications of Nucleic Acid Aptamers for Therapeutic Purposes. Int. J. Mol. Sci. 2017, 18, 1683. [Google Scholar] [CrossRef]
- Kratschmer, C.; Levy, M. Effect of Chemical Modifications on Aptamer Stability in Serum. Nucleic Acid Ther. 2017, 27, 335–344. [Google Scholar] [CrossRef]
- Engelberg, S.; Netzer, E.; Assaraf, Y.G.; Livney, Y.D. Selective Eradication of Human Non-Small Cell Lung Cancer Cells Using Aptamer-Decorated Nanoparticles Harboring a Cytotoxic Drug Cargo. Cell Death Dis. 2019, 10, 702. [Google Scholar] [CrossRef]
- Zhao, Z.; Xu, L.; Shi, X.; Tan, W.; Fang, X.; Shangguan, D. Recognition of Subtype Non-Small Cell Lung Cancer by DNA Aptamers Selected from Living Cells. Analyst 2009, 134, 1808–1814. [Google Scholar] [CrossRef]
- Kim, H.R.; Kim, W.S.; Choi, Y.J.; Choi, C.M.; Rho, J.K.; Lee, J.C. Epithelial-Mesenchymal Transition Leads to Crizotinib Resistance in H2228 Lung Cancer Cells with EML4-ALK Translocation. Mol. Oncol. 2013, 7, 1093–1102. [Google Scholar] [CrossRef]
- Pawlik, A.; Nowak, J.M.; Grzanka, D.; Gackowska, L.; Michalkiewicz, J.; Grzanka, A. Hyperthermia Induces Cytoskeletal Alterations and Mitotic Catastrophe in P53-Deficient H1299 Lung Cancer Cells. Acta Histochem. 2013, 115, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.S.; Jang, W.J.; Chun, K.S.; Jeong, C.H.O. Hsp90 Inhibition by WK88-1 Potently Suppresses the Growth of Gefitinib-Resistant H1975 Cells Harboring the T790M Mutation in EGFR. Oncol. Rep. 2014, 31, 2619–2624. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Elango, R.; Athinarayanan, J.; Subbarayan, V.P.; Lei, D.K.Y.; Alshatwi, A.A. Hesperetin Induces an Apoptosis-Triggered Extrinsic Pathway and a P53- Independent Pathway in Human Lung Cancer H522 Cells. J. Asian Nat. Prod. Res. 2018, 20, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Baust, J.M.; Campbell, L.H.; Harbell, J.W. Best Practices for Cryopreserving, Thawing, Recovering, and Assessing Cells. Vitr. Cell. Dev. Biol.-Anim. 2017, 53, 855–871. [Google Scholar] [CrossRef] [PubMed]
- Martínez Rivas, C.J.; Tarhini, M.; Badri, W.; Miladi, K.; Greige-Gerges, H.; Nazari, Q.A.; Galindo Rodríguez, S.A.; Román, R.Á.; Fessi, H.; Elaissari, A. Nanoprecipitation Process: From Encapsulation to Drug Delivery. Int. J. Pharm. 2017, 532, 66–81. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, G.; Baby, T.; Tengjisi; Chen, D.; Weitz, D.A.; Zhao, C. Stable Polymer Nanoparticles with Exceptionally High Drug Loading by Sequential Nanoprecipitation. Angew. Chem. 2020, 132, 4750–4758. [Google Scholar] [CrossRef]
- Zhang, B.; Jayalath, I.M.; Ke, J.; Sparks, J.L.; Hartley, C.S.; Konkolewicz, D. Chemically Fueled Covalent Crosslinking of Polymer Materials. Chem. Commun. 2019, 55, 2086–2089. [Google Scholar] [CrossRef]
- Aravind, A.; Varghese, S.H.; Veeranarayanan, S.; Mathew, A.; Nagaoka, Y.; Iwai, S.; Fukuda, T.; Hasumura, T.; Yoshida, Y.; Maekawa, T.; et al. Aptamer-Labeled PLGA Nanoparticles for Targeting Cancer Cells. Cancer Nanotechnol. 2012, 3, 1–12. [Google Scholar] [CrossRef]
- Israeli-Lev, G.; Pitchkhadze, M.; Nevo, S.; Fahoum, L.; Meyron-Holtz, E.; Livney, Y.D. Harnessing Proteins to Control Crystal Size and Morphology, for Improved Delivery Performance of Hydrophobic Bioactives, Using Genistein as a Model. Food Hydrocoll. 2017, 63, 97–107. [Google Scholar] [CrossRef]
- Engelberg, Y.; Landau, M. The Human LL-37(17-29) Antimicrobial Peptide Reveals a Functional Supramolecular Structure. Nat. Commun. 2020, 11, 1894. [Google Scholar] [CrossRef]
- Engelberg, S.; Modrejewski, J.; Walter, J.G.; Livney, Y.D.; Assaraf, Y.G. Cancer Cell-Selective, Clathrin-Mediated Endocytosis of Aptamer- Decorated Nanoparticles. Oncotarget 2018, 9, 20993–21006. [Google Scholar] [CrossRef]
- Tang, W.H.; Wang, C.F.; Liao, Y. Di Fetal Bovine Serum Albumin Inhibits Antimicrobial Peptide Activity and Binds Drug Only in Complex with A1-Antitrypsin. Sci. Rep. 2021, 11, 1267. [Google Scholar] [CrossRef]
- Ochoa, S.; Milam, V.T. Modified Nucleic Acids: Expanding the Capabilities of Functional Oligonucleotides. Molecules 2020, 25, 4659. [Google Scholar] [CrossRef]
- Nowak, E.; Kammerer, S.; Küpper, J.H. ATP-Based Cell Viability Assay Is Superior to Trypan Blue Exclusion and XTT Assay in Measuring Cytotoxicity of Anticancer Drugs Taxol and Imatinib, and Proteasome Inhibitor MG-132 on Human Hepatoma Cell Line HepG2. Clin. Hemorheol. Microcirc. 2018, 69, 327–336. [Google Scholar] [CrossRef]
- Yang, L.; Wang, J.; Cheke, R.A.; Tang, S. A Universal Delayed Difference Model Fitting Dose-Response Curves. Dose-Response 2021, 19, 15593258211062785. [Google Scholar] [CrossRef]
- Focke, W.W.; Van Der Westhuizen, I.; Musee, N.; Loots, M.T. Kinetic Interpretation of Log-Logistic Dose-Time Response Curves. Sci. Rep. 2017, 7, 2234. [Google Scholar] [CrossRef]
- Wilson, B.K.; Prud’homme, R.K. Nanoparticle Size Distribution Quantification from Transmission Electron Microscopy (TEM) of Ruthenium Tetroxide Stained Polymeric Nanoparticles. J. Colloid Interface Sci. 2021, 604, 208–220. [Google Scholar] [CrossRef]
- D’Angelo, N.A.; Câmara, M.C.C.; Noronha, M.A.; Grotto, D.; Chorilli, M.; Lourenço, F.R.; Rangel-Yagui, C.d.O.; Lopes, A.M. Development of PEG-PCL-Based Polymersomes through Design of Experiments for Co-Encapsulation of Vemurafenib and Doxorubicin as Chemotherapeutic Drugs. J. Mol. Liq. 2022, 349, 118166. [Google Scholar] [CrossRef]
- Feng, S.S.; Chien, S. Chemotherapeutic Engineering: Application and Further Development of Chemical Engineering Principles for Chemotherapy of Cancer and Other Diseases. Chem. Eng. Sci. 2003, 58, 4087–4114. [Google Scholar] [CrossRef]
- Jiang, W.; Kim, B.Y.S.; Rutka, J.T.; Chan, W.C.W. Nanoparticle-Mediated Cellular Response Is Size-Dependent. Nat. Nanotechnol. 2008, 3, 145–150. [Google Scholar] [CrossRef]
- Kaksonen, M.; Sun, Y.; Drubin, D.G. A Pathway for Association of Receptors, Adaptors, and Actin during Endocytic Internalization. Cell 2003, 115, 475–487. [Google Scholar] [CrossRef]
- Molina-Romero, C.; Rangel-Escareño, C.; Ortega-Gómez, A.; Alanis-Funes, G.J.; Avilés-Salas, A.; Avila-Moreno, F.; Mercado, G.E.; Cardona, A.F.; Hidalgo-Miranda, A.; Arrieta, O. Differential Gene Expression Profiles According to the Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society Histopathological Classification in Lung Adenocarcinoma Subtypes. Hum. Pathol. 2017, 66, 188–199. [Google Scholar] [CrossRef]
- Wilkerson, M.D.; Yin, X.; Walter, V.; Zhao, N.; Cabanski, C.R.; Hayward, M.C.; Miller, C.R.; Socinski, M.A.; Parsons, A.M.; Thorne, L.B.; et al. Differential Pathogenesis of Lung Adenocarcinoma Subtypes Involving Sequence Mutations, Copy Number, Chromosomal Instability, and Methylation. PLoS ONE 2012, 7, e36530. [Google Scholar] [CrossRef]
- Press, D. Aptamer-Hybrid Nanoparticle Bioconjugate Efficiently Delivers MiRNA-29b to Non-Small-Cell Lung Cancer Cells and Inhibits Growth by Downregulating Essential Oncoproteins. Int. J. Nanomed. 2016, 11, 3533–3544. [Google Scholar]
- Powell, D.; Chandra, S.; Dodson, K.; Shaheen, F.; Wiltz, K.; Ireland, S.; Syed, M.; Dash, S.; Wiese, T.; Mandal, T.; et al. Aptamer-Functionalized Hybrid Nanoparticle for the Treatment of Breast Cancer. Eur. J. Pharm. Biopharm. 2017, 114, 108–118. [Google Scholar] [CrossRef]
- Kim, M.W.; Jeong, H.Y.; Kang, S.J.; Jeong, I.H.; Choi, M.J.; You, Y.M.; Im, C.S.; Song, I.H.; Lee, T.S.; Lee, J.S.; et al. Anti-EGF Receptor Aptamer-Guided Co-Delivery of Anti-Cancer SiRNAs and Quantum Dots for Theranostics of Triple-Negative Breast Cancer. Theranostics 2019, 9, 837–852. [Google Scholar] [CrossRef]
- Wan, L.Y.; Yuan, W.F.; Ai, W.B.; Ai, Y.W.; Wang, J.J.; Chu, L.Y.; Zhang, Y.Q.; Wu, J.F. An Exploration of Aptamer Internalization Mechanisms and Their Applications in Drug Delivery. Expert Opin. Drug Deliv. 2019, 16, 207–218. [Google Scholar] [CrossRef]
- Kim, M.Y.; Jeong, S. In Vitro Selection of RNA Aptamer and Specific Targeting of ErbB2 in Breast Cancer Cells. Nucleic Acid Ther. 2011, 21, 173–178. [Google Scholar] [CrossRef]
- Farokhzad, O.C.; Karp, J.M.; Langer, R. Nanoparticle-Aptamer Bioconjugates for Cancer Targeting. Expert Opin. Drug Deliv. 2006, 3, 311–324. [Google Scholar] [CrossRef]
- Zhou, J.; Rossi, J.J. Cell-Specific Aptamer-Mediated Targeted Drug Delivery. Oligonucleotides 2011, 21, 1–10. [Google Scholar] [CrossRef]
- Dhar, S.; Gu, F.X.; Langer, R.; Farokhza, O.C.; Lippard, S.J. Targeted Delivery of Cisplatin to Prostate Cancer Cells by Aptamer Functionalized Pt(IV) Prodrug-PLGA-PEG Nanoparticles. Proc. Natl. Acad. Sci. USA 2008, 105, 17356–17361. [Google Scholar] [CrossRef]
- Jiang, Z.M.; Dai, S.P.; Xu, Y.Q.; Li, T.; Xie, J.; Li, C.; Zhang, Z.H. Crizotinib-Loaded Polymeric Nanoparticles in Lung Cancer Chemotherapy. Med. Oncol. 2015, 32, 193. [Google Scholar] [CrossRef]
- Kettler, K.; Veltman, K.; van de Meent, D.; van Wezel, A.; Hendriks, A.J. Cellular Uptake of Nanoparticles as Determined by Particle Properties, Experimental Conditions, and Cell Type. Environ. Toxicol. Chem. 2014, 33, 481–492. [Google Scholar] [CrossRef]
- Behzadi, S.; Serpooshan, V.; Tao, W.; Hamaly, M.A.; Alkawareek, M.Y.; Dreaden, E.C.; Brown, D.; Alkilany, A.M.; Farokhzad, O.C.; Mahmoudi, M. Cellular Uptake of Nanoparticles: Journey inside the Cell. Chem. Soc. Rev. 2017, 46, 4218–4244. [Google Scholar] [CrossRef]
- Xiao, Z.; Shangguan, D.; Cao, Z.; Fang, X.; Tan, W. Cell-Specific Internalization Study of an Aptamer from Whole Cell Selection. Chem.-A Eur. J. 2008, 14, 1769–1775. [Google Scholar] [CrossRef]
- Adan, A.; Alizada, G.; Kiraz, Y.; Baran, Y.; Nalbant, A. Flow Cytometry: Basic Principles and Applications. Crit. Rev. Biotechnol. 2017, 37, 163–176. [Google Scholar] [CrossRef]
- Tsuji, K.; Ojima, M.; Otabe, K.; Horie, M.; Koga, H.; Sekiya, I.; Muneta, T. Effects of Different Cell-Detaching Methods on the Viability and Cell Surface Antigen Expression of Synovial Mesenchymal Stem Cells. Cell Transplant. 2017, 26, 1089–1102. [Google Scholar] [CrossRef]
- Ferreira, C.S.M.; Cheung, M.C.; Missailidis, S.; Bisland, S.; Gariépy, J. Phototoxic Aptamers Selectively Enter and Kill Epithelial Cancer Cells. Nucleic Acids Res. 2009, 37, 866–876. [Google Scholar] [CrossRef]
- Zhou, J.; Sun, M.; Jin, S.; Fan, L.; Zhu, W.; Sui, X.; Cao, L.; Yang, C.; Han, C. Combined Using of Paclitaxel and Salinomycin Active Targeting Nanostructured Lipid Carriers against Non-Small Cell Lung Cancer and Cancer Stem Cells. Drug Deliv. 2019, 26, 281–289. [Google Scholar] [CrossRef]
- Li, Y.; Peng, Y.; Tan, Y.; Xuan, W.; Fu, T.; Wang, X.Q.; Tan, W. A New Paradigm for Artesunate Anticancer Function: Considerably Enhancing the Cytotoxicity via Conjugating Artesunate with Aptamer. Signal Transduct. Target. Ther. 2021, 6, 2020–2022. [Google Scholar] [CrossRef]
- Ng, E.W.M.; Shima, D.T.; Calias, P.; Cunningham, E.T.; Guyer, D.R.; Adamis, A.P. Pegaptanib, a Targeted Anti-VEGF Aptamer for Ocular Vascular Disease. Nat. Rev. Drug Discov. 2006, 5, 123–132. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barak, D.; Engelberg, S.; Assaraf, Y.G.; Livney, Y.D. Selective Targeting and Eradication of Various Human Non-Small Cell Lung Cancer Cell Lines Using Self-Assembled Aptamer-Decorated Nanoparticles. Pharmaceutics 2022, 14, 1650. https://doi.org/10.3390/pharmaceutics14081650
Barak D, Engelberg S, Assaraf YG, Livney YD. Selective Targeting and Eradication of Various Human Non-Small Cell Lung Cancer Cell Lines Using Self-Assembled Aptamer-Decorated Nanoparticles. Pharmaceutics. 2022; 14(8):1650. https://doi.org/10.3390/pharmaceutics14081650
Chicago/Turabian StyleBarak, Daniel, Shira Engelberg, Yehuda G. Assaraf, and Yoav D. Livney. 2022. "Selective Targeting and Eradication of Various Human Non-Small Cell Lung Cancer Cell Lines Using Self-Assembled Aptamer-Decorated Nanoparticles" Pharmaceutics 14, no. 8: 1650. https://doi.org/10.3390/pharmaceutics14081650
APA StyleBarak, D., Engelberg, S., Assaraf, Y. G., & Livney, Y. D. (2022). Selective Targeting and Eradication of Various Human Non-Small Cell Lung Cancer Cell Lines Using Self-Assembled Aptamer-Decorated Nanoparticles. Pharmaceutics, 14(8), 1650. https://doi.org/10.3390/pharmaceutics14081650