Perfluorocarbon Nanodroplets as Potential Nanocarriers for Brain Delivery Assisted by Focused Ultrasound-Mediated Blood–Brain Barrier Disruption

 , , , , , , and
, , , , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of Fluorinated Surfactants
2.3. Synthesis of FITC-F8TAC13
2.4. Preparation of “Oil-Free” Emulsions for Optimization Experiments
2.5. Preparation of “Baseline” Emulsions
2.6. Preparation of Tagged “Baseline” Emulsions
2.7. Preparation of the “Concentrated” Emulsion for Pharmacokinetics and Tissue Distribution Follow-Up
2.8. Freeze-Drying (FD) Process
2.9. Nanodroplet Characterization
2.10. Transmission Electron Microscopy (TEM) Images
2.11. Determination of PFOB Volume Fraction in Emulsions by 19F-Nuclear Magnetic Resonance (19F-NMR)
2.12. Oil and DiD Titration by HPLC
2.13. Emulsion Stability Determination
2.14. Cell Culture
2.15. Biocompatibility Assay
2.16. Confocal Laser Scanning Microscopy (CLSM)
2.17. Fluorescence-Activated Cell Sorting (FACS)
2.18. In Vivo Studies
2.19. In Vivo Pharmacokinetics and Tissue Distribution
2.20. 19F-NMR PFOB Quantitative Determination in Mice Blood and Organs
2.21. In Vivo Hemispheric BBB Disruption Procedure
2.22. Intracerebral Accumulation of STE Using Fluorescence Microscopy
2.23. Intracerebral Accumulation of STE Using FACS
2.24. Histological Evaluation
3. Results
3.1. Nanoemulsion Preparation and Characterization
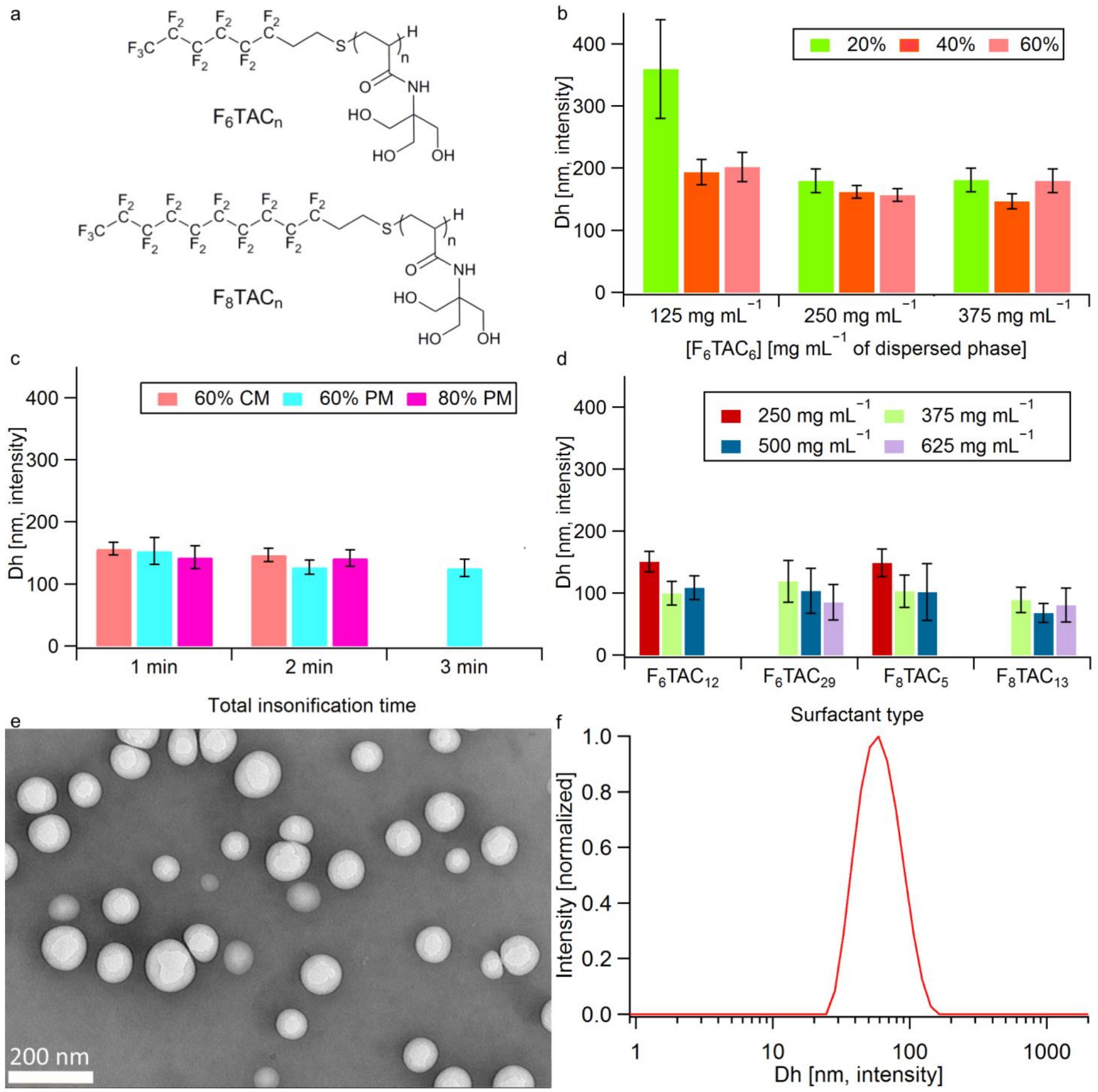
3.1.1. Surfactant Synthesis
3.1.2. Nanoemulsion Synthesis and Droplet Dh Optimization
3.1.3. Oil introduction
3.1.4. Emulsion Freeze-Drying and Labeling
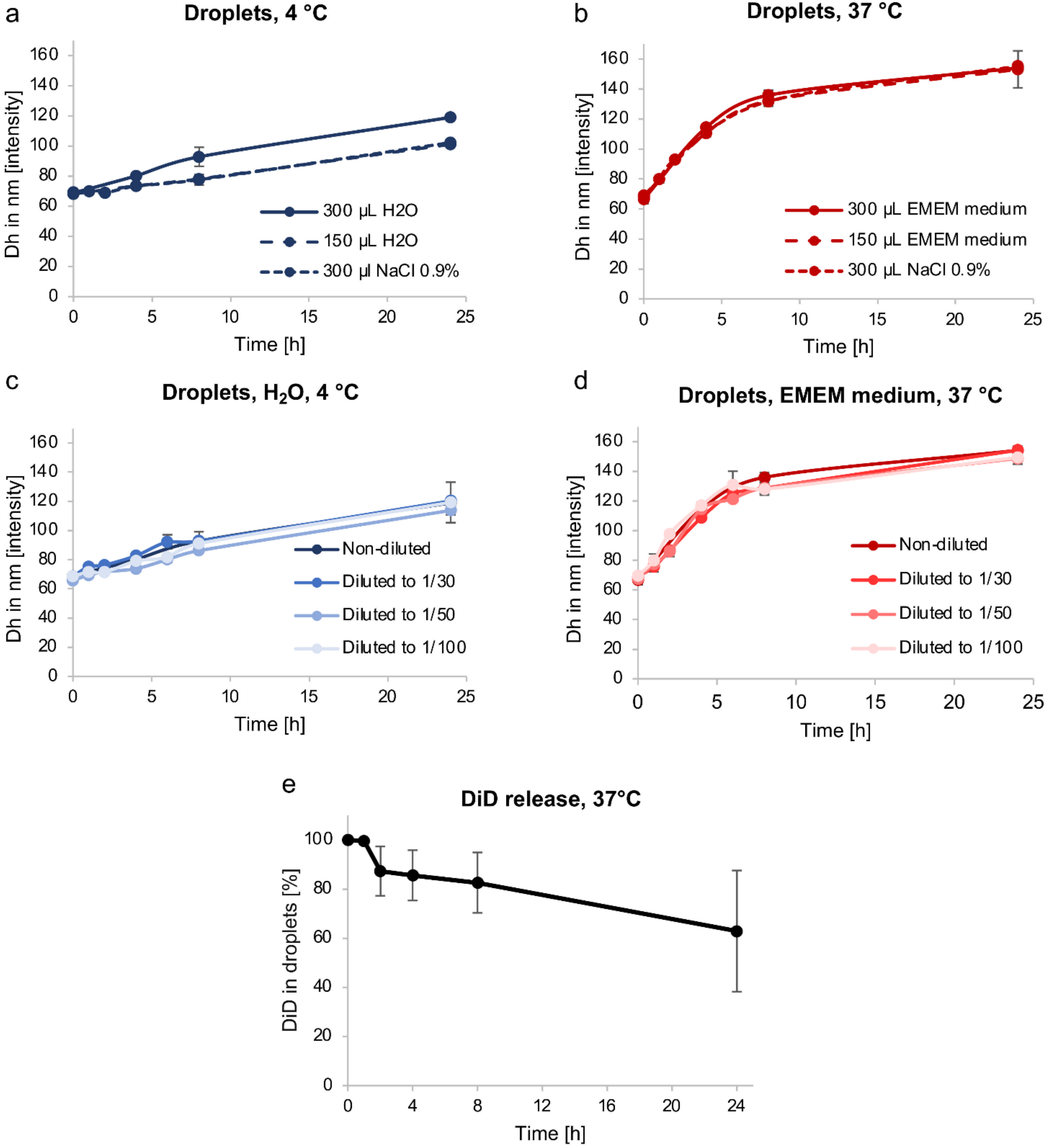
3.2. Emulsion Stability
3.2.1. Droplet Dh Variation over Time
3.2.2. Passive Content Release
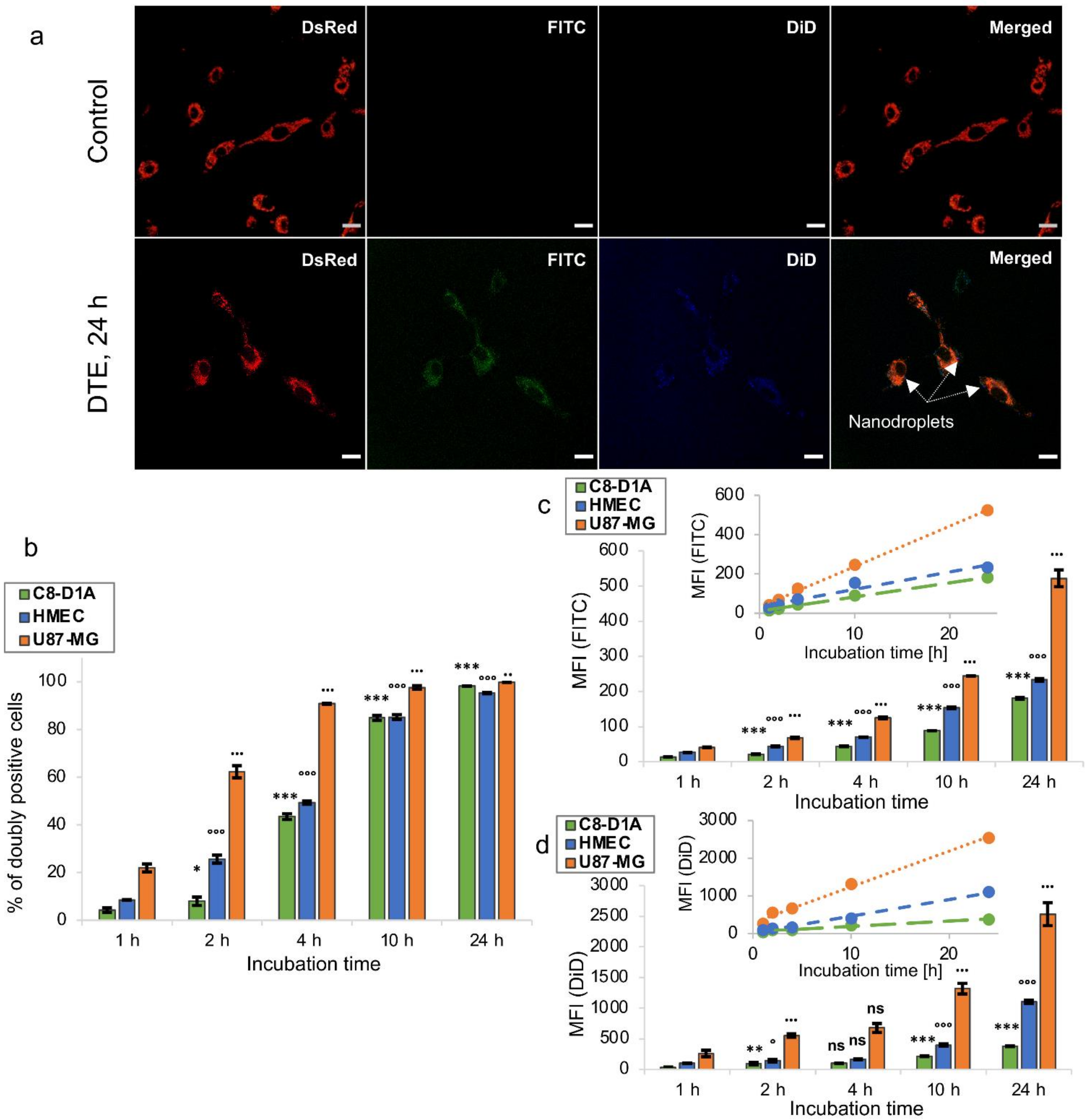
3.3. In Vitro Biocompatibility and Uptake of Nanodroplets
3.3.1. In Vitro Biocompatibility
3.3.2. In Vitro Uptake
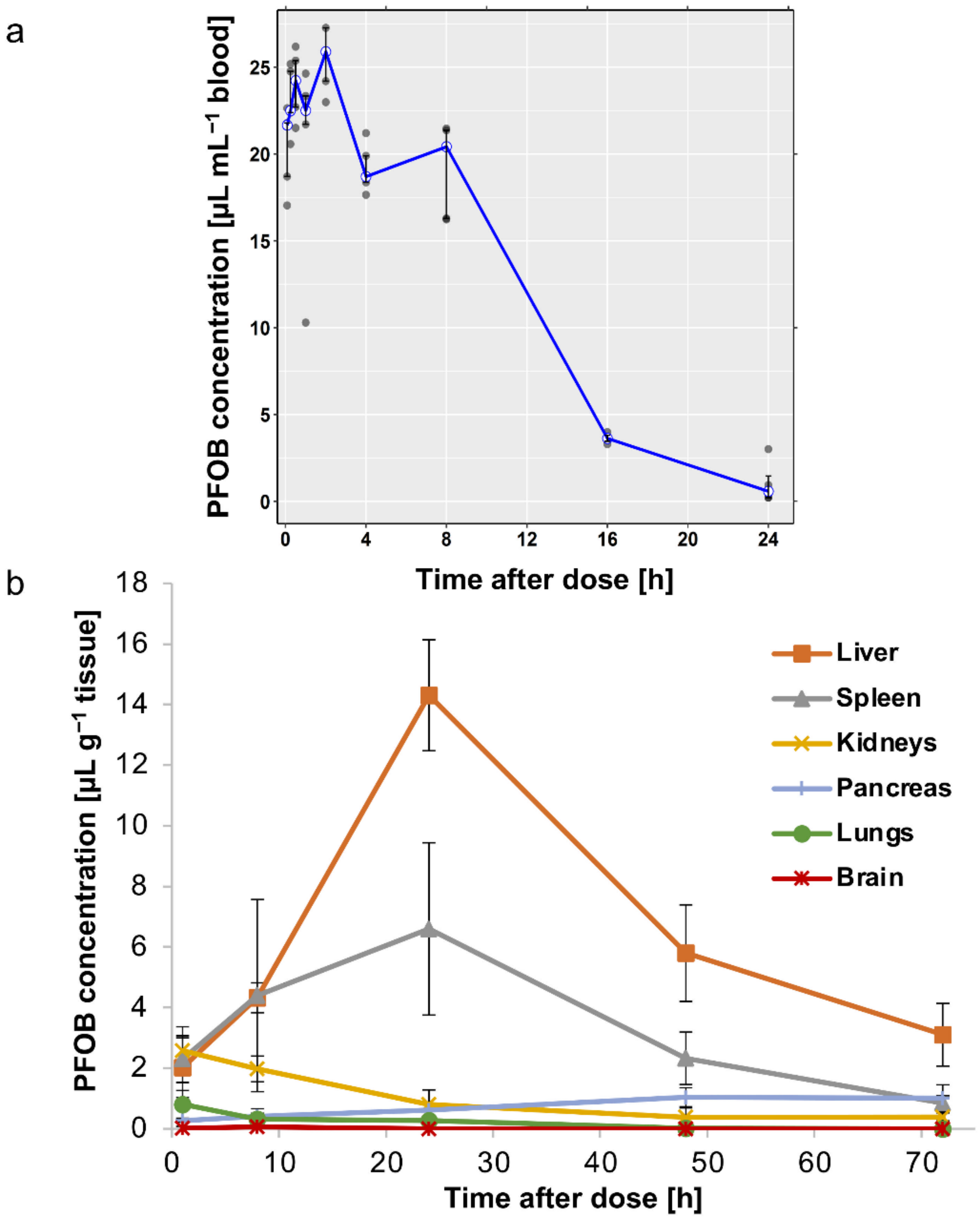
3.4. In Vivo Pharmacokinetics and Tissue Distribution of Nanodroplets
3.4.1. In Vivo Pharmacokinetics
3.4.2. In Vivo Biodistribution
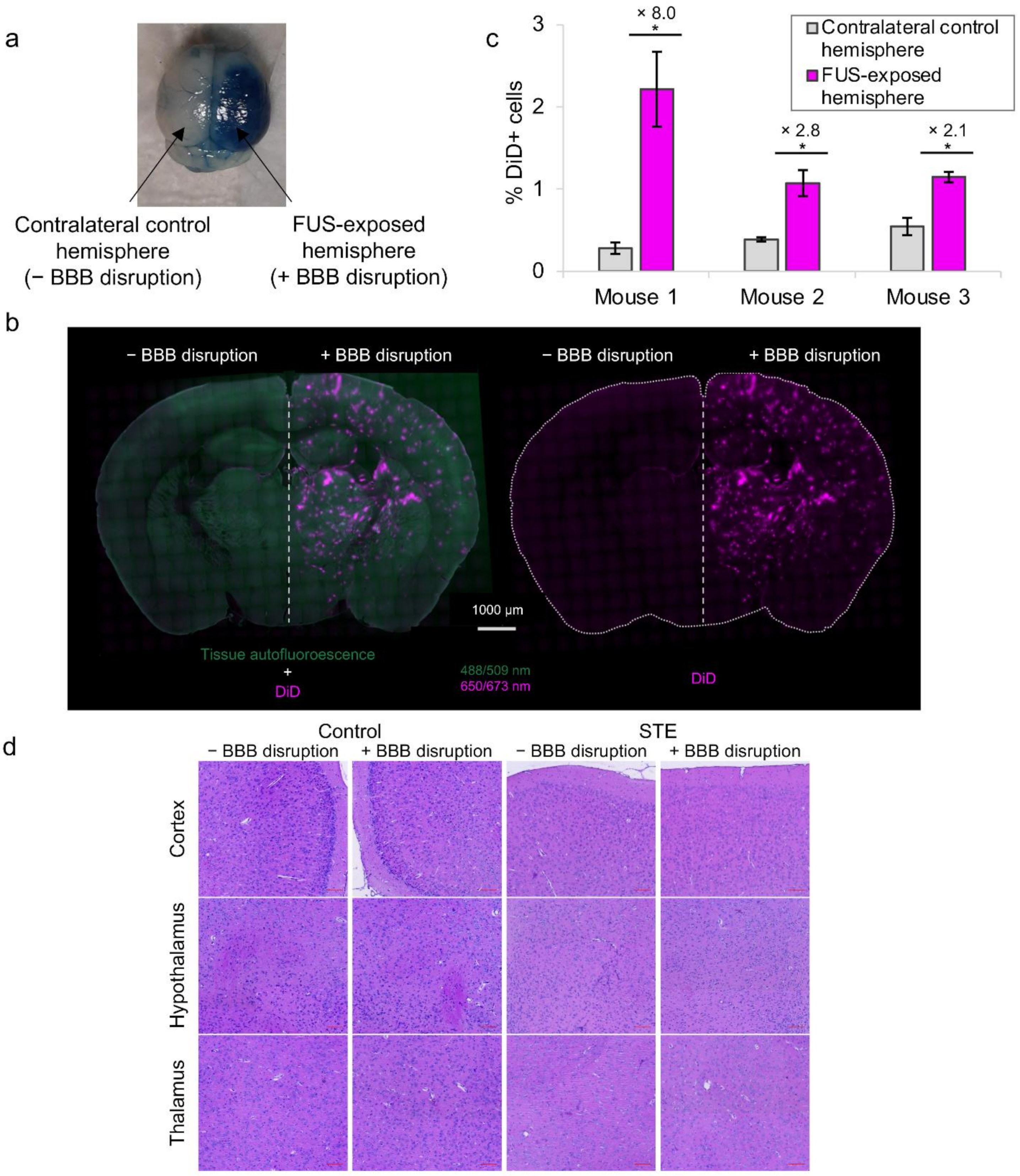
3.5. In Vivo Intracerebral Accumulation of Nanodroplets after BBB Disruption
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 19F-MRI | 19F-magnetic resonance imaging |
| 19F-NMR | 19F-nuclear magnetic resonance |
| AIBN | 2,2′-azobisisobutyronitrile |
| ATBC | Tributyl O-acetyl citrate |
| ATCC | American type culture collection |
| AUC | Area under the curve |
| AUMC | Area under the first moment curve |
| BBB | Blood–brain barrier |
| BSA | Bovine serum albumin |
| C8-D1A | Mouse astrocyte cells |
| C-90 | Capryol® 90 |
| CA | Cellulose acetate |
| Cl | Clearance |
| CLSM | Confocal laser scanning microscopy |
| CM | Continuous mode |
| CNS | Central nervous system |
| DABCO | 1,4-diazabicyclooctane |
| DC | Duty cycle |
| Dh | Hydrodynamic diameter |
| DiD | 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine,4-chlorobenzene sulfonate salt |
| DLS | Dynamic light scattering |
| DMEM | Dulbecco’s modified eagle medium |
| DPn | Degree of polymerization |
| DTE | Double-tagged emulsion |
| EDTA | Ethylene diamine tetraacetic acid |
| EMEM | Eagle’s minimum essential medium |
| Et2O | diethyl ether |
| FACS | Fluorescence-activated cell |
| FCS | Fetal calf serum |
| FD | Freeze-drying |
| FITC | Fluorescein isothiocyanate |
| FUS | Focused ultrasound |
| HMEC-1 | Human microvascular endothelial cells |
| HPLC | High performance liquid chromatography |
| LLOQ | Lower limit of quantification |
| MB | Microbubbles |
| MFI | Mean fluorescence intensity |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| MRT | Mean residencce time |
| NC | Nanocarriers |
| PBS | Phosphate buffer saline |
| PDI | Polydispersity index |
| PFA | Paraformaldehyde |
| PFC | Perflurocarbon |
| PFOB | Perfluorooctyl bromide |
| PM | Pulsed mode |
| SM | Supplementary Materials |
| t1/2 | Terminal half-life |
| TEM | Transmission electron microscopy |
| TFA | trifluoroacetate |
| THAM | Tris(hydroxymethyl)acrylamidomethane |
| U87-MG | Human glioblastoma cell line |
| US | Ultrasound |
| Vd | Volume of distribution |
References
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and Future Treatments in Alzheimer Disease: An Update. J. Central Nerv. Syst. Dis. 2020, 12, 1179573520907397. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Okun, M.S. Diagnosis and Treatment of Parkinson Disease: A Review. JAMA 2020, 323, 548–560. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.B.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Stupp, R.; Taillibert, S.; Kanner, A.A.; Read, W.; Steinberg, D.M.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.K.; Dolman, D.E.M.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. The blood-brain barrier: Bottleneck in brain drug development. NeuroRX 2005, 2, 3–14. [Google Scholar] [CrossRef]
- Terstappen, G.C.; Meyer, A.H.; Bell, R.D.; Zhang, W. Strategies for delivering therapeutics across the blood–brain barrier. Nat. Rev. Drug Discov. 2021, 20, 362–383. [Google Scholar] [CrossRef]
- Tsou, Y.; Zhang, X.; Zhu, H.; Syed, S.; Xu, X. Drug Delivery to the Brain across the Blood–Brain Barrier Using Nanomaterials. Small 2017, 13, 1701921. [Google Scholar] [CrossRef]
- Saraiva, C.; Praça, C.; Ferreira, R.; Santos, T.; Ferreira, L.; Bernardino, L. Nanoparticle-mediated brain drug delivery: Overcoming blood–brain barrier to treat neurodegenerative diseases. J. Control. Release 2016, 235, 34–47. [Google Scholar] [CrossRef]
- Đorđević, S.; Gonzalez, M.M.; Conejos-Sánchez, I.; Carreira, B.; Pozzi, S.; Acúrcio, R.C.; Satchi-Fainaro, R.; Florindo, H.F.; Vicent, M.J. Current hurdles to the translation of nanomedicines from bench to the clinic. Drug Deliv. Transl. Res. 2021, 12, 500–525. [Google Scholar] [CrossRef]
- Iqbal, J.; Abbasi, B.A.; Ahmad, R.; Mahmood, T.; Ali, B.; Khalil, A.T.; Kanwal, S.; Shah, S.A.; Alam, M.M.; Badshah, H.; et al. Nanomedicines for developing cancer nanotherapeutics: From benchtop to bedside and beyond. Appl. Microbiol. Biotechnol. 2018, 102, 9449–9470. [Google Scholar] [CrossRef] [PubMed]
- Burgess, A.; Hynynen, K. Microbubble-Assisted Ultrasound for Drug Delivery in the Brain and Central Nervous System. Adv. Exp. Med. Biol. 2016, 880, 293–308. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-Y.; Aurup, C.; Sanchez, C.S.; Grondin, J.; Zheng, W.; Kamimura, H.; Ferrera, V.P.; Konofagou, E.E. Efficient Blood-Brain Barrier Opening in Primates with Neuronavigation-Guided Ultrasound and Real-Time Acoustic Mapping. Sci. Rep. 2018, 8, 7978. [Google Scholar] [CrossRef] [PubMed]
- Mainprize, T.; Lipsman, N.; Huang, Y.; Meng, Y.; Bethune, A.; Ironside, S.; Heyn, C.; Alkins, R.; Trudeau, M.; Sahgal, A.; et al. Blood-Brain Barrier Opening in Primary Brain Tumors with Non-invasive MR-Guided Focused Ultrasound: A Clinical Safety and Feasibility Study. Sci. Rep. 2019, 9, 321. [Google Scholar] [CrossRef]
- Carpentier, A.; Canney, M.; Vignot, A.; Reina, V.; Beccaria, K.; Horodyckid, C.; Karachi, C.; Leclercq, D.; Lafon, C.; Chapelon, J.-Y.; et al. Clinical trial of blood-brain barrier disruption by pulsed ultrasound. Sci. Transl. Med. 2016, 8, 343re2. [Google Scholar] [CrossRef]
- Idbaih, A.; Canney, M.; Belin, L.; Desseaux, C.; Vignot, A.; Bouchoux, G.; Asquier, N.; Law-Ye, B.; Leclercq, D.; Bissery, A.; et al. Safety and Feasibility of Repeated and Transient Blood–Brain Barrier Disruption by Pulsed Ultrasound in Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2019, 25, 3793–3801. [Google Scholar] [CrossRef]
- Timbie, K.F.; Mead, B.P.; Price, R.J. Drug and gene delivery across the blood–brain barrier with focused ultrasound. J. Control. Release 2015, 219, 61–75. [Google Scholar] [CrossRef]
- Ghali, M.G.Z.; Srinivasan, V.M.; Kan, P. Focused Ultrasonography-Mediated Blood-Brain Barrier Disruption in the Enhancement of Delivery of Brain Tumor Therapies. World Neurosurg. 2019, 131, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Hynynen, K.; Lipsman, N. Applications of focused ultrasound in the brain: From thermoablation to drug delivery. Nat. Rev. Neurol. 2020, 17, 7–22. [Google Scholar] [CrossRef]
- Marty, B.; Larrat, B.; Van Landeghem, M.; Robic, C.; Robert, P.; Port, M.; Le Bihan, D.; Pernot, M.; Tanter, M.; Lethimonnier, F.; et al. Dynamic Study of Blood–Brain Barrier Closure after its Disruption using Ultrasound: A Quantitative Analysis. J. Cereb. Blood Flow Metab. 2012, 32, 1948–1958. [Google Scholar] [CrossRef]
- Conti, A.; Mériaux, S.; Larrat, B. About the Marty model of blood-brain barrier closure after its disruption using focused ultrasound. Phys. Med. Biol. 2019, 64, 14NT02. [Google Scholar] [CrossRef] [PubMed]
- Lea-Banks, H.; O'Reilly, M.; Hynynen, K. Ultrasound-responsive droplets for therapy: A review. J. Control. Release 2018, 293, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Jin, K.; Pang, Q.; Shen, S.; Yan, Z.; Jiang, T.; Zhu, X.; Yu, L.; Pang, Z.; Jiang, X. On-Demand Drug Release from Dual-Targeting Small Nanoparticles Triggered by High-Intensity Focused Ultrasound Enhanced Glioblastoma-Targeting Therapy. ACS Appl. Mater. Interfaces 2017, 9, 31612–31625. [Google Scholar] [CrossRef]
- Rapoport, N.; Nam, K.-H.; Gupta, R.; Gao, Z.; Mohan, P.; Payne, A.; Todd, N.; Liu, X.; Kim, T.; Shea, J.; et al. Ultrasound-mediated tumor imaging and nanotherapy using drug loaded, block copolymer stabilized perfluorocarbon nanoemulsions. J. Control. Release 2011, 153, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Krafft, M.P.; Riess, J.G. Therapeutic oxygen delivery by perfluorocarbon-based colloids. Adv. Colloid Interface Sci. 2021, 294, 102407. [Google Scholar] [CrossRef] [PubMed]
- Pavia, A.A.; Pucci, B.; Riess, J.G.; Zarif, L. New fluorinated biocompatible non-ionic telomeric amphiphiles bearing trishydroxymethyl groups. Bioorg. Med. Chem. Lett. 1991, 1, 103–106. [Google Scholar] [CrossRef]
- Maurizis, J.C.; Azim, M.; Rapp, M.; Pucci, B.; Pavia, A.; Madelmont, J.C.; Veyre, A. Disposition in rat of a new fluorinated, biocompatible, non-ionic telomeric carrier. Xenobiotica 1994, 24, 535–541. [Google Scholar] [CrossRef]
- Zarif, L.; Riess, J.G.; Pucci, B.; Pavia, A.A. Biocompatibility of Alkyl and Perfluoroalkyl Telomeric Surfactants Derived from Tham. Biomater. Artif. Cells Immobil. Biotechnol. 1993, 21, 597–608. [Google Scholar] [CrossRef]
- Pucci, B.; Maurizis, J.; Pavia, A. Telomeres et cotelomeres d'interet biologique et biomedical—IV. Les telomeres du tris(hydroxymethyl)-acrylamidomethane nouveaux agents amphiphiles non ioniques. Eur. Polym. J. 1991, 27, 1101–1106. [Google Scholar] [CrossRef]
- Desgranges, S.; Lorton, O.; Gui-Levy, L.; Guillemin, P.; Celicanin, Z.; Hyacinthe, J.-N.; Breguet, R.; Crowe, L.A.; Becker, C.D.; Soulié, M.; et al. Micron-sized PFOB liquid core droplets stabilized with tailored-made perfluorinated surfactants as a new class of endovascular sono-sensitizers for focused ultrasound thermotherapy. J. Mater. Chem. B 2018, 7, 927–939. [Google Scholar] [CrossRef]
- Astafyeva, K.; Somaglino, L.; Desgranges, S.; Berti, R.; Patinote, C.; Langevin, D.; Lazeyras, F.; Salomir, R.; Polidori, A.; Continopepin, C.; et al. Perfluorocarbon nanodroplets stabilized by fluorinated surfactants: Characterization and potentiality as theranostic agents. J. Mater. Chem. B 2015, 3, 2892–2907. [Google Scholar] [CrossRef] [PubMed]
- Alves, I.D.; Carré, M.; Montero, M.-P.; Castano, S.; Lecomte, S.; Marquant, R.; Lecorché, P.; Burlina, F.; Schatz, C.; Sagan, S.; et al. A proapoptotic peptide conjugated to penetratin selectively inhibits tumor cell growth. Biochim. et Biophys. Acta (BBA) Biomembr. 2014, 1838, 2087–2098. [Google Scholar] [CrossRef] [PubMed]
- Braguer, D.; Correard, F.; Maximova, K.; Villard, C.; Roy, M.; Al-Kattan, A.; Sentis, M.; Gingras, M.; Kabashin, A.; Esteve, M.-A. Gold nanoparticles prepared by laser ablation in aqueous biocompatible solutions: Assessment of safety and biological identity for nanomedicine applications. Int. J. Nanomed. 2014, 9, 5415–5430. [Google Scholar] [CrossRef]
- Felix, M.-S.; Borloz, E.; Metwally, K.; Dauba, A.; Larrat, B.; Matagne, V.; Ehinger, Y.; Villard, L.; Novell, A.; Mensah, S.; et al. Ultrasound-Mediated Blood-Brain Barrier Opening Improves Whole Brain Gene Delivery in Mice. Pharmaceutics 2021, 13, 1245. [Google Scholar] [CrossRef] [PubMed]
- Tran, V.L.; Novell, A.; Tournier, N.; Gerstenmayer, M.; Schweitzer-Chaput, A.; Mateos, C.; Jego, B.; Bouleau, A.; Nozach, H.; Winkeler, A.; et al. Impact of blood-brain barrier permeabilization induced by ultrasound associated to microbubbles on the brain delivery and kinetics of cetuximab: An immunoPET study using 89Zr-cetuximab. J. Control. Release 2020, 328, 304–312. [Google Scholar] [CrossRef]
- Hugon, G.; Goutal, S.; Dauba, A.; Breuil, L.; Larrat, B.; Winkeler, A.; Novell, A.; Tournier, N. [18F]2-Fluoro-2-deoxy-sorbitol PET Imaging for Quantitative Monitoring of Enhanced Blood-Brain Barrier Permeability Induced by Focused Ultrasound. Pharmaceutics 2021, 13, 1752. [Google Scholar] [CrossRef]
- Contino, C.; Ollier, M.; Maurizis, J.C.; Lacombe, J.M.; Pucci, B. Synthesis of cotelomers derived from tris(hydroxymethyl)acrylamidomethane (THAM) bearing cytosine arabinoside moieties. Preliminary investigation of their biological activity. Tetrahedron Lett. 1996, 37, 9049–9052. [Google Scholar] [CrossRef]
- Ohta, S.; Kikuchi, E.; Ishijima, A.; Azuma, T.; Sakuma, I.; Ito, T. Investigating the optimum size of nanoparticles for their delivery into the brain assisted by focused ultrasound-induced blood–brain barrier opening. Sci. Rep. 2020, 10, 18220. [Google Scholar] [CrossRef]
- Shen, Y.; Guo, J.; Chen, G.; Chin, C.T.; Chen, X.; Chen, J.; Wang, F.; Chen, S.; Dan, G. Delivery of Liposomes with Different Sizes to Mice Brain after Sonication by Focused Ultrasound in the Presence of Microbubbles. Ultrasound Med. Biol. 2016, 42, 1499–1511. [Google Scholar] [CrossRef]
- Tadros, T.F. Emulsion Formation, Stability, and Rheology. In Emulsion Formation and Stability; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2013; pp. 1–75. ISBN 978-3-527-64794-1. [Google Scholar]
- Gupta, A.; Eral, H.B.; Hatton, T.A.; Doyle, P.S. Nanoemulsions: Formation, properties and applications. Soft Matter 2016, 12, 2826–2841. [Google Scholar] [CrossRef]
- Li, P.-H.; Chiang, B.-H. Process optimization and stability of d-limonene-in-water nanoemulsions prepared by ultrasonic emulsification using response surface methodology. Ultrason. Sonochem. 2012, 19, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Lehmler, H.-J.; Xu, L.; Vyas, S.M.; Ojogun, V.A.; Knutson, B.L.; Ludewig, G. Synthesis, physicochemical properties and in vitro cytotoxicity of nicotinic acid ester prodrugs intended for pulmonary delivery using perfluorooctyl bromide as vehicle. Int. J. Pharm. 2008, 353, 35–44. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kim, J.O.; Rashid, R.; Kim, D.W.; Yousaf, A.M.; Mustapha, O.; Din, F.U.; Park, J.H.; Yong, C.S.; Oh, Y.-K.; Youn, Y.S.; et al. Comparative study on solid self-nanoemulsifying drug delivery and solid dispersion system for enhanced solubility and bioavailability of ezetimibe. Int. J. Nanomed. 2015, 10, 6147–6159. [Google Scholar] [CrossRef] [PubMed]
- Borhade, V.; Nair, H.; Hegde, D. Design and Evaluation of Self-Microemulsifying Drug Delivery System (SMEDDS) of Tacrolimus. AAPS PharmSciTech 2008, 9, 13–21. [Google Scholar] [CrossRef]
- Al Rifai, N.; Desgranges, S.; Le Guillou-Buffello, D.; Giron, A.; Urbach, W.; Nassereddine, M.; Charara, J.; Contino-Pépin, C.; Taulier, N. Ultrasound-triggered delivery of paclitaxel encapsulated in an emulsion at low acoustic pressures. J. Mater. Chem. B 2020, 8, 1640–1648. [Google Scholar] [CrossRef]
- Rowe, R.C. (Ed.) Handbook of Pharmaceutical Excipients, 6th ed.; APhA; (PhP) Pharmaceutical Press: London, UK, 2009; ISBN 978-0-85369-792-3. [Google Scholar]
- Gattefossé Regulatory Data Sheet (RDS)—Pharmaceutical Market (Human and Veterinary Medicines) CAPRYOL 90 (Code: 3254). 2018. Available online: https://www.gattefosse.com/back/files/RDS%20Pharma_CAPRYOL_90_7x.pdf (accessed on 7 June 2022).
- Abdelwahed, W.; Degobert, G.; Fessi, H. Freeze-drying of nanocapsules: Impact of annealing on the drying process. Int. J. Pharm. 2006, 324, 74–82. [Google Scholar] [CrossRef]
- Li, F.; Wang, T.; He, H.B.; Tang, X. The properties of bufadienolides-loaded nano-emulsion and submicro-emulsion during lyophilization. Int. J. Pharm. 2008, 349, 291–299. [Google Scholar] [CrossRef]
- Mohammady, M.; Mohammadi, Y.; Yousefi, G. Freeze-Drying of Pharmaceutical and Nutraceutical Nanoparticles: The Effects of Formulation and Technique Parameters on Nanoparticles Characteristics. J. Pharm. Sci. 2020, 109, 3235–3247. [Google Scholar] [CrossRef]
- Gallarate, M.; Mittone, E.; Carlotti, M.E.; Trotta, M.; Piccerelle, P. Formulation of Dry Emulsion for Topical Applications. J. Dispers. Sci. Technol. 2009, 30, 823–833. [Google Scholar] [CrossRef]
- Corveleyn, S.; Remon, J. Formulation of a lyophilized dry emulsion tablet for the delivery of poorly soluble drugs. Int. J. Pharm. 1998, 166, 65–74. [Google Scholar] [CrossRef]
- Choi, M.J.; Briançon, S.; Bazile, D.; Royere, A.; Min, S.G.; Fessi, H. Effect of Cryoprotectant and Freeze-Drying Process on the Stability of W/O/W Emulsions. Dry. Technol. 2007, 25, 809–819. [Google Scholar] [CrossRef]
- Gupta, S.; Chavhan, S.; Sawant, K.K. Self-nanoemulsifying drug delivery system for adefovir dipivoxil: Design, characterization, in vitro and ex vivo evaluation. Colloids Surf. A Physicochem. Eng. Asp. 2011, 392, 145–155. [Google Scholar] [CrossRef]
- Xiang, Y.; Bernards, N.; Hoang, B.; Zheng, J.; Matsuura, N. Perfluorocarbon nanodroplets can reoxygenate hypoxic tumors in vivo without carbogen breathing. Nanotheranostics 2019, 3, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Perrault, S.D.; Walkey, C.; Jennings, T.; Fischer, H.C.; Chan, W.C.W. Mediating Tumor Targeting Efficiency of Nanoparticles Through Design. Nano Lett. 2009, 9, 1909–1915. [Google Scholar] [CrossRef]
- Salatin, S.; Khosroushahi, A.Y. Overviews on the cellular uptake mechanism of polysaccharide colloidal nanoparticles. J. Cell. Mol. Med. 2017, 21, 1668–1686. [Google Scholar] [CrossRef]
- Nguyen, V.T.A.; De Pauw-Gillet, M.-C.; Gauthier, M.; Sandre, O. Magnetic Polyion Complex Micelles for Cell Toxicity Induced by Radiofrequency Magnetic Field Hyperthermia. Nanomaterials 2018, 8, 1014. [Google Scholar] [CrossRef]
- Flaim, S.F. Pharmacokinetics and Side Effects of Perfluorocarbon-Based Blood Substitutes. Artif. Cells Blood Substitutes, Biotechnol. 1994, 22, 1043–1054. [Google Scholar] [CrossRef]
- Keipert, P.E.; Otto, S.; Flaim, S.F.; Weers, J.G.; Schutt, E.A.; Pelura, T.J.; Klein, D.H.; Yaksh, T.L. Influence of Perflubron Emulsion Particle Size on Blood Half-Life and Febrile Response in Rats. Artif. Cells Blood Substitutes Biotechnol. 1994, 22, 1169–1174. [Google Scholar] [CrossRef]
- Spahn, D.R. Blood substitutes Artificial oxygen carriers: Perfluorocarbon emulsions. Crit. Care 1999, 3, R93–R97. [Google Scholar] [CrossRef][Green Version]
- Airan, R.D.; Meyer, R.A.; Ellens, N.P.K.; Rhodes, K.R.; Farahani, K.; Pomper, M.G.; Kadam, S.D.; Green, J.J. Noninvasive Targeted Transcranial Neuromodulation via Focused Ultrasound Gated Drug Release from Nanoemulsions. Nano Lett. 2017, 17, 652–659. [Google Scholar] [CrossRef]
- Crowe, T.P.; Greenlee, M.H.W.; Kanthasamy, A.G.; Hsu, W.H. Mechanism of intranasal drug delivery directly to the brain. Life Sci. 2018, 195, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Hynynen, K.; McDannold, N.; Sheikov, N.A.; Jolesz, F.A.; Vykhodtseva, N. Local and reversible blood–brain barrier disruption by noninvasive focused ultrasound at frequencies suitable for trans-skull sonications. NeuroImage 2005, 24, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Jiang, C. Evolution of blood–brain barrier in brain diseases and related systemic nanoscale brain-targeting drug delivery strategies. Acta Pharm. Sin. B 2020, 11, 2306–2325. [Google Scholar] [CrossRef] [PubMed]
- Hua, S.; De Matos, M.B.C.; Metselaar, J.M.; Storm, G. Current Trends and Challenges in the Clinical Translation of Nanoparticulate Nanomedicines: Pathways for Translational Development and Commercialization. Front. Pharmacol. 2018, 9, 790. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Kim, M.J.; Jung, H.H.; Chang, W.S.; Choi, H.S.; Rachmilevitch, I.; Zadicario, E.; Chang, J.W. Safety and feasibility of multiple blood-brain barrier disruptions for the treatment of glioblastoma in patients undergoing standard adjuvant chemotherapy. J. Neurosurg. 2021, 134, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Rezai, A.R.; Ranjan, M.; D’Haese, P.-F.; Haut, M.W.; Carpenter, J.; Najib, U.; Mehta, R.I.; Chazen, J.L.; Zibly, Z.; Yates, J.R.; et al. Noninvasive hippocampal blood−brain barrier opening in Alzheimer’s disease with focused ultrasound. Proc. Natl. Acad. Sci. USA 2020, 117, 9180–9182. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No oil | ATBC | ATBC | ATBC | ATBC | C-90 | C-90 | C-90 | C-90 | C-90 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Oil (v/v) b | 0% | 5% | 10% | 10% a | 5% a | 5% | 10% | 15% | 20% | 20% a |
| Dh (nm) | 61 ± 1 | 52 ± 4 | n/a | 49 ± 1 | 47 ± 1 | 49 ± 3 | 51 ± 2 | 43 ± 1 | n/a | 42 ± 1 |
| PDI | 0.218 | 0.296 | >0.3 | 0.165 | 0.167 | 0.231 | 0.155 | 0.148 | >0.3 | 0.167 |
| Pharmacokinetic Parameters | Values |
|---|---|
| t1/2 (blood terminal half-life) | 3.11 h |
| Cmax (maximum concentration) | 25.90 µL mL−1 |
| Tmax (time to maximum concentration) | 2 h |
| Vd (Volume of distribution) | 0.93 mL |
| Cl (Clearance) | 0.14 mL h−1 |
| AUC0–24 h (Area Under the Curve) | 283.00 h µL mL−1 |
| AUC0–∞ (Area Under the Curve) | 285.70 h µL mL−1 |
| MRT (Mean Residence Time) | 6.65 h |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bérard, C.; Desgranges, S.; Dumas, N.; Novell, A.; Larrat, B.; Hamimed, M.; Taulier, N.; Estève, M.-A.; Correard, F.; Contino-Pépin, C. Perfluorocarbon Nanodroplets as Potential Nanocarriers for Brain Delivery Assisted by Focused Ultrasound-Mediated Blood–Brain Barrier Disruption. Pharmaceutics 2022, 14, 1498. https://doi.org/10.3390/pharmaceutics14071498
Bérard C, Desgranges S, Dumas N, Novell A, Larrat B, Hamimed M, Taulier N, Estève M-A, Correard F, Contino-Pépin C. Perfluorocarbon Nanodroplets as Potential Nanocarriers for Brain Delivery Assisted by Focused Ultrasound-Mediated Blood–Brain Barrier Disruption. Pharmaceutics. 2022; 14(7):1498. https://doi.org/10.3390/pharmaceutics14071498
Chicago/Turabian StyleBérard, Charlotte, Stéphane Desgranges, Noé Dumas, Anthony Novell, Benoit Larrat, Mourad Hamimed, Nicolas Taulier, Marie-Anne Estève, Florian Correard, and Christiane Contino-Pépin. 2022. "Perfluorocarbon Nanodroplets as Potential Nanocarriers for Brain Delivery Assisted by Focused Ultrasound-Mediated Blood–Brain Barrier Disruption" Pharmaceutics 14, no. 7: 1498. https://doi.org/10.3390/pharmaceutics14071498
APA StyleBérard, C., Desgranges, S., Dumas, N., Novell, A., Larrat, B., Hamimed, M., Taulier, N., Estève, M.-A., Correard, F., & Contino-Pépin, C. (2022). Perfluorocarbon Nanodroplets as Potential Nanocarriers for Brain Delivery Assisted by Focused Ultrasound-Mediated Blood–Brain Barrier Disruption. Pharmaceutics, 14(7), 1498. https://doi.org/10.3390/pharmaceutics14071498