
Anti-Survival Effect of SI306 and Its Derivatives on Human Glioblastoma Cells


 ,
,  ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
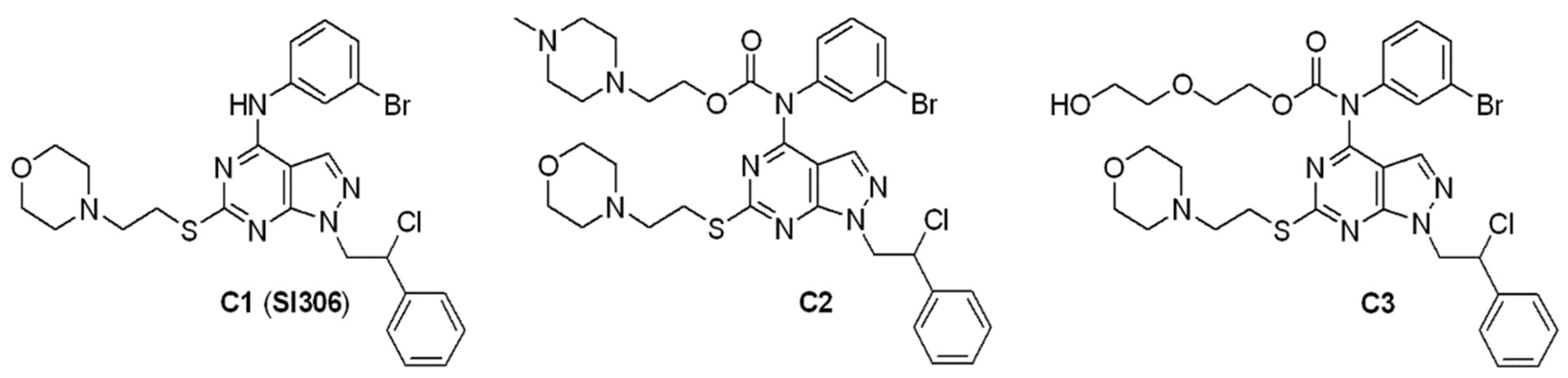
2.1. Chemistry
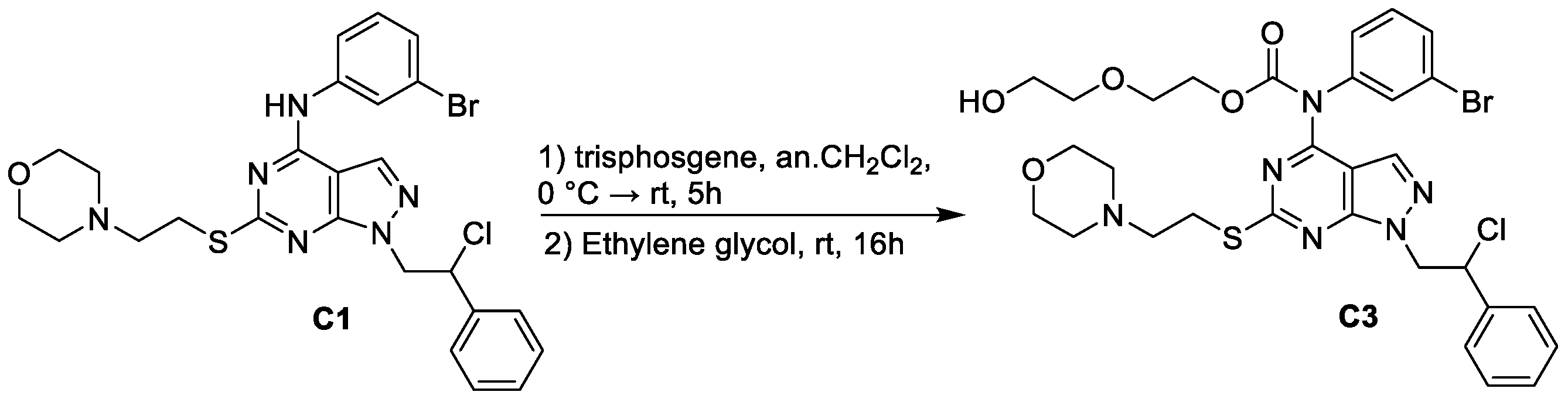
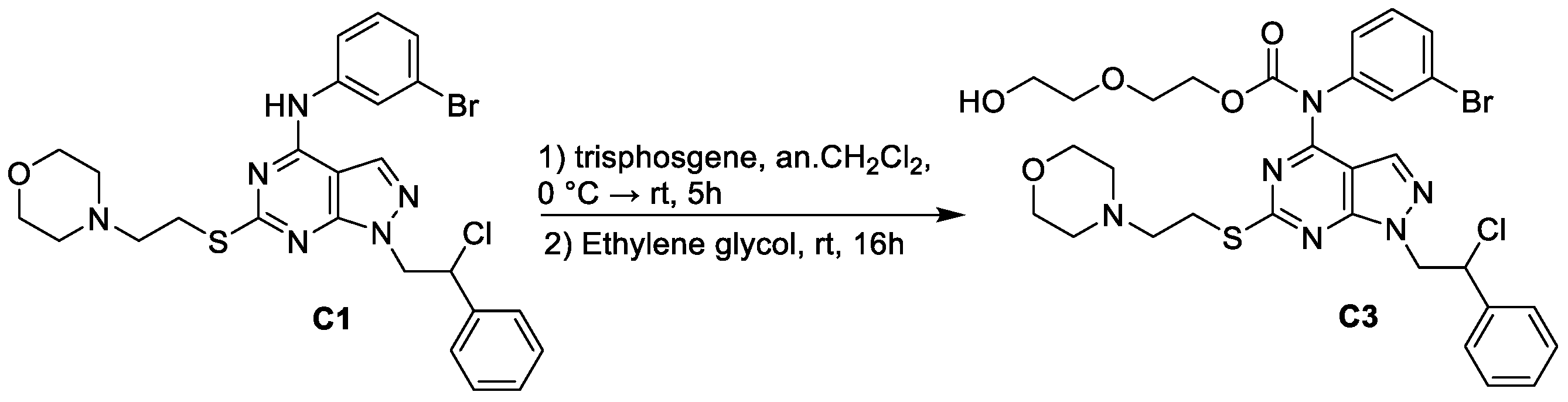
2.2. Synthesis of 2-(2-hydroxyethoxy)ethyl (3-bromophenyl)(1-(2-chloro-2-phenylethyl)-6-((2-morpholinoethyl)thio)-1H-pyrazolo[3,4-d]pyrimidin-4-yl)carbamate (C3)
2.3. Cell Cultures
2.4. Treatments
2.5. Cell Viability Assay
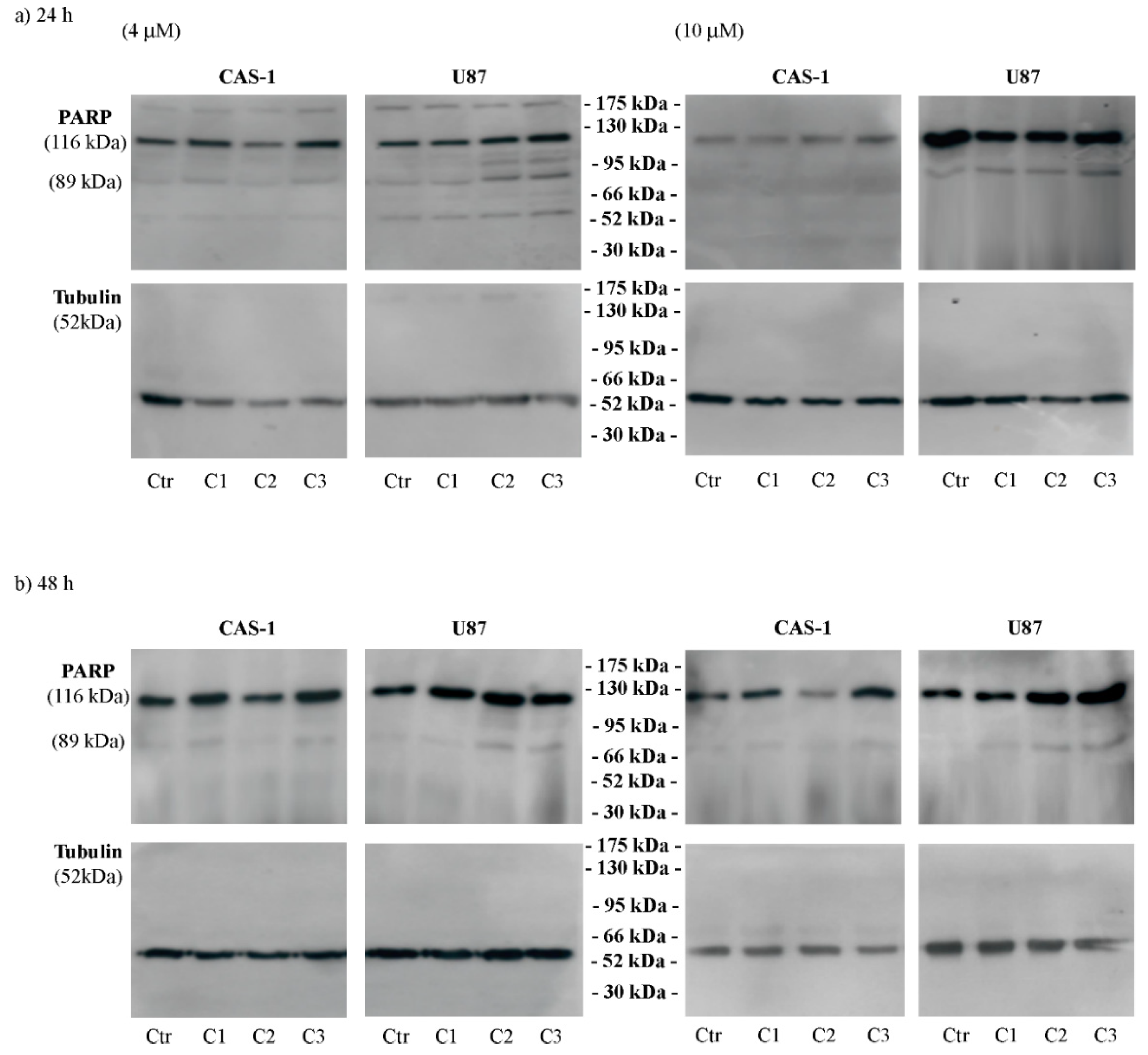
2.6. Blot Analyses
2.7. Statistical Analyses
3. Results
3.1. Chemistry
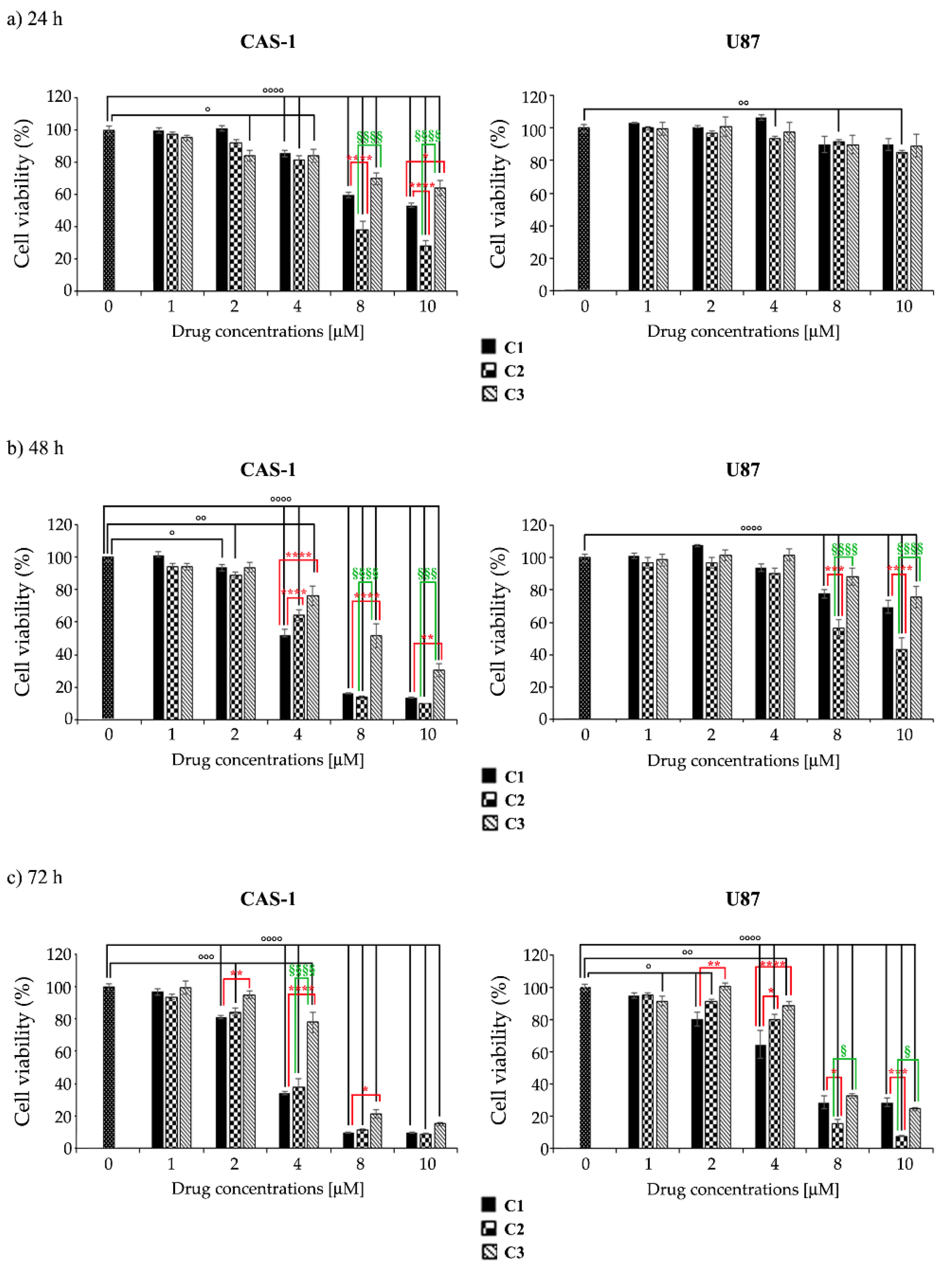
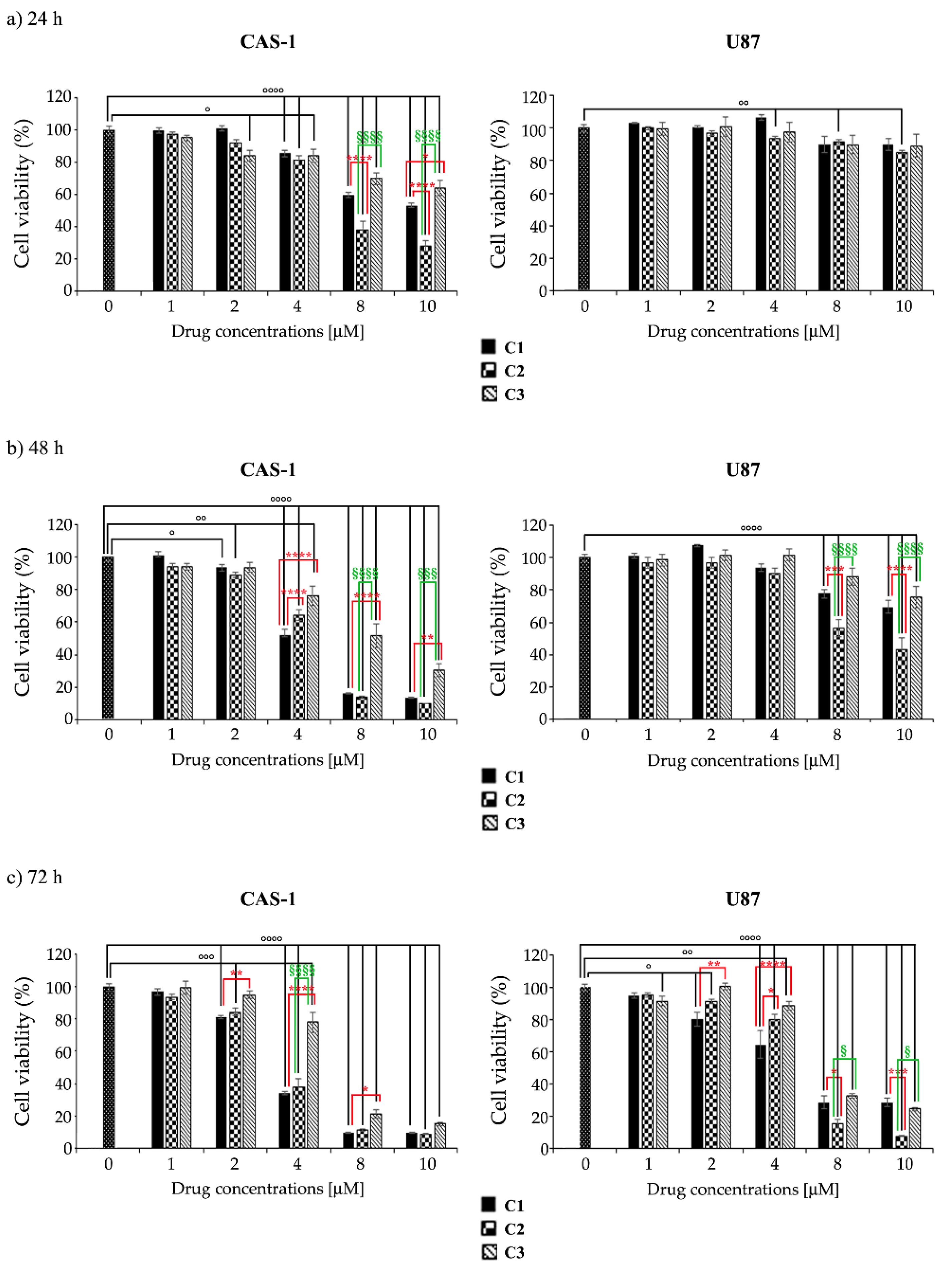
3.2. U87 Cells Are Less Sensitive Than CAS1 to the Effect of SI306- and SI306-Derived Drugs and Show a Major Susceptibility to Compound 2
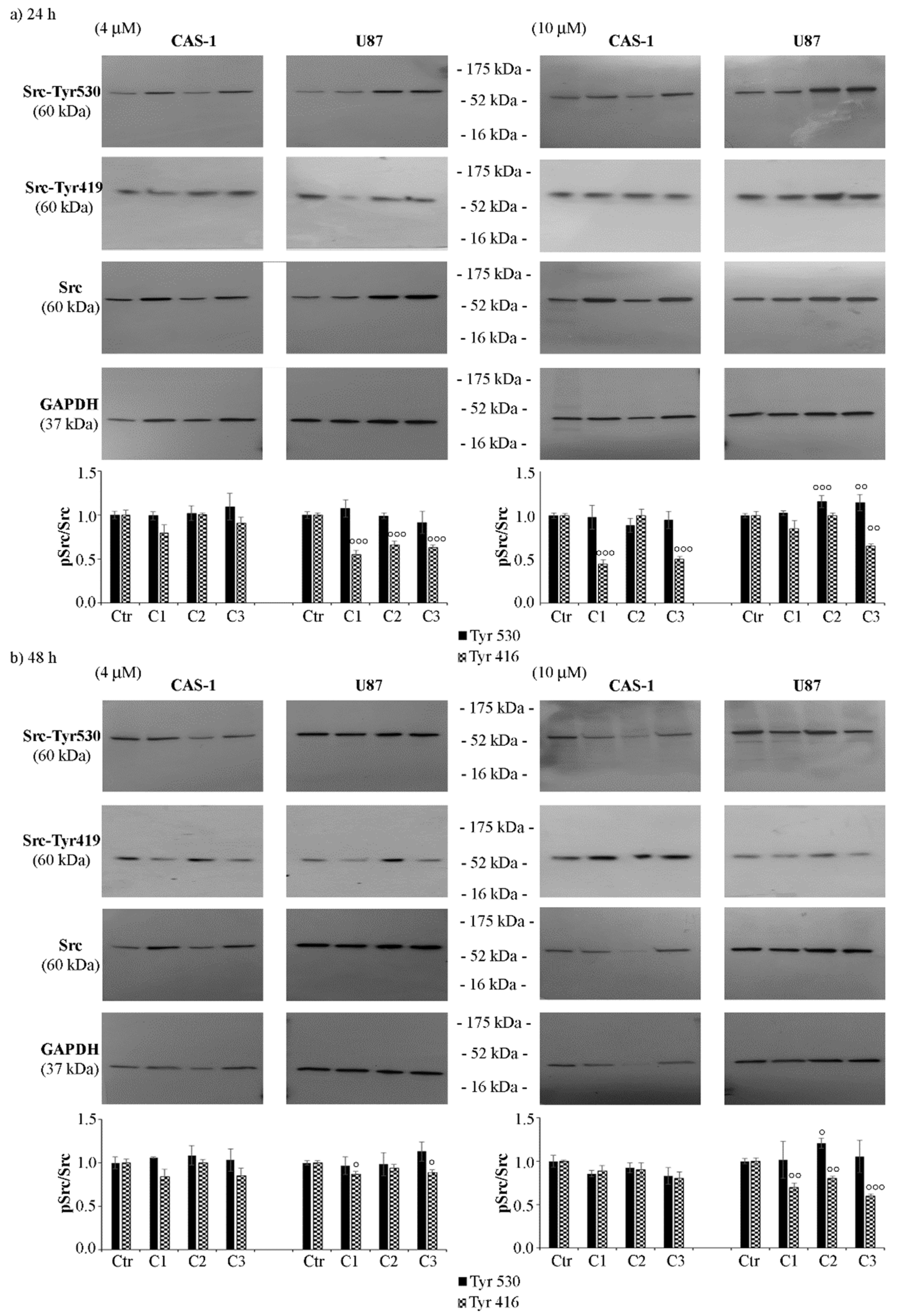
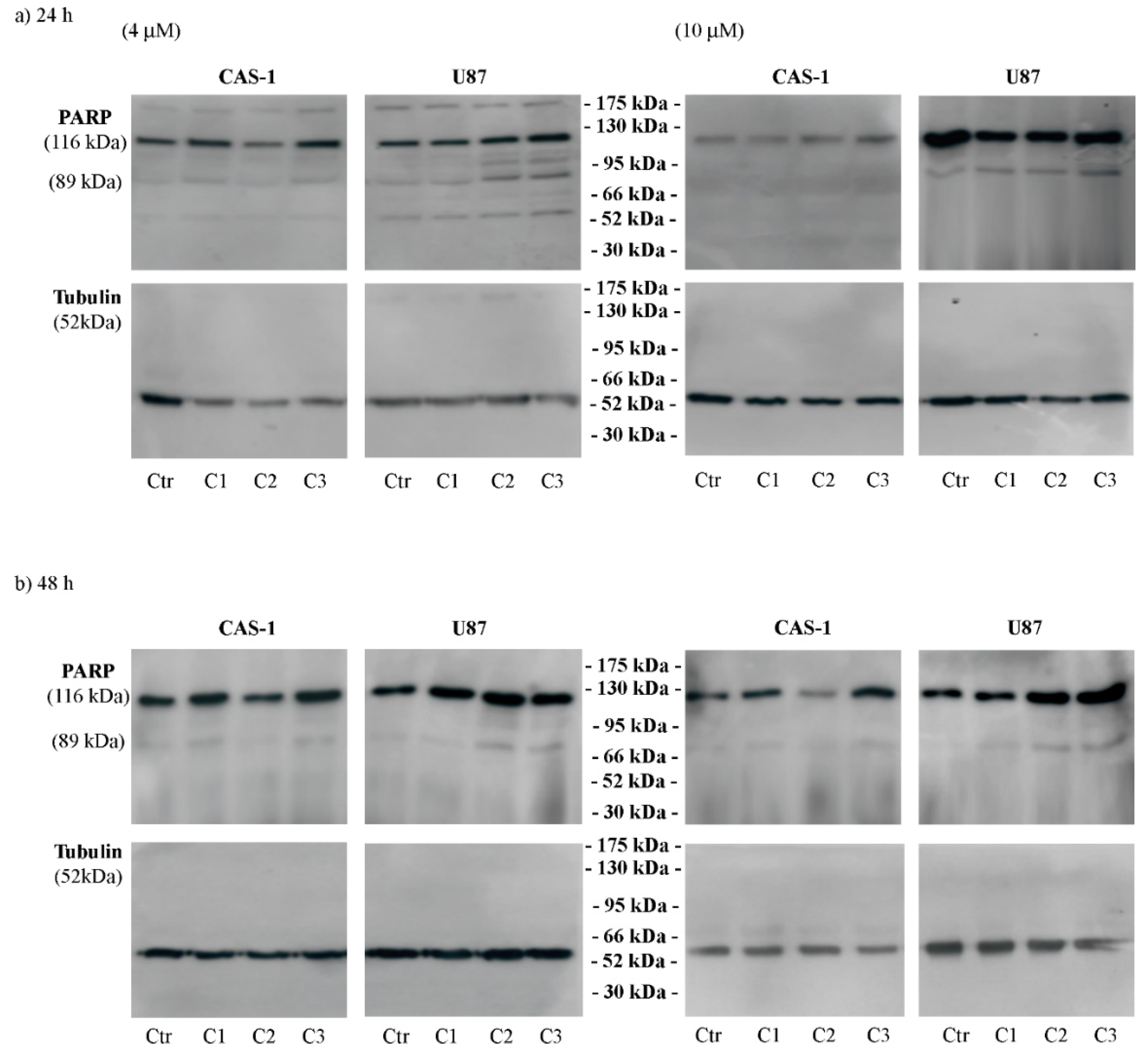
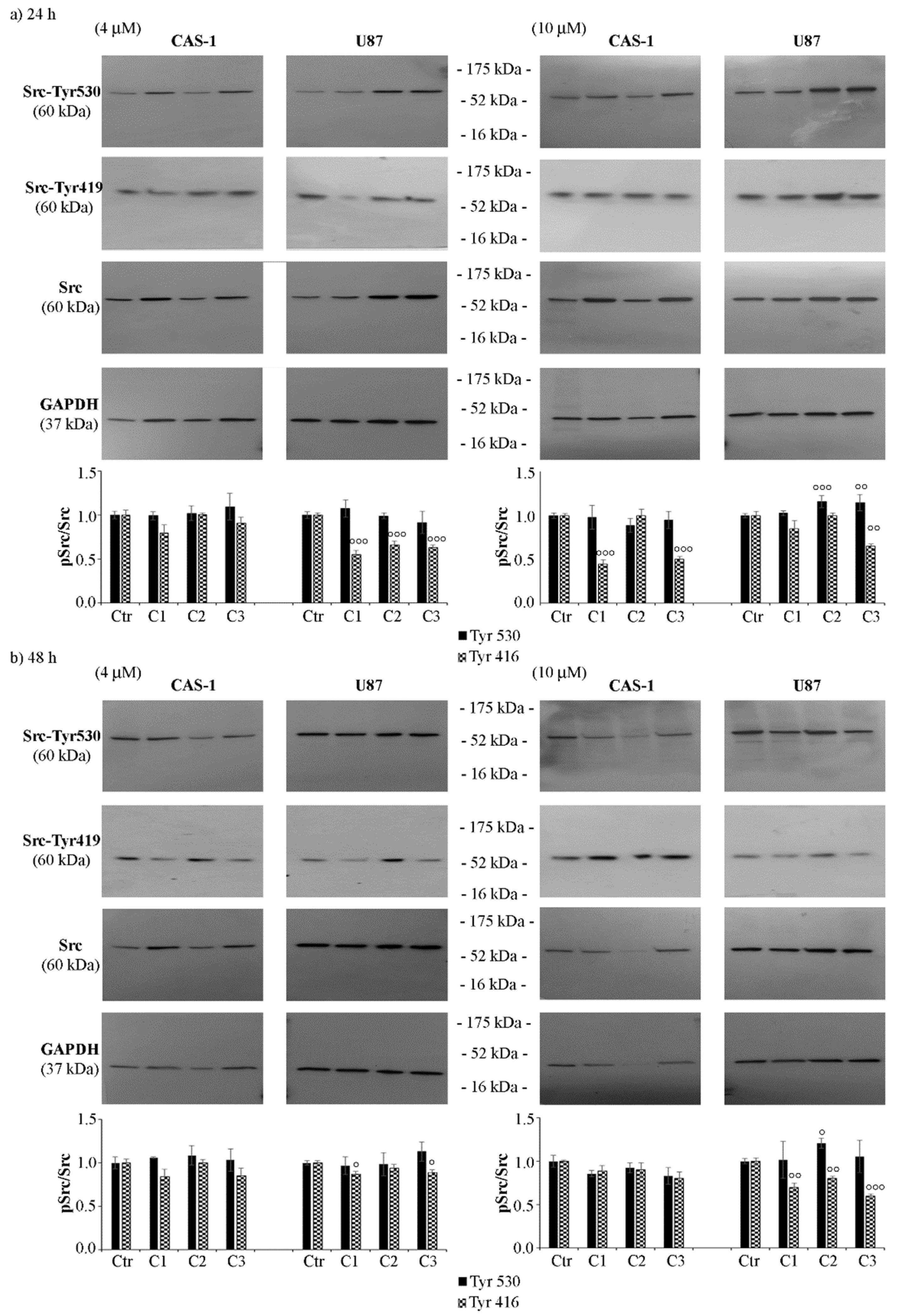
3.3. SI306 Derivatives Inhibit Src Activity in U87 Cells
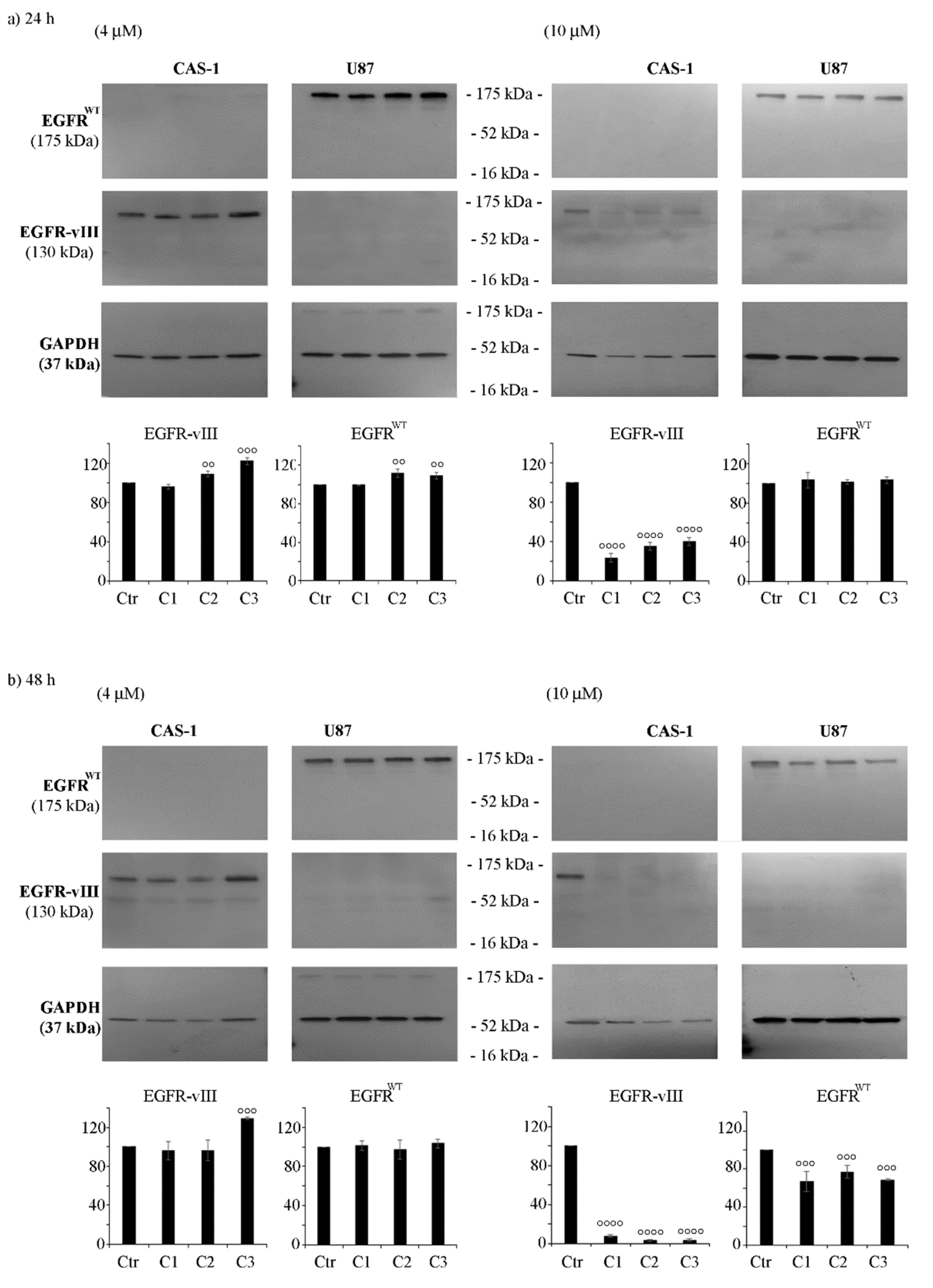
3.4. CAS-1 Cells Express EGFR-vIII While U87 Cells Express EGFRWT and the Exposure to SI306 or Its Derivatives Has a Different Impact on Their Expression Levels
4. Discussion
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.C.; Ashley, D.M.; López, G.Y.; Malinzak, M.; Friedman, H.S.; Khasraw, M. Management of glioblastoma: State of the art and future directions. CA Cancer J. Clin. 2020, 70, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Olar, A.; Aldape, K.D. Using the molecular classification of glioblastoma to inform personalized treatment. J. Pathol. 2014, 232, 165–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, T.A.; Schmassmann, P.; Shekarian, T.; Boulay, J.L.; Ritz, M.F.; Zanganeh, S.; Vom Berg, J.; Hutter, G. Microglia-Centered Combinatorial Strategies Against Glioblastoma. Front. Immunol. 2020, 11, 571951. [Google Scholar] [CrossRef]
- Majc, B.; Novak, M.; Kopitar-Jerala, N.; Jewett, A.; Breznik, B. Immunotherapy of Glioblastoma: Current Strategies and Challenges in Tumor Model Development. Cells 2021, 10, 265. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.K.; Sulman, E.P.; Wen, P.Y.; Kurz, S.C. Novel Therapies for Glioblastoma. Curr. Neurol. Neurosci. Rep. 2020, 20, 19. [Google Scholar] [CrossRef] [PubMed]
- Cirotti, C.; Contadini, C.; Barilà, D. SRC Kinase in Glioblastoma News from an Old Acquaintance. Cancers 2020, 12, 1558. [Google Scholar] [CrossRef] [PubMed]
- Calgani, A.; Vignaroli, G.; Zamperini, C.; Coniglio, F.; Festuccia, C.; Di Cesare, E.; Gravina, G.L.; Mattei, C.; Vitale, F.; Schenone, S.; et al. Suppression of SRC Signaling Is Effective in Reducing Synergy between Glioblastoma and Stromal Cells. Mol. Cancer Ther. 2016, 15, 1535–1544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceccherini, E.; Indovina, P.; Zamperini, C.; Dreassi, E.; Casini, N.; Cutaia, O.; Forte, I.M.; Pentimalli, F.; Esposito, L.; Polito, M.S.; et al. SRC family kinase inhibition through a new pyrazolo[3,4-d]pyrimidine derivative as a feasible approach for glioblastoma treatment. J. Cell Biochem. 2015, 116, 856–863. [Google Scholar] [CrossRef] [PubMed]
- Fallacara, A.L.; Zamperini, C.; Podolski-Renić, A.; Dinić, J.; Stanković, T.; Stepanović, M.; Mancini, A.; Rango, E.; Iovenitti, G.; Molinari, A.; et al. A New Strategy for Glioblastoma Treatment: In Vitro and In Vivo Preclinical Characterization of Si306, a Pyrazolo[3,4-d]Pyrimidine Dual Src/P-Glycoprotein Inhibitor. Cancers 2019, 11, 848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greco, C.; Taresco, V.; Pearce, A.K.; Vasey, C.E.; Smith, S.; Rahman, R.; Alexander, C.; Cavanagh, R.J.; Musumeci, F.; Schenone, S. Development of Pyrazolo[3,4-d]pyrimidine Kinase Inhibitors as Potential Clinical Candidates for Glioblastoma Multiforme. ACS Med. Chem. Lett. 2020, 11, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, F.; Fallacara, A.L.; Brullo, C.; Grossi, G.; Botta, L.; Calandro, P.; Chiariello, M.; Kissova, M.; Crespan, E.; Maga, G.; et al. Identification of new pyrrolo[2,3-d]pyrimidines as Src tyrosine kinase inhibitors in vitro active against Glioblastoma. Eur. J. Med. Chem. 2017, 127, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Vignaroli, G.; Iovenitti, G.; Zamperini, C.; Coniglio, F.; Calandro, P.; Molinari, A.; Fallacara, A.L.; Sartucci, A.; Calgani, A.; Colecchia, D.; et al. Prodrugs of Pyrazolo[3,4-d]pyrimidines: From Library Synthesis to Evaluation as Potential Anticancer Agents in an Orthotopic Glioblastoma Model. J. Med. Chem. 2017, 60, 6305–6320. [Google Scholar] [CrossRef] [PubMed]
- Cammarata, F.P.; Torrisi, F.; Forte, G.I.; Minafra, L.; Bravatà, V.; Pisciotta, P.; Savoca, G.; Calvaruso, M.; Petringa, G.; Cirrone, G.A.P.; et al. Proton Therapy and Src Family Kinase Inhibitor Combined Treatments on U87 Human Glioblastoma Multiforme Cell Line. Int. J. Mol. Sci. 2019, 20, 4745. [Google Scholar] [CrossRef] [Green Version]
- Kostić, A.; Jovanović Stojanov, S.; Podolski-Renić, A.; Nešović, M.; Dragoj, M.; Nikolić, I.; Tasić, G.; Schenone, S.; Pešić, M.; Dinić, J. Pyrazolo[3,4-d]pyrimidine Tyrosine Kinase Inhibitors Induce Oxidative Stress in Patient-Derived Glioblastoma Cells. Brain Sci 2021, 11, 884. [Google Scholar] [CrossRef] [PubMed]
- Nešović, M.; Divac Rankov, A.; Podolski-Renić, A.; Nikolić, I.; Tasić, G.; Mancini, A.; Schenone, S.; Pešić, M.; Dinić, J. Src Inhibitors Pyrazolo[3,4-d]pyrimidines, Si306 and Pro-Si306, Inhibit Focal Adhesion Kinase and Suppress Human Glioblastoma Invasion In Vitro and In Vivo. Cancers 2020, 12, 1570. [Google Scholar] [CrossRef]
- Greenwald, R.B.; Choe, Y.H.; McGuire, J.; Conover, C.D. Effective drug delivery by PEGylated drug conjugates. Adv. Drug Deliv. Rev. 2003, 55, 217–250. [Google Scholar] [CrossRef]
- Wu, T.; Chen, K.; He, S.; Liu, X.; Zheng, X.; Jiang, Z.X. Drug Development through Modification of Small Molecular Drugs with Monodisperse Poly(ethylene glycol)s. Org. Process Res. Dev. 2020, 24, 1364–1372. [Google Scholar] [CrossRef]
- Cvrljevic, A.N.; Akhavan, D.; Wu, M.; Martinello, P.; Furnari, F.B.; Johnston, A.J.; Guo, D.; Pike, L.; Cavenee, W.K.; Scott, A.M.; et al. Activation of Src induces mitochondrial localisation of de2-7EGFR (EGFRvIII) in glioma cells: Implications for glucose metabolism. J. Cell Sci. 2011, 124, 2938–2988. [Google Scholar] [CrossRef] [Green Version]
- Eskilsson, E.; Rosland, G.V.; Talasila, K.M.; Knappskog, S.; Keunen, O.; Sottoriva, A.; Foerster, S.; Solecki, G.; Taxt, T.; Jirik, R.; et al. EGFRvIII mutations can emerge as late and heterogenous events in glioblastoma development and promote angiogenesis through Src activation. Neuro-Oncology 2016, 18, 1644–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, Z.; Aksoy, O.; Zheng, T.; Fan, Q.W.; Weiss, W.A. Epidermal growth factor receptor and EGFRvIII in glioblastoma: Signaling pathways and targeted therapies. Oncogene 2018, 37, 1561–1575. [Google Scholar] [CrossRef] [PubMed]
- Banisadr, A.; Eick, M.; Beri, P.; Parisian, A.D.; Yeoman, B.; Placone, J.K.; Engler, A.J.; Furnari, F. EGFRvIII uses intrinsic and extrinsic mechanisms to reduce glioma adhesion and increase migration. J. Cell Sci. 2020, 133, jcs247189. [Google Scholar] [CrossRef] [PubMed]
- Rutkowska, A.; Stoczyńska-Fidelus, E.; Janik, K.; Włodarczyk, A.; Rieske, P. EGFRvIII: An Oncogene with Ambiguous Role. J. Oncol. 2019, 2019, 1092587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tintori, C.; Fallacara, A.L.; Radi, M.; Zamperini, C.; Dreassi, E.; Crespan, E.; Maga, G.; Schenone, S.; Musumeci, F.; Brullo, C.; et al. Combining X-ray crystallography and molecular modeling toward the optimization of pyrazolo[3,4-d]pyrimidines as potent c-Src inhibitors active in vivo against neuroblastoma. J. Med. Chem. 2015, 58, 347–361. [Google Scholar] [CrossRef] [PubMed]
- Vignaroli, G.; Zamperini, C.; Dreassi, E.; Radi, M.; Angelucci, A.; Sanità, P.; Crespan, E.; Kissova, M.; Maga, G.; Schenone, S.; et al. Pyrazolo[3,4-d]pyrimidine prodrugs: Strategic optimization of the aqueous solubility of dual Src/Abl inhibitors. ACS Med. Chem. Lett. 2013, 4, 622–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marengo, B.; De Ciucis, C.; Ricciarelli, R.; Passalacqua, M.; Nitti, M.; Zingg, J.M.; Marinari, U.M.; Pronzato, M.A.; Domenicotti, C. PKCδ sensitizes neuroblastoma cells to L-buthionine-sulfoximine and etoposide inducing reactive oxygen species overproduction and DNA damage. PLoS ONE 2011, 6, e14661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteleone, L.; Speciale, A.; Valenti, G.E.; Traverso, N.; Ravera, S.; Garbarino, O.; Leardi, R.; Farinini, E.; Roveri, A.; Ursini, F.; et al. PKCα Inhibition as a Strategy to Sensitize Neuroblastoma Stem Cells to Etoposide by Stimulating Ferroptosis. Antioxidants 2021, 10, 691. [Google Scholar] [CrossRef] [PubMed]
- Boulares, A.H.; Yakovlev, A.G.; Ivanova, V.; Stoica, B.A.; Wang, G.; Iyer, S.; Smulson, M. Role of poly(ADP-ribose) polymerase (PARP) cleavage in apoptosis. Caspase 3-resistant PARP mutant increases rates of apoptosis in transfected cells. J. Biol. Chem. 1999, 274, 22932–22940. [Google Scholar] [CrossRef] [Green Version]
- Tintori, C.; Laurenzana, I.; La Rocca, F.; Falchi, F.; Carraro, F.; Ruiz, A.; Esté, J.A.; Kissova, M.; Crespan, E.; Maga, G.; et al. Identification of Hck inhibitors as hits for the development of antileukemia and anti-HIV agents. ChemMedChem 2013, 8, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Ayrapetov, M.K.; Wang, Y.H.; Lin, X.; Gu, X.; Parang, K.; Sun, G. Conformational basis for SH2-Tyr(P)527 binding in Src inactivation. J. Biol. Chem. 2006, 281, 23776–23784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boczek, E.E.; Luo, Q.; Dehling, M.; Röpke, M.; Mader, S.L.; Seidl, A.; Kaila, V.R.I.; Buchner, J. Autophosphorylation activates c-Src kinase through global structural rearrangements. J. Biol. Chem. 2019, 294, 13186–13197. [Google Scholar] [CrossRef]
- Lewis-Tuffin, L.J.; Feathers, R.; Hari, P.; Durand, N.; Li, Z.; Rodriguez, F.J.; Bakken, K.; Carlson, B.; Schroeder, M.; Sarkaria, J.N.; et al. Src family kinases differentially influence glioma growth and motility. Mol. Oncol. 2015, 9, 1783–1798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frederick, L.; Wang, X.Y.; Eley, G.; James, C.D. Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas. Cancer Res. 2000, 60, 1383–1387. [Google Scholar] [PubMed]
- Stettner, M.R.; Wang, W.; Nabors, L.B.; Bharara, S.; Flynn, D.C.; Grammer, J.R.; Gillespie, G.Y.; Gladson, C.L. Lyn kinase activity is the predominant cellular SRC kinase activity in glioblastoma tumor cells. Cancer Res. 2005, 65, 5535–5543. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Bernasconi, P.; Clauser, K.R.; Mani, D.R.; Finn, S.P.; Beroukhim, R.; Burns, M.; Julian, B.; Peng, X.P.; Hieronymus, H.; et al. Bead-based profiling of tyrosine kinase phosphorylation identifies SRC as a potential target for glioblastoma therapy. Nat. Biotechnol. 2009, 27, 77–83. [Google Scholar] [CrossRef] [Green Version]
- Knight, Z.A.; Lin, H.; Shokat, K.M. Targeting the cancer kinome through polypharmacology. Nat. Rev. Cancer 2010, 10, 130–137. [Google Scholar] [CrossRef]
- Agarwal, S.; Mittapalli, R.K.; Zellmer, D.M.; Gallardo, J.L.; Donelson, R.; Seiler, C.; Decker, S.A.; Santacruz, K.S.; Pokorny, J.L.; Sarkaria, J.N.; et al. Active efflux of Dasatinib from the brain limits efficacy against murine glioblastoma: Broad implications for the clinical use of molecularly targeted agents. Mol. Cancer Ther. 2012, 11, 2183–2192. [Google Scholar] [CrossRef] [Green Version]
- Lassman, A.B.; Pugh, S.L.; Gilbert, M.R.; Aldape, K.D.; Geinoz, S.; Beumer, J.H.; Christner, S.M.; Komaki, R.; DeAngelis, L.M.; Gaur, R.; et al. Phase 2 trial of dasatinib in target-selected patients with recurrent glioblastoma (RTOG 0627). Neuro-Oncology 2015, 17, 992–998. [Google Scholar] [CrossRef] [Green Version]
- Galanis, E.; Anderson, S.K.; Twohy, E.L.; Carrero, X.W.; Dixon, J.G.; Tran, D.D.; Jeyapalan, S.A.; Anderson, D.M.; Kaufmann, T.J.; Feathers, R.W.; et al. A phase 1 and randomized, placebo-controlled phase 2 trial of bevacizumab plus dasatinib in patients with recurrent glioblastoma: Alliance/North Central Cancer Treatment Group N0872. Cancer 2019, 125, 3790–3800. [Google Scholar] [CrossRef]
- Molinari, A.; Fallacara, A.L.; Di Maria, S.; Zamperini, C.; Poggialini, F.; Musumeci, F.; Schenone, S.; Angelucci, A.; Colapietro, A.; Crespan, E.; et al. Efficient optimization of pyrazolo[3,4-d]pyrimidines derivatives as c-Src kinase inhibitors in neuroblastoma treatment. Bioorg. Med. Chem. Lett. 2018, 28, 3454–3457. [Google Scholar] [CrossRef] [PubMed]
- Radi, M.; Tintori, C.; Musumeci, F.; Brullo, C.; Zamperini, C.; Dreassi, E.; Fallacara, A.L.; Vignaroli, G.; Crespan, E.; Zanoli, S.; et al. Design, synthesis, and biological evaluation of pyrazolo[3,4-d]pyrimidines active in vivo on the Bcr-Abl T315I mutant. J. Med. Chem. 2013, 56, 5382–5394. [Google Scholar] [CrossRef] [PubMed]
- Colla, R.; Izzotti, A.; De Ciucis, C.; Fenoglio, D.; Ravera, S.; Speciale, A.; Ricciarelli, R.; Furfaro, A.L.; Pulliero, A.; Passalacqua, M.; et al. Glutathione-mediated antioxidant response and aerobic metabolism: Two crucial factors involved in determining the multi-drug resistance of high-risk neuroblastoma. Oncotarget 2016, 7, 70715–70737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alhalabi, O.T.; Fletcher, M.N.C.; Hielscher, T.; Kessler, T.; Lokumcu, T.; Baumgartner, U.; Wittmann, E.; Schlue, S.; Rahman, M.G.; Hai, L.; et al. A novel patient stratification strategy to enhance the therapeutic efficacy of dasatinib in glioblastoma. Neuro-Oncology 2022, 24, 39–51. [Google Scholar] [CrossRef]
- Nishikawa, R.; Ji, X.D.; Harmon, R.C.; Lazar, C.S.; Gill, G.N.; Cavenee, W.K.; Huang, H.J. A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc. Natl. Acad. Sci. USA 1994, 91, 7727–7731. [Google Scholar] [CrossRef] [Green Version]
- Shinojima, N.; Tada, K.; Shiraishi, S.; Kamiryo, T.; Kochi, M.; Nakamura, H.; Makino, K.; Saya, H.; Hirano, H.; Kuratsu, J.; et al. Prognostic value of epidermal growth factor receptor in patients with glioblastoma multiforme. Cancer Res. 2003, 63, 6962–6970. [Google Scholar]
- Congdon, K.L.; Gedeon, P.C.; Suryadevara, C.M.; Caruso, H.G.; Cooper, L.J.; Heimberger, A.B.; Sampson, J.H. Epidermal growth factor receptor and variant III targeted immunotherapy. Neuro-Oncology 2014, 16 (Suppl. 8), viii20-5. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CAS-1 | U87 | |||||
|---|---|---|---|---|---|---|
| Compound | 24 h | 48 h | 72 h | 24 h | 48 h | 72 h |
| C1 | 11.74 ± 0.34 | 3.88 ± 0.15 | 3.03 ± 0.1 | 52.46 ± 1.24 | 18.63 ± 0.32 | 5.28 ± 0.35 |
| C2 | 6.05 ± 0.15 *** | 3.66 ± 0.28 *** | 3.13 ± 0.16 *** | 46.46 ± 0.8 *** | 9.24 ± 0.14 **** | 3.77 ± 0.15 ** |
| C3 | 13.92 ± 0.42 °°°° | 6.97 ± 0.27 ***/°°° | 4.47 ± 0.2 ***/°°° | 54.51 ± 1.16 °°°° | 28.36 ± 0.2 ****/°°°° | 5.79 ± 0.11 °°° |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monteleone, L.; Marengo, B.; Musumeci, F.; Grossi, G.; Carbone, A.; Valenti, G.E.; Domenicotti, C.; Schenone, S. Anti-Survival Effect of SI306 and Its Derivatives on Human Glioblastoma Cells. Pharmaceutics 2022, 14, 1399. https://doi.org/10.3390/pharmaceutics14071399
Monteleone L, Marengo B, Musumeci F, Grossi G, Carbone A, Valenti GE, Domenicotti C, Schenone S. Anti-Survival Effect of SI306 and Its Derivatives on Human Glioblastoma Cells. Pharmaceutics. 2022; 14(7):1399. https://doi.org/10.3390/pharmaceutics14071399
Chicago/Turabian StyleMonteleone, Lorenzo, Barbara Marengo, Francesca Musumeci, Giancarlo Grossi, Anna Carbone, Giulia E. Valenti, Cinzia Domenicotti, and Silvia Schenone. 2022. "Anti-Survival Effect of SI306 and Its Derivatives on Human Glioblastoma Cells" Pharmaceutics 14, no. 7: 1399. https://doi.org/10.3390/pharmaceutics14071399
APA StyleMonteleone, L., Marengo, B., Musumeci, F., Grossi, G., Carbone, A., Valenti, G. E., Domenicotti, C., & Schenone, S. (2022). Anti-Survival Effect of SI306 and Its Derivatives on Human Glioblastoma Cells. Pharmaceutics, 14(7), 1399. https://doi.org/10.3390/pharmaceutics14071399