2. Experimental Section
2.1. Materials and Methods
Synthesis. General. Unless otherwise stated, all materials were obtained from the market and used as specified. Reactions were carried out under argon or nitrogen and monitored by analytical thin-layer chromatography using glass-backed plates (5 × 10 cm) pre-coated with 60 F254 silica gel (supplied by Merck & Co., Inc., Whitehouse Station in Readington Township, NJ, USA). Flame-dried glassware was cooled and used in an argon or nitrogen atmosphere for reactions requiring anhydrous conditions. The resulting chromatograms were observed with a UV lamp (λ = 254 nm) and then charred with a heat gun and immersed in an ethanol solution containing sulphuric acid (3%
v/
v) or vanillin (5%
w/
v) with phosphomolybdic acid (2.5%
w/
v). Solvents used for the reaction included THF, diethyl ether (ether), DMF, toluene, dichloromethane, and pyridine, which were dried and distilled under argon or nitrogen before use. Flash chromatography was routinely used to separate and purify product mixtures using silica gel 60 in sizes 230–400 mesh supplied by Merck, with elution systems given in volume/volume ratios.
1H and
13C NMR spectroscopy was performed by Varian Mercury-300 (300 MHz), Varian Mercury-400 (400 MHz), Bruker Avance Neo AV4400, AV4600, and DMX-600 (600 MHz) collections; chemical shift values are reported in parts per million (δ) units relative to TMS. Multiples are expressed as s (singlet), br s (generalized singlet), d (doublet), t (triplet), q (quadruplet), and dd (doublet of doublet), dt (doublet of triplet), and m (multiplet). Coupling constants (J) are reported in hertz. Using an Agilent 1100 MSD mass spectrometer, electrospray mass spectra (ESMS) were recorded as m/z values, and to obtain HRMS, a Bruker (Impact HD, Tokyo, Japan) Autoflex Max TOF/TOF (MALDI) was used. All compounds with >95% purity were determined by an Agilent 1100 series HPLC system using a C18 column (Thermo Golden, 4.6 mm × 250 mm); see
Supplementary Materials for detailed conditions. The IUPAC nomenclature of the compounds was determined using ACD/Name Pro software.
2.2. 4-(2-(1-(4-Chlorophenyl)cyclohexane-1-carbonyl)-14,14-dimethyl-12-oxo-5,8,13-trioxa-2,11-diazapentadecyl)benzoic acid (2)
To compound 1 (300 mg, 0.48 mmol) in 100 mL MeOH, 10 mL 0.5M LiOH(aq) was added. The reaction mixture was stirred at room temperature for 15 h. The solvent was removed and the residue was redissolved in 100 mL CH2Cl2. The insoluble residue was then filtered off. The filtrate was washed with CH2Cl2, dried over Na2SO4(s) and the solvent was removed under vacuum. The product was obtained as white powder (compound 2, 263 mg, 0.43 mmol, 90%). 1H NMR (400 MHz, CD3OD) δ 7.86 (d, J = 7.4 Hz, 2H), 7.35 (d, J = 8.8 Hz, 2H), 7.30 (d, J = 8.8 Hz, 2H), 6.93 (br, 2H), 4.31 (br, 2H), 3.64 (d, J = 13.6 Hz, 2H), 3.56–3.39 (m, 6H), 3.19 (t, J = 5.7 Hz, 2H), 2.31 (br, 2H), 1.95–1.52 (m, 8H), 1.40 (s, 9H), 1.30 (br, 2H). 13C NMR (101 MHz, CD3OD) δ 177.04, 174.94, 158.40, 146.06, 140.22, 138.33, 133.51, 130.62, 130.19, 128.19, 127.02, 80.09, 71.40, 71.05, 69.70, 53.75, 52.49, 47.32, 41.28, 38.00, 28.76, 26.97, 24.96. HRMS(ESI) calculated for C32H44ClN2O7+: 603.2832, found: 603.2827 (M + H+)+.
2.3. (14R,16R,33R,2S,4S,10E,12Z,14S)-86-Chloro-14-hydroxy-85,14-dimethoxy-33,2,7,10-tetramethyl-12,6-dioxo-7-aza-1(6,4)-oxazinana-3(2,3)-oxirana-8(1,3)-benzenacyclotetradecaphane-10,12-dien-4-yl N-(3-((4-(2-(1-(4-chlorophenyl)cyclohexane-1-carbonyl)-14,14-dimethyl-12-oxo-5,8,13-trioxa-2,11-diazapentadecyl)benzoyl)thio)propanoyl)-N-methyl-L-alaninate (3)
To a stirred solution of compound 2 (150 mg, 0.24 mmol) dissolved in DMF (20 mL), (14S,16S,32R,33S,2R,4S,10E,12E,14R)-86-chloro-14-hydroxy-85,14-dimethoxy-33,2,7,10-tetramethyl-12,6-dioxo-7-aza-1(6,4)-oxazinana-3(2,3)-oxirana-8(1,3)-benzenacyclotetradecaphane-10,12-dien-4-yl N-(3-mercaptopropanoyl)-N-methyl-D-alaninate (DM-1) (100 mg, 0.13 mmol), 4-Dimethylaminopyridine (DMAP, 30 mg, 0.24 mmol), and N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDCl, 30 mg, 0.18 mmol) were added. The mixture was stirred at room temperature in the dark for 15 h. After completion of the reaction, the mixture was extracted with CH2Cl2 (100 mL) and saturated aqueous NaHCO3 (100 mL) and H2O (4 × 100 mL). The combined organic layers were dried over MgSO4, concentrated under reduced pressure. Purification of the crude residue by reversed-phase chromatography on Lichroprep@RP-18 was eluted with ACN gradient grade to get compound 3 (70 mg, 0.05 mmol, 40%). 1H NMR (600 MHz, CDCl3) δ 7.60 (s, 2H), 7.29 (d, J = 8.6 Hz, 2H), 7.20 (d, J = 8.7 Hz, 2H), 6.91 (s, 1H), 6.77 (d, J = 11.3 Hz, 2H), 6.66 (s, 1H), 6.44 (dd, J = 15.2, 10.9 Hz, 1H), 6.23 (s, 1H), 5.63 (dd, J = 15.6, 9.2 Hz, 1H), 5.44 (q, J = 6.7 Hz, 1H), 4.89 (s, 1H), 4.73 (dd, J = 12.1, 3.1 Hz, 1H), 4.33–4.18 (m, 2H), 3.95 (s, 3H), 3.77 (d, J = 12.9 Hz, 1H), 3.67 (s, 1H), 3.59–3.40 (m, 7H), 3.40–3.30 (m, 5H), 3.30–3.22 (m, 3H), 3.16–3.03 (m, 5H), 3.02 (d, J = 9.7 Hz, 1H), 2.81 (s, 4H), 2.69 (d, J = 6.3 Hz, 1H), 2.57 (dd, J = 14.4, 12.1 Hz, 1H), 2.34 (t, J = 7.0 Hz, 1H), 2.27–2.12 (m, 3H), 1.69 (s, 10H), 1.54 (d, J = 15.3 Hz, 1H), 1.41 (s, 10H), 1.29 (dd, J = 10.4, 6.6 Hz, 6H), 1.26–1.19 (m, 2H), 0.79 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 170.90, 170.82, 168.85, 156.06, 155.88, 152.31, 142.09, 141.03, 139.70, 133.54, 132.47, 129.18, 127.54, 127.38, 126.93, 126.51, 125.33, 122.86, 118.73, 113.18, 88.57, 80.99, 79.44, 78.39, 74.24, 70.44, 70.36, 69.00, 67.47, 60.00, 56.76, 56.66, 52.45, 51.19, 46.69, 40.45, 38.99, 37.29, 36.20, 35.34, 34.18, 32.46, 30.63, 28.53, 25.95, 24.28, 23.77, 15.68, 14.70, 13.46, 12.13, 0.13. HRMS(ESI) calculated for C67H89Cl2N5O16S+: 1322.5475, found: 1322.5461 (M + H+)+.
2.4. (14R,16R,33R,2S,4S,10E,12Z,14S)-86-Chloro-14-hydroxy-85,14-dimethoxy-33,2,7,10-tetramethyl-12,6-dioxo-7-aza-1(6,4)-oxazinana-3(2,3)-oxirana-8(1,3)-benzenacyclotetradecaphane-10,12-dien-4-yl N-(4-((4-(2-(1-(4-chlorophenyl)cyclohexane-1-carbonyl)-14,14-dimethyl-12-oxo-5,8,13-trioxa-2,11-diazapentadecyl)benzoyl)thio)-4-methylpentanoyl)-N-methyl-D-alaninate (4)
To a stirred solution of compound 2 (150 mg, 0.24 mmol) dissolved in DMF (20 mL), (14R,16R,33R,2S,4S,10E,12Z,14S)-86-chloro-14-hydroxy-85,14-dimethoxy-33,2,7,10-tetramethyl-12,6-dioxo-7-aza-1(6,4)-oxazinana-3(2,3)-oxirana-8(1,3)-benzenacyclotetradecaphane-10,12-dien-4-yl N-(4-mercapto-4-methylpentanoyl)-N-methyl-D-alaninate (DM-4) (110 mg, 0.14 mmol) and 4-Dimethylaminopyridine (DMAP, 30 mg, 0.24 mmol) and N-(3-Dimethylaminopropyl)-N´-ethylcarbodiimide hydrochloride (EDCl, 30 mg, 0.18 mmol) were added. The mixture was stirred at room temperature in the dark for 15 h. After completion of the reaction, the mixture was extracted with CH2Cl2 (100 mL) and saturated aqueous NaHCO3 (100 mL) and H2O (4 × 100 mL). The combined organic layers were dried over MgSO4, and concentrated under reduced pressure. Purification of the crude residue by reversed-phase chromatography on Lichroprep@RP-18 was eluted with ACN gradient grade to get compound 4 (55 mg, 0.04 mmol, 28%). 1H NMR (600 MHz, CDCl3) δ 7.52 (s, 2H), 7.30 (d, J = 8.7 Hz, 2H), 7.21 (d, J = 8.7 Hz, 2H), 6.91 (s, 1H), 6.80 (s, 1H), 6.76 (d, J = 11.1 Hz, 1H), 6.53 (s, 1H), 6.41 (dd, J = 15.4, 11.1 Hz, 1H), 6.24 (s, 1H), 5.69 (dd, J = 15.3, 9.0 Hz, 1H), 5.41 (q, J = 6.6 Hz, 1H), 4.91 (s, 1H), 4.74 (dd, J = 12.1, 3.1 Hz, 1H), 4.25 (t, J = 12.4 Hz, 2H), 3.95 (s, 2H), 3.66 (d, J = 13.0 Hz, 2H), 3.57–3.41 (m, 7H), 3.35 (s, 4H), 3.32–3.24 (m, 3H), 3.15 (d, J = 11.3 Hz, 1H), 3.03 (s, 2H), 2.96 (s, 2H), 2.80 (s, 3H), 2.57–2.45 (m, 2H), 2.38 (t, J = 12.3 Hz, 2H), 2.25–2.08 (m, 3H), 1.70 (s, 10H), 1.53 (s, 5H), 1.47–1.35 (m, 12H), 1.35–1.22 (m, 10H), 0.96 (d, J = 6.6 Hz, 1H), 0.91–0.81 (m, 3H), 0.77 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 175.12, 172.01, 171.11, 168.81, 156.07, 155.83, 152.35, 141.99, 141.48, 139.23, 136.52, 133.49, 132.47, 129.19, 127.74, 127.04, 126.45, 125.54, 122.18, 118.57, 113.15, 88.53, 81.00, 79.40, 78.18, 74.18, 70.43, 70.37, 68.97, 67.36, 60.03, 56.73, 56.55, 52.43, 51.12, 46.82, 40.46, 38.90, 37.17, 36.22, 36.06, 35.17, 32.37, 30.74, 30.08, 29.45, 28.87, 28.54, 28.06, 25.96, 23.77, 15.58, 14.66, 13.35, 12.18, 0.13. HRMS(ESI) calculated for C70H95Cl2N5NaO16S+: 1364.5944, found: 1364.5928 (M+ H+)+.
2.5. 4-(12-(4-((N-(4-(3,5-bis((bis(pyridin-2-ylmethyl)amino)methyl)phenoxy)butyl)-1-(4-chlorophenyl)cyclohexane-1-carboxamido)methyl)phenyl)-2-(1-(4-chlorophenyl)cyclohexane-1-carbonyl)-12-oxo-5,8-dioxa-2,11-diazadodecyl)benzoic acid (6)
(a) To compound 1 (1.2 g, 1.94 mmol) in 50 mL CH2Cl2, 2 mL of TFA was added. The reaction mixture was stirred at room temperature for 3 h. The NaHCO3(aq.) solution was added and then extracted with 100 mL CH2Cl2. The combined organic extracts were washed with brine, dried over Na2SO4(s), filtered, and evaporated. The product was obtained as oil (methyl 4-((N-(2-(2-(2-aminoethoxy)ethoxy)ethyl)-1-(4-chlorophenyl)cyclohexane-1-carboxamido)methyl)benzoate, 0.96g, 1.85 mmol, 95%). (b) A solution of compound 5 (1.16 g, 1.23 mmol) in 10 mL DMF was heated to 40 °C. 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI, 0.2 g, 1.28 mmol) and hydroxybenzotriazole (HOBt, 0.2g, 1.48 mmol) were added and the reaction was allowed to stir at 40 °C for 30 min. Methyl 4-((N-(2-(2-(2-aminoethoxy)ethoxy)ethyl)-1-(4-chlorophenyl)cyclohexane-1-carboxamido)methyl)benzoate (0.96 g, 1.85 mmol) was added followed by addition of N-methylmorpholine (NMM, 0.5ml, 4.5mmol). The reaction was stirred at 40 °C for 15 h, after which time it was diluted with H2O. The aqueous solution was separated and extracted with 200 mL of CH2Cl2. The combined organic extracts were washed with brine, dried over Na2SO4(s), filtered, and evaporated. Purification of the crude residue by flash chromatography on pH = 7 silica gel eluting with MeOH/CH2Cl2 (1:9) gave rise to ester compounds (0.71 g, 0.49 mmol, 40%). (c) Then, to ester compounds (0.71 g, 0.49 mmol) in 100 mL of MeOH, 10 mL of 0.5M LiOH(aq) was added. The reaction mixture was stirred at room temperature for 15 h. The solvent was removed and the residue was redissolved in 100 mL CH2Cl2. The insoluble residue was then filtered off. The filtrate was dried over Na2SO4(s) and the solvent removed under vacuum. The product was obtained as white powder (compound 6, 0.58 g, 0.41 mmol, 82%). 1H NMR (300 MHz, CDCl3) δ 8.55 (d, J = 4.9 Hz, 4H), 7.93 (s, 1H), 7.73–7.45 (m, 12H), 7.25–7.09 (m, 12H), 7.04–6.56 (m, 6H), 4.12 (q, J = 7.1 Hz, 1H), 3.81 (s, 11H), 3.73–3.50 (m, 17H), 3.48 (s, 1H), 3.34 (br, 2H), 3.05 (br, 3H), 2.97–2.41 (m, 9H), 2.21 (br, 5H), 2.04 (s, 1H), 1.25 (t, J = 7.1 Hz, 5H). 13C NMR (101 MHz, CD3OD) δ 161.01, 149.88, 146.48, 141.99, 139.11, 133.90, 131.04, 130.59, 129.00, 128.61, 128.11, 127.47, 125.08, 124.24, 115.38, 71.84, 71.01, 70.06, 61.29, 60.24, 54.11, 52.85, 41.41, 38.36, 27.67, 27.36, 25.35. HRMS(ESI) calculated for C84H94Cl2N9O8+: 1426.6602, found: 1426.6613(M + H+)+.
2.6. (14R,16R,33R,2S,4S,10E,12Z,14S)-86-Chloro-14-hydroxy-85,14-dimethoxy-33,2,7,10-tetramethyl-12,6-dioxo-7-aza-1(6,4)-oxazinana-3(2,3)-oxirana-8(1,3)-benzenacyclotetradecaphane-10,12-dien-4-yl N-(3-((4-(12-(4-((N-(4-(3,5-bis((bis(pyridin-2-ylmethyl)amino)methyl)phenoxy)butyl)-1-(4-chlorophenyl)cyclohexane-1-carboxamido)methyl)phenyl)-2-(1-(4-chlorophenyl)cyclohexane-1-carbonyl)-12-oxo-5,8-dioxa-2,11-diazadodecyl)benzoyl)thio)propanoyl)-N-methyl-L-alaninate (7)
To a stirred solution of compound 6 (100 mg, 0.070 mmol) dissolved in DMF (3 mL), (14S,16S,32R,33S,2R,4S,10E,12E,14R)-86-chloro-14-hydroxy-85,14-dimethoxy-33,2,7,10-tetramethyl-12,6-dioxo-7-aza-1(6,4)-oxazinana-3(2,3)-oxirana-8(1,3)-benzenacyclotetradecaphane-10,12-dien-4-yl N-(3-mercaptopropanoyl)-N-methyl-D-alaninate (DM-1) (60 mg, 0.081 mmol, 1.1 equiv.), 4-dimethylaminopyridine (DMAP, 15 mg, 0.122 mmol, 1.7 equiv.) and N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDCl, 15 mg, 0.096 mmol, 1.3 equiv.) were added. The mixture was stirred at room temperature in the dark for 15 h. After completion of the reaction, the mixture was extracted with CH2Cl2 (100 mL), saturated aqueous NaHCO3 (100 mL), and H2O (4 × 100 mL). The combined organic layers were dried over MgSO4, and concentrated under reduced pressure. Purification of the crude residue by reversed-phase chromatography on Lichroprep@RP-18 was eluted with ACN gradient grade to get compound 7 (46 mg, 0.021 mmol, 30%). 1H NMR (400 MHz, CDCl3) δ 8.50–8.44 (m, 4H), 7.68 (d, J = 7.9 Hz, 2H), 7.64–7.52 (m, 10H), 7.17 (t, J = 8.7 Hz, 5H), 7.11 (ddd, J = 6.8, 4.9, 1.9 Hz, 5H), 6.95 (br, 2H), 6.87 (br, 2H), 6.77 (d, J = 9.5 Hz, 4H), 6.68–6.61 (m, 1H), 6.43 (dd, J = 15.3, 11.1 Hz, 1H), 6.26 (d, J = 1.2 Hz, 1H), 5.63 (dd, J = 15.3, 9.1 Hz, 1H), 5.43 (t, J = 6.9 Hz, 1H), 4.72 (dd, J = 12.1, 3.1 Hz, 1H), 4.54 (s, 1H), 4.27 (ddd, J = 12.5, 10.5, 2.0 Hz, 2H), 4.09 (s, 1H), 3.93 (s, 4H), 3.89 (s, 2H), 3.78 (s, 10H), 3.59 (t, J = 15.3 Hz, 14H), 3.48 (d, J = 9.0 Hz, 2H), 3.38–3.28 (m, 5H), 3.23 (dt, J = 13.5, 6.7 Hz, 3H), 3.07 (d, J = 17.6 Hz, 5H), 3.00 (d, J = 9.6 Hz, 2H), 2.88 (d, J = 8.3 Hz, 1H), 2.79 (s, 4H), 2.72–2.60 (m, 1H), 2.56 (dd, J = 14.4, 12.1 Hz, 1H), 2.29 (s, 6H), 2.24–2.09 (m, 6H), 1.99 (q, J = 6.5 Hz, 1H), 1.56–1.49 (m, 2H), 1.49–1.37 (m, 2H), 1.34–1.16 (m, 18H), 0.90–0.82 (m, 1H), 0.78 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 170.81, 170.76, 168.83, 159.81, 155.85, 152.32, 149.03, 144.52, 142.05, 141.04, 140.68, 139.55, 136.55, 133.42, 132.44, 130.01, 129.14, 127.65, 127.35, 126.88, 126.42, 125.34, 122.85, 122.08, 118.69, 113.58, 113.19, 88.68, 80.90, 78.33, 74.21, 70.42, 70.03, 68.73, 67.43, 60.10, 59.98, 58.68, 56.69, 56.63, 53.54, 52.40, 51.11, 46.65, 39.84, 38.94, 36.96, 36.35, 36.01, 35.30, 34.15, 32.44, 31.99, 30.55, 29.86, 29.78, 29.70, 29.64, 29.60, 29.57, 29.44, 29.40, 29.34, 27.30, 25.93, 25.89, 25.65, 24.27, 23.75, 22.77, 15.64, 14.66, 14.21, 13.45, 12.10. HRMS(ESI) calculated for C119H139 Cl3 N12NaO17S+: 2167.9060, found: 2167.9042 (M + Na+)+.
2.7. (14S,16S,32R,33S,2R,4S,10E,12E,14R)-86-Chloro-14-hydroxy-85,14-dimethoxy-33,2,7,10-tetramethyl-12,6-dioxo-7-aza-1(6,4)-oxazinana-3(2,3)-oxirana-8(1,3)-benzenacyclotetradecaphane-10,12-dien-4-yl N-(3-((4-(12-(4-((N-(4-(3,5-bis((bis(pyridin-2-ylmethyl)amino)methyl)phenoxy)butyl)-1-(4-chlorophenyl)cyclohexane-1-carboxamido)methyl)phenyl)-2-(1-(4-chlorophenyl)cyclohexane-1-carbonyl)-12-oxo-5,8-dioxa-2,11-diazadodecyl)benzoyl)thio)propanoyl)-N-methyl-D-alaninate*2[Zn(NO3)2]: Zn8_DM1
To a stirred solution of compound 7 (46 mg, 0.021 mmol) in 2 mL EtOH, 1 mL of Zn(NO3)2 (12.4 mg, 0.042 mmol) in EtOH at room temperature was added. After 10 min, the mixture was concentrated under reduced pressure, and the products were precipitated in ether to recover 50 mg of white solid (Zn8_DM1, 0.019 mmol, 90%). 1H NMR (600 MHz, D6-DMSO) δ 8.73–8.63 (m, 3H), 8.45 (s, 1H), 8.15–8.05 (m, 3H), 7.78 (s, 2H), 7.69–7.61 (m, 3H), 7.56 (s, 3H), 7.49 (s, 1H), 7.39 (d, J = 8.3 Hz, 5H), 7.23 (s, 5H), 7.11 (s, 1H), 7.08–6.97 (m, 2H), 6.91 (s, 3H), 6.66 (d, J = 11.5 Hz, 1H), 6.59 (d, J = 14.7 Hz, 1H), 6.56 (d, J = 1.8 Hz, 1H), 5.93 (d, J = 1.5 Hz, 1H), 5.49 (t, J = 12.2 Hz, 1H), 5.35 (q, J = 6.9 Hz, 1H), 4.55 (s, 2H), 4.46 (d, J = 12.1 Hz, 2H), 4.42–4.30 (m, 4H), 4.30–4.11 (m, 3H), 4.11–4.02 (m, 2H), 3.96–3.70 (m, 10H), 3.64 (d, J = 12.4 Hz, 1H), 3.58–3.40 (m, 8H), 3.25 (s, 7H), 3.16–3.03 (m, 2H), 3.03–2.82 (m, 5H), 2.78 (d, J = 9.6 Hz, 1H), 2.68 (s, 3H), 2.56 (s, 1H), 2.45 (t, J = 13.1 Hz, 1H), 2.27 (s, 2H), 2.13 (s, 2H), 2.04–1.93 (m, 2H), 1.77–1.47 (m, 17H), 1.47–1.38 (m, 2H), 1.31–1.14 (m, 11H), 1.11 (d, J = 6.2 Hz, 3H), 1.08–1.00 (m, 2H), 0.84 (t, J = 6.9 Hz, 1H), 0.75 (s, 3H). 13C NMR (151 MHz, D6-DMSO) δ 174.32, 173.76, 170.37, 168.12, 155.02, 154.32, 151.25, 147.93, 144.68, 144.46, 141.05, 140.92, 140.77, 138.90, 133.71, 132.80, 131.05, 129.66, 128.81, 128.12, 127.21, 126.49, 126.15, 124.89, 124.62, 117.63, 116.94, 113.80, 88.25, 80.01, 77.72, 73.21, 69.72, 69.53, 68.93, 67.73, 66.95, 59.97, 56.92, 56.47, 56.15, 56.05, 55.70, 51.60, 50.50, 45.43, 37.78, 36.31, 35.13, 34.60, 33.39, 31.86, 31.29, 29.50, 29.09, 29.03, 28.87, 28.84, 28.75, 28.70, 28.59, 26.57, 25.28, 25.13, 23.73, 23.21, 22.10, 18.57, 15.15, 14.45, 13.96, 13.04, 11.20. HRMS(ESI) calculated for C119H139Cl3 N12O17SZn2+: 1104.9241, found: 1104.9232(M + Zn2+)2+.
2.8. (14R,16R,32S,33R,2S,4R,10E,12E,14S)-86-Chloro-14-hydroxy-85,14-dimethoxy-33,2,7,10-tetramethyl-12,6-dioxo-7-aza-1(6,4)-oxazinana-3(2,3)-oxirana-8(1,3)-benzenacyclotetradecaphane-10,12-dien-4-yl N-(4-((4-(12-(4-((N-(4-(3,5-bis((bis(pyridin-2-ylmethyl)amino)methyl)phenoxy)butyl)-1-(4-chlorophenyl)cyclohexane-1-carboxamido)methyl)phenyl)-2-(1-(4-chlorophenyl)cyclohexane-1-carbonyl)-12-oxo-5,8-dioxa-2,11-diazadodecyl)benzoyl)thio)-4-methylpentanoyl)-N-methyl-L-alaninate (9)
To a stirred solution of compound 6 (100 mg, 0.070 mmol) dissolved in DMF (3 mL), (14R,16R,32S,33R,2S,4R,10E,12E,14S)-86-chloro-14-hydroxy-85,14-dimethoxy-33,2,7,10-tetramethyl-12,6-dioxo-7-aza-1(6,4)-oxazinana-3(2,3)-oxirana-8(1,3)-benzenacyclotetradecaphane-10,12-dien-4-yl N-(4-mercapto-4-methylpentanoyl)-N-methyl-L-alaninate (DM-4) (70 mg, 0.089 mmol, 1.2 equiv.), 4-dimethylaminopyridine (DMAP, 15 mg, 0.122 mmol, 1.7 equiv.), and N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDCl, 15 mg, 0.096 mmol, 1.3 equiv.) were added. The mixture was stirred at room temperature in the dark for 15 h. After completion of the reaction, the mixture was extracted with CH2Cl2 (100 mL), saturated aqueous NaHCO3 (100 mL), and H2O (4 × 100 mL). The combined organic layers were dried over MgSO4, and concentrated under reduced pressure. Purification of the crude residue by reversed-phase chromatography on Lichroprep@RP-18 was eluted with ACN gradient grade to get compound 9 (20 mg, 0.009 mmol, 12%). 1H NMR (400 MHz, CDCl3) δ 8.54–8.43 (m, 4H), 7.70 (d, J = 7.8 Hz, 2H), 7.65–7.54 (m, 8H), 7.51 (d, J = 8.1 Hz, 2H), 7.29 (d, J = 1.9 Hz, 1H), 7.27 (d, J = 2.1 Hz, 1H), 7.24 (s, 1H), 7.20 (d, J = 2.0 Hz, 2H), 7.18 (d, J = 2.1 Hz, 2H), 7.16 (s, 1H), 7.13 (d, J = 1.8 Hz, 1H), 7.12 (t, J = 1.9 Hz, 2H), 7.11 (d, J = 1.9 Hz, 1H), 6.96 (br, 2H), 6.89 (br, 2H), 6.78 (dd, J = 11.4, 7.6 Hz, 4H), 6.53 (d, J = 1.8 Hz, 1H), 6.41 (dd, J = 15.3, 11.1 Hz, 1H), 6.26 (s, 1H), 5.69 (dd, J = 15.3, 9.0 Hz, 1H), 5.41 (q, J = 6.8 Hz, 1H), 5.33 (td, J = 4.5, 2.2 Hz, 1H), 4.74 (dd, J = 12.1, 3.0 Hz, 1H), 4.55 (s, 1H), 4.31–4.15 (m, 2H), 4.10 (s, 1H), 3.94 (s, 2H), 3.78 (s, 8H), 3.71–3.51 (m, 16H), 3.48 (d, J = 9.0 Hz, 2H), 3.32 (s, 3H), 3.25 (d, J = 6.7 Hz, 2H), 3.18–3.08 (m, 2H), 3.01 (d, J = 9.7 Hz, 2H), 2.95 (s, 2H), 2.79 (s, 2H), 2.59–2.43 (m, 2H), 2.43–2.30 (m, 3H), 2.26–2.14 (m, 5H), 2.10 (dd, J = 14.3, 3.0 Hz, 1H), 2.05–1.96 (m, 2H), 1.94 (s, 7H), 1.52 (s, 4H), 1.42 (d, J = 9.2 Hz, 4H), 1.36–1.18 (m, 20H), 1.16 (t, J = 7.3 Hz, 1H), 1.09 (t, J = 6.9 Hz, 1H), 0.91–0.81 (m, 2H), 0.76 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 172.19, 171.30, 169.03, 160.07, 156.06, 152.56, 149.28, 142.22, 141.73, 140.93, 139.36, 136.78, 133.65, 132.69, 130.25, 129.40, 128.04, 127.30, 127.07, 126.63, 125.78, 123.09, 122.43, 122.31, 118.79, 113.82, 113.41, 88.86, 81.18, 78.38, 74.40, 70.67, 70.31, 67.57, 60.36, 60.25, 58.93, 56.92, 56.78, 52.62, 51.37, 47.02, 45.72, 40.10, 39.11, 37.20, 36.55, 36.26, 35.38, 32.60, 32.24, 30.92, 30.29, 30.11, 30.03, 29.94, 29.82, 29.65, 29.58, 29.05, 28.30, 27.55, 26.15, 25.88, 24.00, 23.02, 15.78, 14.87, 14.45, 13.58, 12.39. HRMS(ESI) calculated for C122H145 Cl3 N12NaO17S+: 2209.9529, found: 2209.9518 (M + Na+)+.
2.9. (14R,16R,32S,33R,2S,4R,10E,12E,14S)-86-Chloro-14-hydroxy-85,14-dimethoxy-33,2,7,10-tetramethyl-12,6-dioxo-7-aza-1(6,4)-oxazinana-3(2,3)-oxirana-8(1,3)-benzenacyclotetradecaphane-10,12-dien-4-yl N-(4-((4-(12-(4-((N-(4-(3,5-bis((bis(pyridin-2-ylmethyl)amino)methyl)phenoxy)butyl)-1-(4-chlorophenyl)cyclohexane-1-carboxamido)methyl)phenyl)-2-(1-(4-chlorophenyl)cyclohexane-1-carbonyl)-12-oxo-5,8-dioxa-2,11-diazadodecyl)benzoyl)thio)-4-methylpentanoyl)-N-methyl-L-alaninate*2[Zn(NO3)2]: Zn10_DM4
To a stirred solution of compound 9 (20 mg, 0.009 mmol) in 2 mL, 1 mL of Zn(NO3)2 (5.3 mg, 0.018 mmol) in EtOH at room temperature was added. After 10 min, the mixture was concentrated under reduced pressure and products were precipitated in ether to recover 23 mg of white solid (Zn10_DM4, 0.008 mmol, 90%). 1H NMR (600 MHz, D6-DMSO) δ 8.68 (d, J = 5.3 Hz, 3H), 8.45 (s, 1H), 8.19–7.99 (m, 3H), 7.78 (s, 2H), 7.65 (t, J = 6.4 Hz, 3H), 7.56 (s, 3H), 7.39 (t, J = 10.0 Hz, 5H), 7.32–7.14 (m, 6H), 7.07 (s, 1H), 7.02 (d, J = 17.6 Hz, 2H), 6.96–6.89 (m, 2H), 6.87 (s, 1H), 6.66–6.58 (m, 1H), 6.53 (dd, J = 15.0, 11.2 Hz, 1H), 6.39 (s, 1H), 5.91 (s, 1H), 5.61 (dd, J = 15.0, 9.1 Hz, 1H), 5.40–5.23 (m, 2H), 4.63–4.41 (m, 3H), 4.41–4.30 (m, 3H), 4.17 (s, 2H), 4.11–3.96 (m, 2H), 3.86 (s, 6H), 3.78 (d, J = 15.8 Hz, 4H), 3.63 (s, 1H), 3.57–3.41 (m, 8H), 3.25 (s, 6H), 3.09 (s, 1H), 3.04–2.83 (m, 4H), 2.77 (d, J = 9.6 Hz, 1H), 2.67 (s, 2H), 2.57 (d, J = 14.9 Hz, 1H), 2.41 (t, J = 13.0 Hz, 1H), 2.29 (d, J = 15.6 Hz, 4H), 2.13 (s, 3H), 2.05–1.90 (m, 4H), 1.78–1.48 (m, 20H), 1.44 (td, J = 11.9, 5.0 Hz, 3H), 1.35 (d, J = 17.9 Hz, 3H), 1.31–1.16 (m, 13H), 1.13 (d, J = 6.7 Hz, 2H), 1.10 (d, J = 6.4 Hz, 2H), 1.05 (t, J = 7.0 Hz, 1H), 0.85 (t, J = 7.0 Hz, 1H), 0.73 (s, 2H). 13C NMR (151 MHz, D6-DMSO) δ 174.57, 173.99, 171.41, 171.10, 168.36, 155.33, 154.58, 151.53, 148.19, 144.78, 141.16, 141.03, 138.45, 133.98, 132.95, 131.34, 129.93, 129.10, 128.81, 127.45, 126.57, 125.47, 125.16, 124.88, 121.62, 117.91, 117.21, 114.02, 88.50, 80.28, 77.82, 73.42, 69.97, 69.80, 69.21, 67.06, 60.27, 57.21, 56.65, 56.39, 56.31, 55.98, 51.97, 51.18, 50.78, 45.93, 40.33, 37.88, 36.62, 35.92, 35.40, 34.83, 32.04, 31.56, 30.64, 29.94, 29.50, 29.36, 29.30, 29.25, 29.14, 29.11, 29.02, 28.97, 28.85, 27.30, 26.83, 25.54, 25.39, 23.49, 22.37, 18.84, 15.25, 14.67, 14.22, 13.30, 11.63. HRMS(ESI) calculated for C122H145Cl3N12O17SZn2+: 1125.9475, found 1125.9469 (M + Zn2+)2+.
2.10. Cell Culture/Viability Assay/Data Analysis
HCC1806 cells were grown in RPMI 1640 medium, and MTS assays were performed to check cell viability. Cells (2500–3000 cells/well) were grown in flat-bottomed 96-well plates for 24 h, and then serial dilutions of the compound were added, and cells were allowed to incubate for a further 72 h. At the end of the 72-h incubation period, the medium was removed, and 100 μL of a solution including a mixture of MTS and PMS was added. The cells were incubated for 1.5 h at 37 °C in a humidified incubator with 5% CO2 to allow viable cells to convert tetrazolium salts to formazan. Conversion to formazan was measured by absorbance (490 nm) using a BioTek PowerWave-X absorbance microplate meter. The data collected were normalized using a DMSO-treated control (100% viability) and a background control (0% viability) to verify growth inhibition. At the same time, IC50 values were calculated using GraphPad Prism version 4 software (San Diego, CA, USA), i.e., the reduction in cell viability compared to the DMSO-treated control resulted in the amount of compound that resulted in a 50% reduction in cell viability compared to the DMSO-treated control.
2.11. PS SPR Binding Assay
Phospholipids, 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) and 1,2-dioleoyl-sn-glycero-3-[phosphate-L-serine] (DOPS) were obtained as chloroform solutions from Avanti Polar Lipids (Alabaster, AL, USA). These stock solutions were mixed in the specified proportions. A 0.4 mL aliquot of the lipid solution at a 10 mg/mL concentration was added to a round bottom flask and evaporated under a stream of N2 to provide a thin lipid film. The lipid films were rehydrated in PBS buffer for at least 1 h at room temperature. The resulting suspension was extruded through a 100 nm polycarbonate filter using an Avanti MiniExtruder according to the manufacturer’s instructions. Zeta potential (ZP) and liposome size distribution were recorded by dynamic light scattering (DLS) and microelectrophoresis using a Zetasizer Nano ZS instrument. The binding kinetics between conjugates and liposomes were recorded at 25 °C using a Biacore T200 biosensor equipped with an L1 sensor chip (GE Healthcare, Chicago, IL, USA). The new sensor chip was pretreated with running buffer (5% DMSO in phosphate buffered saline (PBS) at a pH of 7.4) and two consecutive 30 s pulses of 2:3 v/v 50 mM HCl/isopropanol at a flow rate of 30 μL/min. New liposome capture plates were prepared for each binding cycle. In PBS buffer, liposomes are diluted to 0.5–1 mM and captured to saturation in isolated flow cells at 2–5 μL/min (30–150 s). The conjugate is first diluted with running buffer and injected onto the liposome surface in one injection. The conjugation and dissociation phases were examined for 60 s at a flow rate of 30 μL/min. At the end of each binding cycle, the surface was regenerated by injecting 2:3 v/v 50 mM hydrochloric acid/isopropanol and equilibrated with running buffer before the next injection of the test compound. Non-specific binding was removed by subtracting the SPR signal from the reference flow cell (DOPC immobilized surface). The sensory maps were fully fitted using a bivalent analyte model with BIAcore T200 evaluation software 3.0.
2.12. IVIS Imaging of HCC1806 Tumors
HCC1806 tumor-bearing mice were used when the average tumor volume reached approximately 500–600 mm3. Tumor volume (mm3) was calculated by the formula. Volume = (length × width2)/2, measured with digital calipers. The non-targeting dye 794 at a dose of 2 mg/kg was administered intravenously. All treated mice were imaged at 24 and 48 h using the IVIS spectroscopy system. Briefly, mice were anaesthetized by inhalation of 2.5% isoflurane and placed on the platform of the IVIS instrument (IVIS® Spectrum, PerkinElmer, Waltham, MA, USA) with the following imaging conditions set: excitation filter, 745 nm; emission filter, 820 nm; exposure time, 1 s, bin, 8 (medium); f/stop, 2; field of view, 22.7 cm. Fluorescence intensity was quantified, and images were processed using Living Image 4.5 software (PerkinElmer, Alameda, CA, USA).
2.13. Pharmacokinetic Studies of Conjugates
Six-week-old male ICR mice from BioLASCO Taiwan were divided into three groups and administered intravenously at a 5 mg/kg dose. Blood samples were taken from each animal at time points 0.003, 0.083, 0.25, 0.5, 1, 2, 4, 6, 8, and 24 h and stored on ice (0–4 °C). The plasma was separated from the blood by centrifugation (in a Beckman Allegra 6R at 3000 rpm, 4 °C for 15 min) and stored under freezing conditions (−20 °C). Plasma and harvested tumor samples were stored at −80 °C until use. Fifty microliters of mouse plasma or tumor samples homogenized with ddH2O at a dilution ratio of 1:3 (w/v) with MiniBeadbeater-16 (BioSpec Products Inc., Bartlesville, OK, USA) were mixed with 100 μL of acetonitrile containing 250 ng/mL BPR0L187. The mixture was vortexed for 30 s and then centrifuged at 15,000× g for 20 min. The supernatant was transferred to a clean test tube, and 15 µL of supernatant was injected into the LC/MS/MS. Plasma samples were analyzed by liquid chromatography-tandem mass spectrometry (LC/MS/MS). The chromatography system was an Agilent 1200 series liquid chromatography system and an Agilent ZORBAX Eclipse XDB-C8 column (5 µm, 3.0 × 150 mm) connected with an MDS Sciex API4000 tandem mass spectrometer equipped with an ESI in forwarding scan mode at 600 °C. Data acquisition was performed by multiple reaction monitoring (MRM). A gradient system was used to separate the analytes from the IS. Mobile phase A was a 10 mM aqueous ammonium acetate solution containing 0.1% formic acid. Mobile phase B was acetonitrile. The gradient profile was as follows. 0.0–1.1 min, 50% B; 1.2–3.7 min, 55% B-90% B; 3.8–5.0 min, 90% B-50% B. The flow rate was 1.5 mL/min. The autosampler was set to inject 15 µL of the sample every 5 min.
4. Results and Discussion
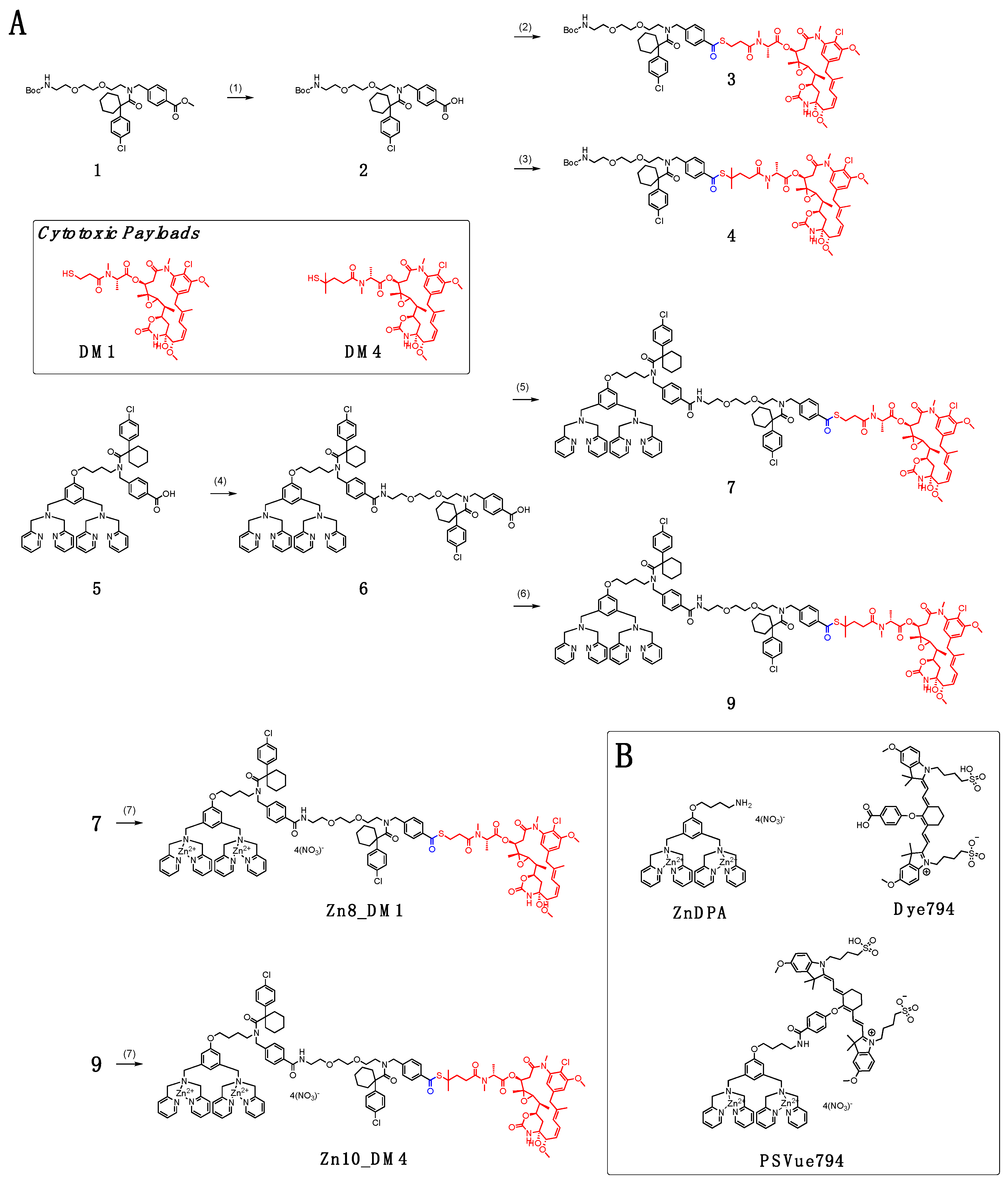
The release of thioester-linked maytansinoid can be modulated by installing cyclohexyl-para-chlorophenyl steric hindered functional groups in the proximity. The longevity of the intact conjugate in systemic circulation serves as a key parameter during treatment in facilitating the selective accumulation of such a conjugate at the tumor site. As for targeting prodrugs, we envision that maytansinoid cytotoxicity is initially masked in the systemic circulation, and through the engagement of the targeting ligand, the payload was then uncaged at the tumor site.
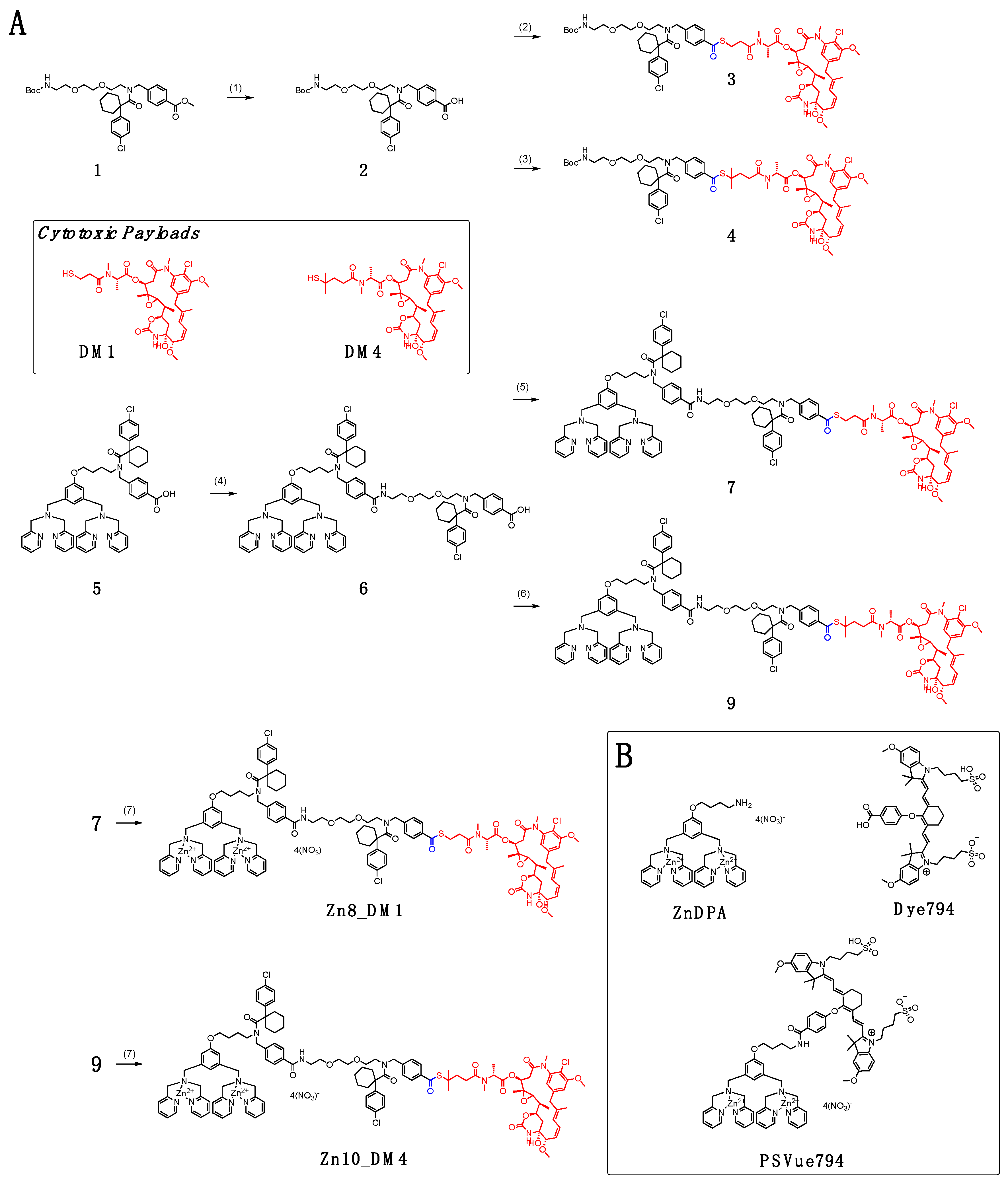
In
Scheme 1A, conjugation between Boc-protected amino polyethylene glycol (PEG) with cyclohexyl-para-chlorophenyl periphery carboxylic acid
2 and maytansinoids DM1 and DM4 afforded linker payloads
3 and
4, respectively. We installed a small-unit linear PEG to bridge the targeting ligand and the cytotoxic payload. Because large-unit pegylation can significantly increase the solubility of the conjugate, it might result in steric interference, leading to loss of the targeting ligand’s binding ability [
27].
To incorporate the linker-maytansinoids into the small molecule drug conjugate (SMDC) form, we first coupled the dipicolylamine (DPA) carboxylic acid
5 with the amino PEG harnessing cyclohexyl-para-chlorophenyl periphery that yielded the carboxylic acid precursor
6. We have shown that cyclohexyl-para-chlorophenyl in
6 resulted in an advantageous payload release profile and reduced toxicity of the released ZnDPA ligand [
18]. With the activation of the carboxylic acid
6 by EDCl and HOBt in dry
N,
N-dimethylformamide, synthesis of new DPA-maytansinoid conjugates with pegylated linkers was then carried out with coupling of DM1 or DM4 to furnish conjugate
7 and
9, respectively. Incubating each DPA–drug conjugate
7 or
9 with two equivalents of zinc nitrate at room temperature in 1:1 dichloromethane/methanol provided the eventual ZnDPA conjugates,
Zn8_DM1 and
Zn10_DM4.
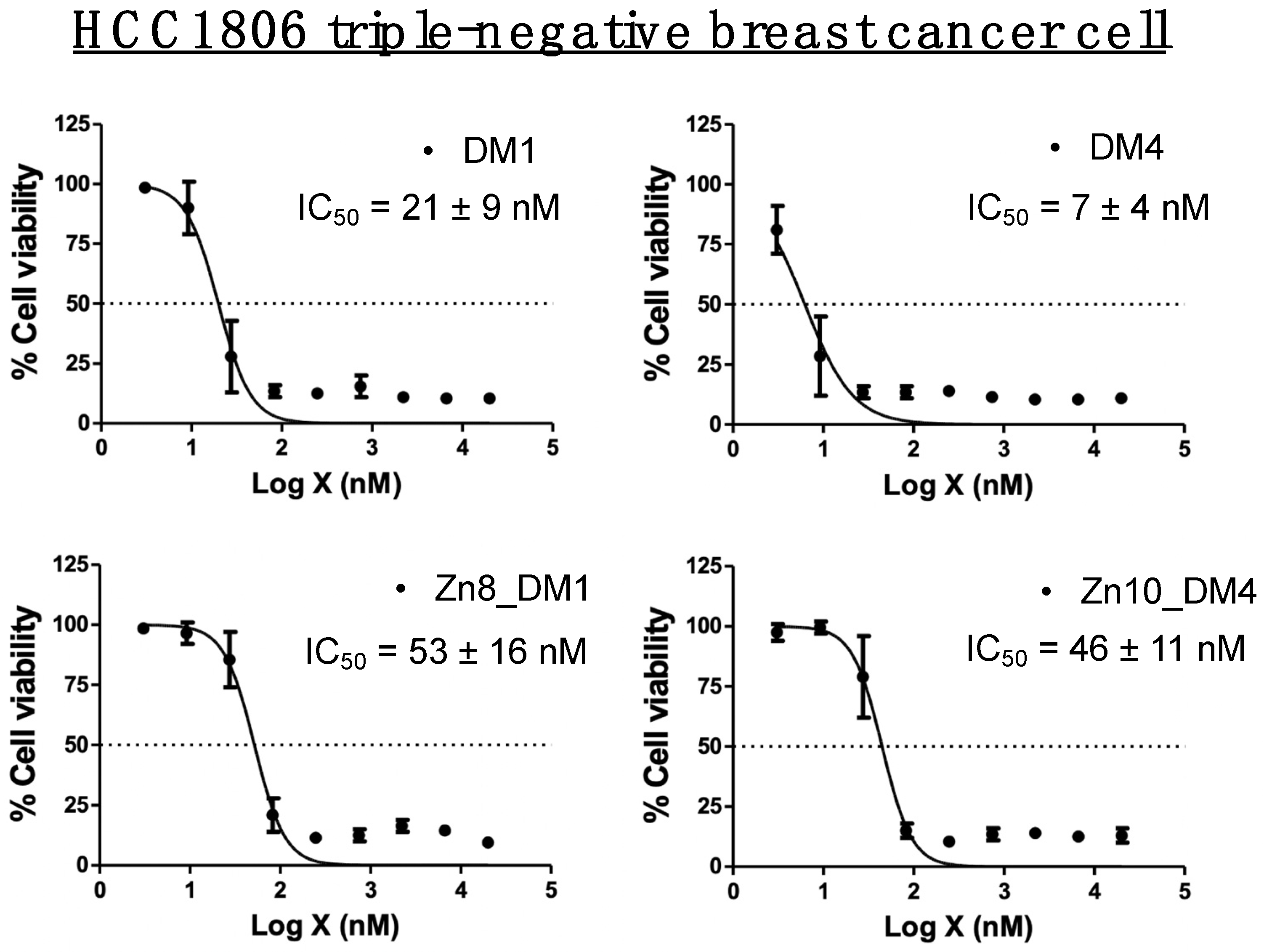
We first examined plasma stabilities and in vitro cytotoxicities of maytansinoids and their respective conjugates
Zn8_DM1 and
Zn10_DM4 against the HCC1806 triple-negative breast cancer cell line (
Figure 1). No cleavage of the maytansinoids from either the linker-drug
3 and
4 or conjugates
Zn8_DM1 and
Zn10_DM4 were observed after 24 h incubation with plasma (
Supporting Figure S1). The cytotoxic payloads DM1 and DM4 inhibited HCC1806 at 21 nM and 7 nM, respectively. By this targeting prodrug strategy, new conjugates
Zn8_DM1 (53 nM) and
Zn10_DM4 (46 nM) exhibited 2.5–6 fold attenuation in cytotoxicities against HCC1806 triple-negative breast cancer cells relative to their parent payloads, suggesting that the thioester-linked maytansinoids shielded their cytotoxic properties in vitro (
Figure 1).
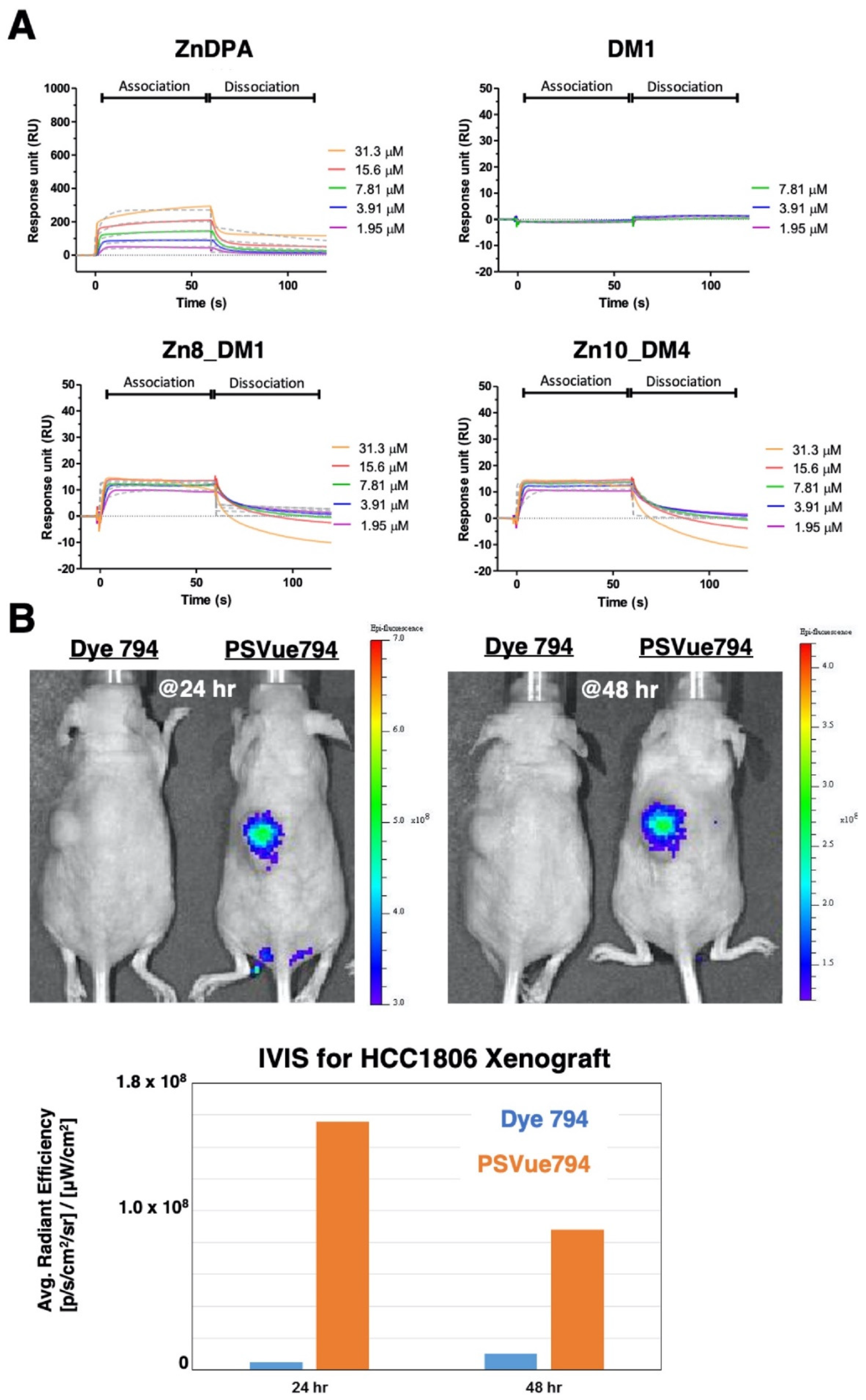
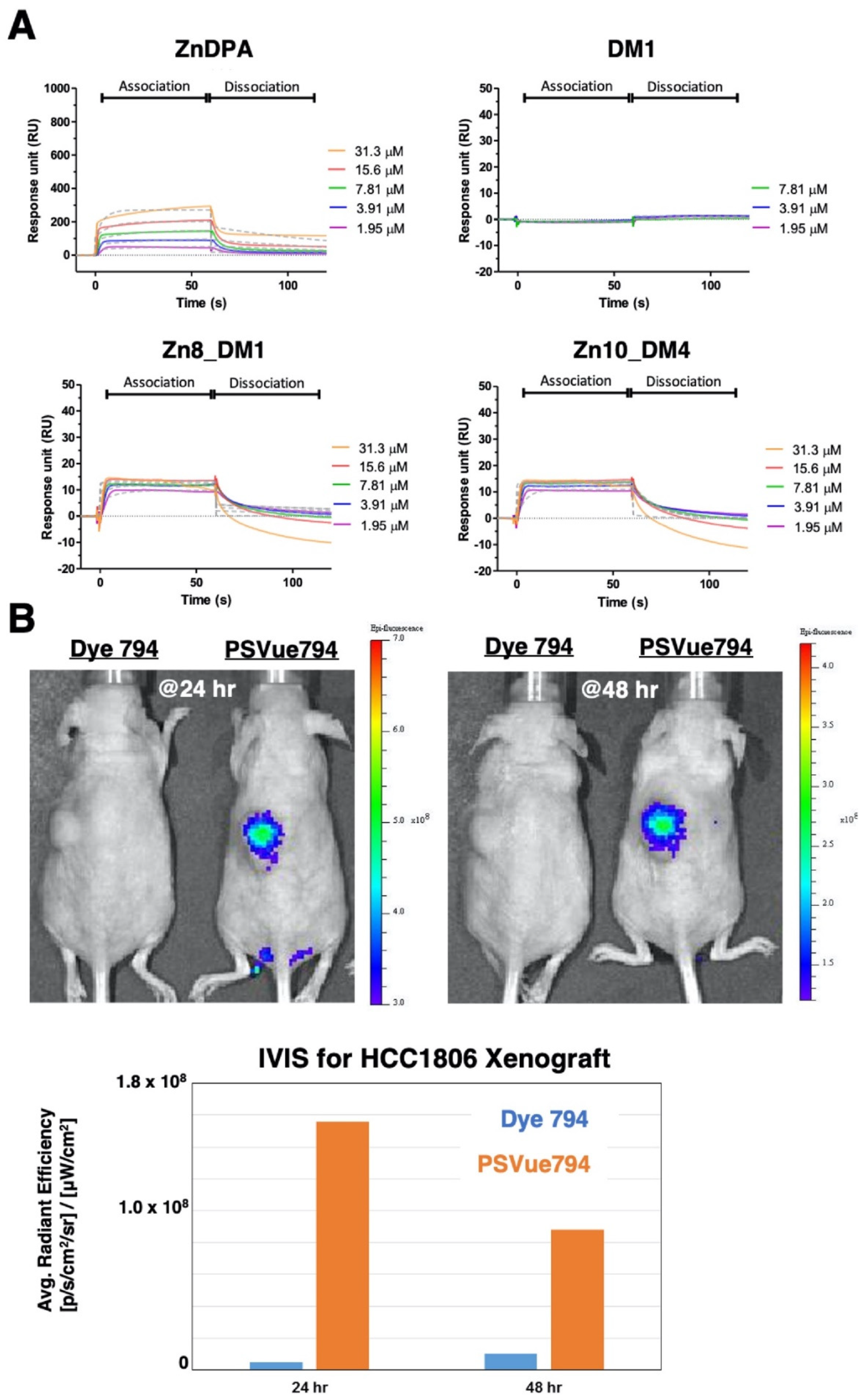
To probe the utility of such thioester-linked maytansinoids in SMDC, we first assessed whether this addition of linker-drug allowed the ZnDPA motif to retain its’ binding property towards phosphatidylserine (PS). As the attachment of a therapeutic payload near the small molecule targeting ligand might diminish receptor binding, we performed in vitro binding studies through surface plasmon resonance (SPR) measurements between
Zn8_DM1 or
Zn10_DM4 and PS-coated liposomes (DOPC/ DOPS (3:1,
v/
v)) according to procedures reported in the literature [
28]. The maytansinoid DM1 did not show association properties with the PS-coated liposomes (
Figure 2A). The ZnDPA motif played an essential role for PS recognition. We showed that the employment of linker-maytansinoids in
Zn8_DM1 or
Zn10_DM4 still allowed recognition between the ZnDPA moiety and PS-coated liposomes (
Figure 2A). To examine PS expression in the HCC1806 xenograft model, a near infra-red fluorophore-conjugated ZnDPA (PSVue
®794) was intravenously injected in vivo in comparison to the fluorophore (Dye 794) lacking the targeting moiety. We observed an efficient and lasting accumulation of PS-seeking ZnDPA probe over dye_794 at the tumor site (
Figure 2B), supporting the expression of PS at the HCC1806 tumor and providing important insight into the ZnDPA-based therapeutic delivery strategy.
Leveraging the favorable plasma stabilities of the linker maytansinoids, we then evaluated the in vivo systemic stability of
Zn8_DM1 and
Zn10_DM4. With a single intravenous dose of 5 mg/kg in the ICR mice, we profiled the in vivo systemic pharmacokinetic properties of
Zn8_DM1 and
Zn10_DM4 (
Table 1). In general, small volume distributions V
ss (0.2~0.6 L/kg) of the maytansinoid conjugates were observed, suggesting that systemic distributions of
Zn8_DM1 and
Zn10_DM4 were limited to the circulation. With a slow and similar clearance (CL) rate at 0.5 mL/min/kg among the conjugates,
Zn8_DM1 and
Zn10_DM4 exhibited a large AUC ratio between intact conjugates and released payloads within 24 h in circulation. This data alleviated the concerns on the premature release of the respective cytotoxic payload from
Zn8_DM1 or
Zn10_DM4 and should facilitate adequate delivery of the cytotoxic cargoes at the tumor site. In particular,
Zn10_DM4 exhibited a higher intact conjugate AUC over that of
Zn8_DM1, which might be attributed to the steric hindering with methyl groups on the adjacent carbon next to the sulfur group in DM4. By installing newly constructed thioester-linked maytansinoids in the ZnDPA-based conjugates, we demonstrated stable systemic exposure of
Zn8_DM1 or
Zn10_DM4 in vivo with PS-targeting ability and prodrug properties.
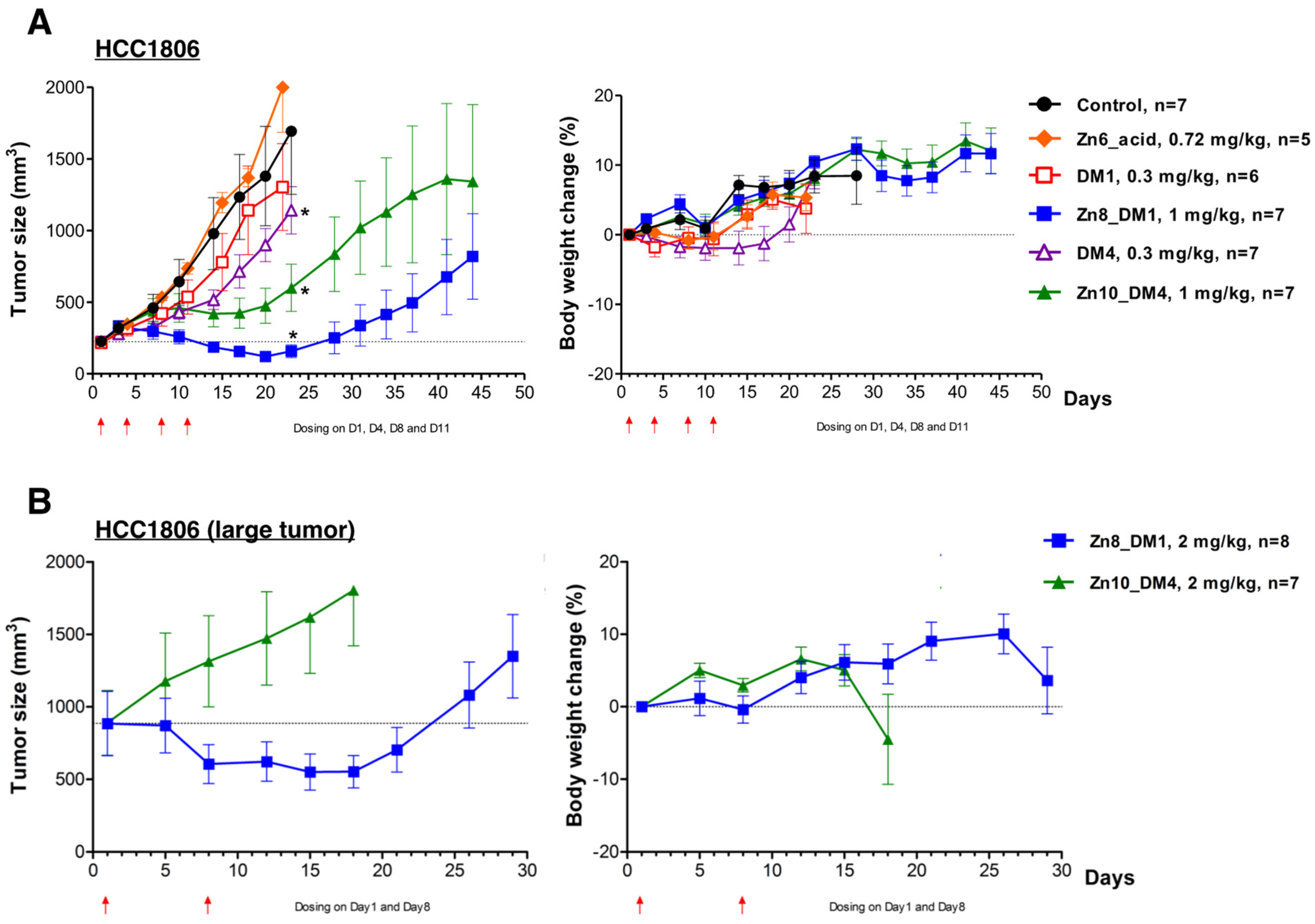
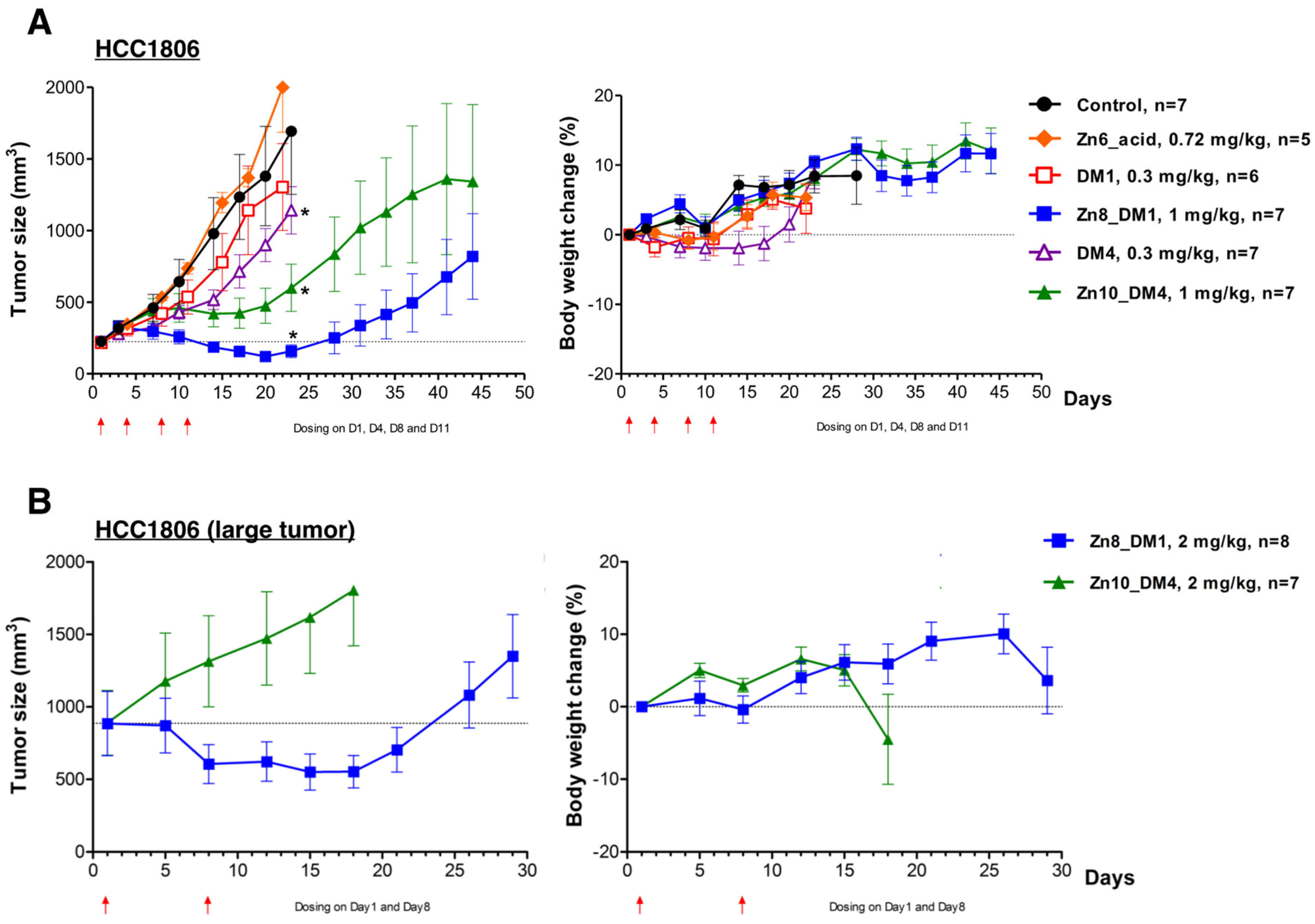
ZnDPA-based thioester-linked maytansinoids
Zn8_DM1 or
Zn10_DM4 exhibited significant activities against HCC1806 human triple-negative breast cancer growth compared to vehicle control (
Figure 3A). With equimolar doses of cytotoxic payloads (0.3 mg/kg for DM1 or DM4 and 1 mg/kg for
Zn8_DM1 or
Zn10_DM4), we observed that these maytansinoid conjugates exhibited better efficacies than their respective payloads alone (
Figure 3A). On the other hand,
Zn6_acid, the part of ZnDPA-linker without the bioactive maytansinoid payloads, was intravenously given in 4 doses at 0.72 mg/kg, and did not reduce the tumor burden in mice (
Figure 3A). Notably, a week after the stoppage of treatment,
Zn8_DM1 showed shrinkage of HCC1806 tumor growth and had more significant antitumor activity when compared to
Zn10_DM4, which had a tumor growth inhibition (TGI) of 74% only. Although 0.3 mg/kg of DM4 elicited marginal improvement in its antitumor activity during the course of treatment when compared to that of DM1, in its respective conjugated form,
Zn8_DM1 showed better efficacy than that of
Zn10_DM4. We reasoned that the introduction of methyl groups to the adjacent carbon next to the sulfur group in DM4 might generate more steric hindrance for efficient DM4 cleavage from
Zn10_DM4 in the tumor microenvironment. With a low-frequency regimen at 2 mg/kg per week for two weeks, a therapy experiment was conducted against larger (~900 mm
3) HCC1806 tumors. Gratifyingly, our data showed that
Zn8_DM1 could readily arrest or shrink the growth of larger tumors (
Figure 3B), providing insight into reducing tumor burden and facilitating surgical removal procedures for the treatment of TNBC cancer.
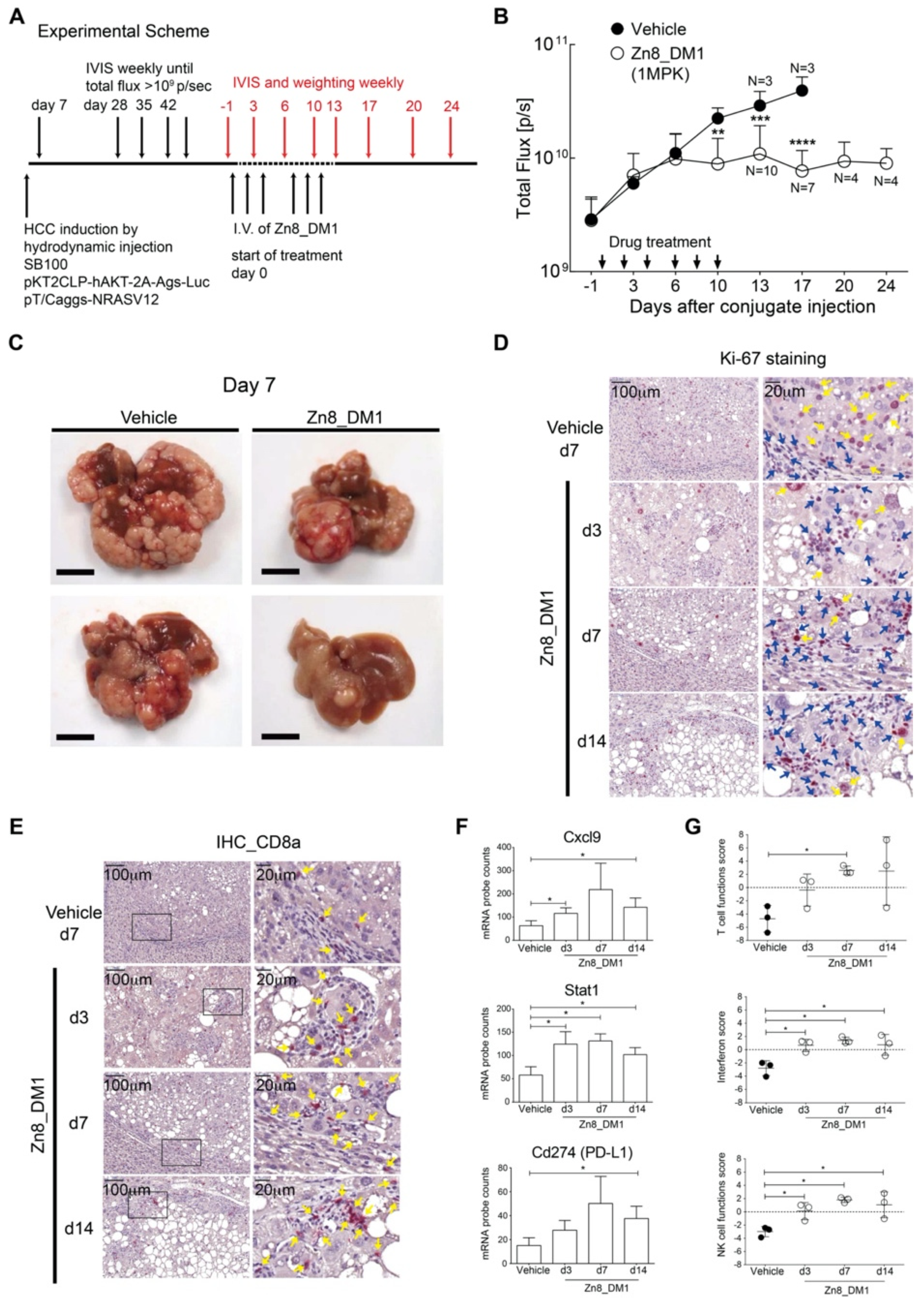
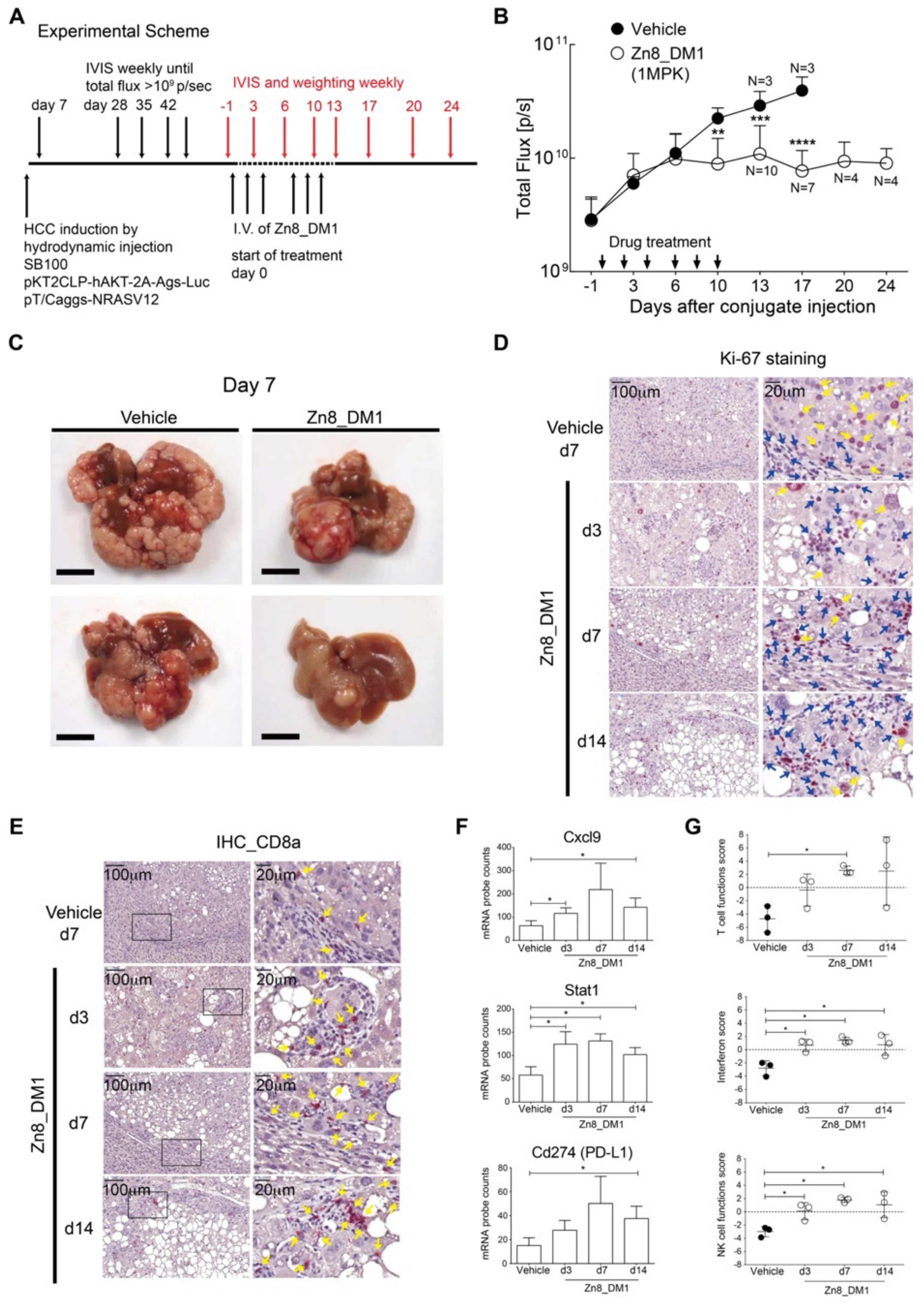
In an oncogene-induced, sorafenib-resistant hepatocellular carcinoma mouse model with relevant tumor-associated immunological profiles in the TME, we then evaluated the efficacies of
Zn8_DM1 by non-invasive monitoring of tumor progression with a bioluminescence reporter (
Figure 4A) [
26]. Notably,
Zn8_DM1 achieved a potent anti-HCC tumor growth evident by reducing the flux of luciferase activity at 3 times of 1 mg/kg a week for 2 weeks (
Figure 4B). Indeed, livers harvested from
Zn8_DM1-treated mice a week post last treatment dose exhibited less tumor burden than those of the vehicle-treated group (
Figure 4C). Next, a marked reduction in Ki-67 staining was found with
Zn8_DM1-treated tumor samples (
Figure 4D), suggesting
Zn8_DM1 treatment could modulate Ki-67-positive cancer cell proliferation. Moreover, inflamed “hot” tumor potentiated by T-cell infiltration with immune activation properties has been shown to improve clinical response rates to (PD-L)1/PD-1 immunotherapy treatment [
29,
30,
31]. A significant increase in cytotoxic CD8
+ T-cell infiltration was found in
Zn8_DM1-treated tumors (
Figure 4E). These cytotoxic T cells harnessed their killing ability via programming tumor cells to undergo apoptosis.
We next evaluated
Zn8_DM1-mediated changes in gene expression in the TME. To address the influence of
Zn8_DM1 on the immune landscape of HCC TME, total RNA from
Zn8_DM1- or vehicle-treated tumor tissues was isolated to determine the absolute copies of inflammation-related mRNAs. We found a significant upregulation of CXCL9 expression in the
Zn8_DM1-treated tumor tissues (
Figure 4F). Chemokine (C-X-C motif) ligand 9 (CXCL9) was identified as a chemoattractant for CD8
+ T cells through the interaction with C-X-C motif chemokine receptor 3 (CXCR3). The CXCL9–CXCR3 axis was correlated with improved PD-1 immunotherapy outcome [
32]. On the other hand, gene expression of the signal transducer and activator of transcription 1 (STAT1), a critical inflammatory regulator of TME that potentiates the immune checkpoint blockade (ICB) treatment [
29], was elevated (
Figure 4F). Findings of STAT1-deficient T cells possessed less serine esterase activity with impaired production of IFNγ [
33], suggesting its role in regulating cytotoxic T cell function. Furthermore, ICB-responsive inflammatory TME with elevated PD-L1 gene expression was also observed in
Zn8_DM1-treated tumor (
Figure 4F). Our results provided evidence that
Zn8_DM1 treatment could result in the concurrent increase of gene expression of key factors that aided the establishment of an immune-inflamed “hot” tumor [
30]. With significant improvement in T-cell function score, NK cell function score, and interferon score,
Zn8_DM1-treatment facilitated intrinsic functional enhancement among these parameters in the TME (
Figure 4G).


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}