
Prediction of Pharmacokinetics of IDP-73152 in Humans Using Physiologically-Based Pharmacokinetics


Abstract
1. Introduction
2. Materials and Methods
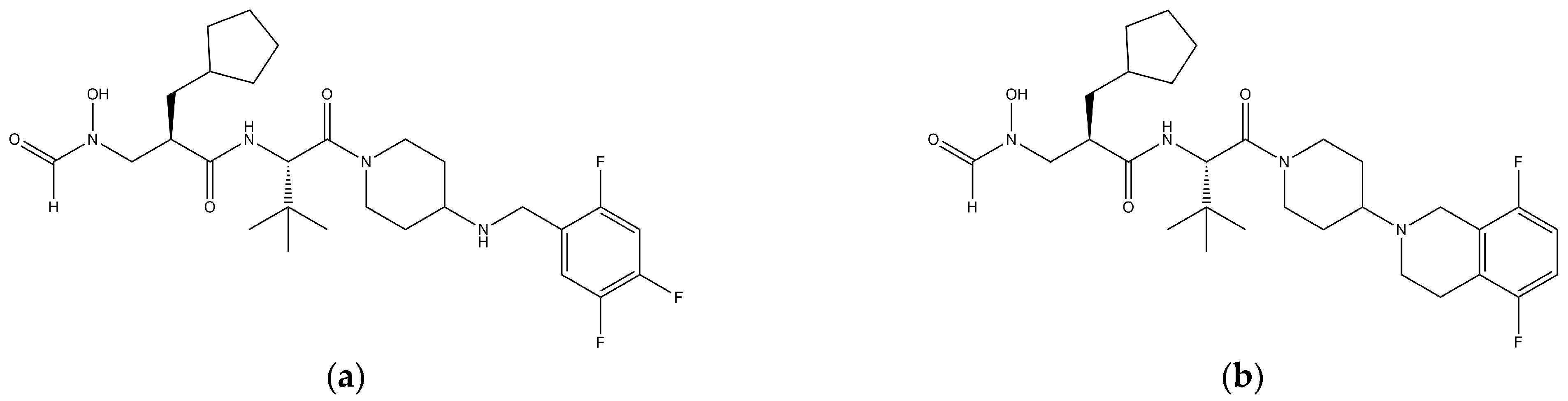
2.1. Materials
2.2. In Vitro PK Studies
2.2.1. Caco-2 Cell Permeability
2.2.2. Free Faction of IDP-73152 in the Plasma, Microsomal and Hepatocyte Incubation
2.2.3. Blood Partitioning
2.2.4. Metabolic Stability and Blood Stability
2.3. In Vivo PK Studies
2.3.1. PK Studies in Rats
2.3.2. PK Studies in Mice and Dogs
2.3.3. Human Study
2.4. PBPK Modeling of IDP-73152
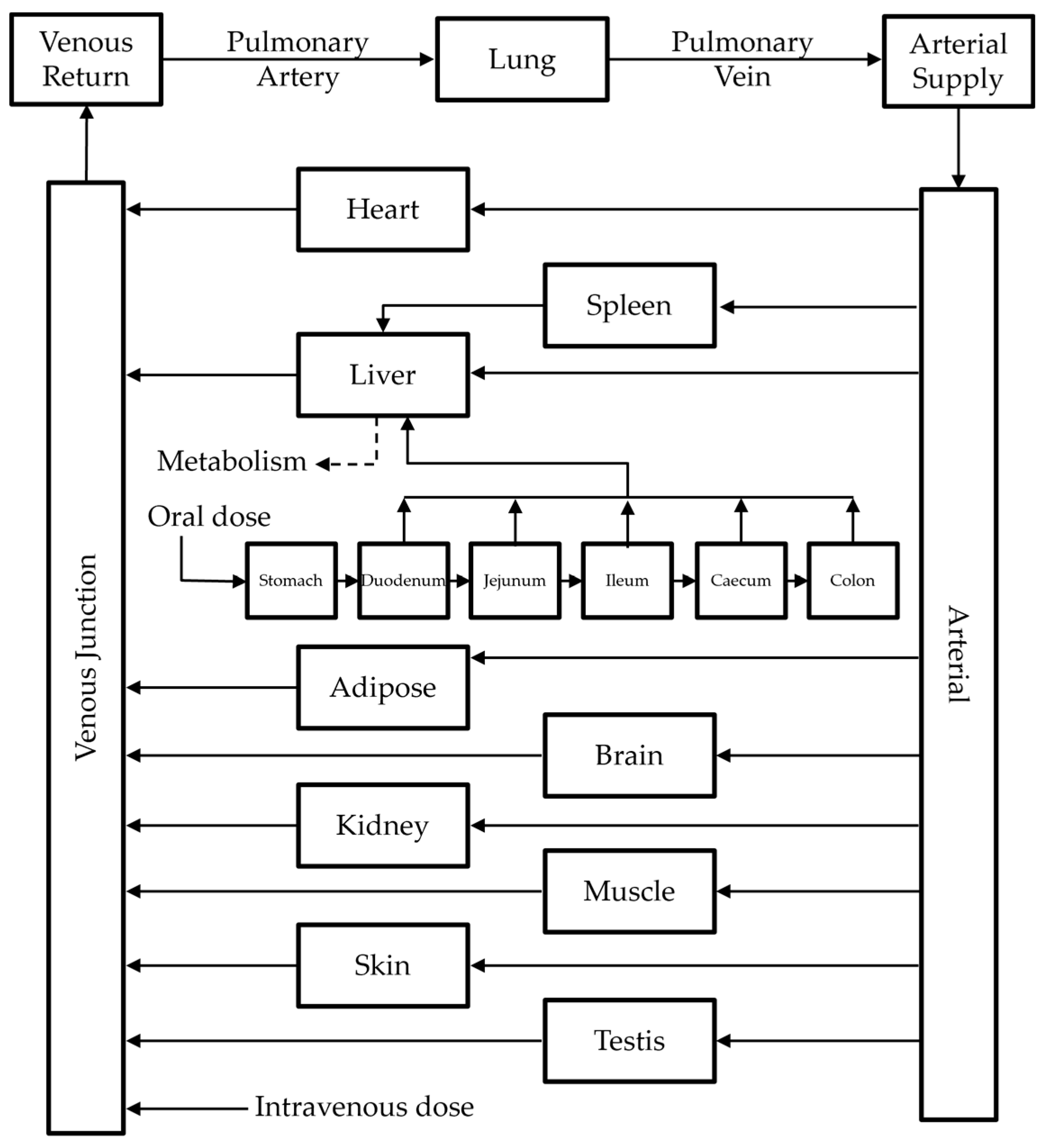
2.4.1. Model Structure
2.4.2. Model Development
2.4.3. Model Extension to Mice and Dogs
2.4.4. Model Extension to Humans
2.4.5. Determination for the Adequacy of PBPK Model
2.5. Data and PK Analysis
3. Results
3.1. In Vitro PK Studies
3.1.1. Caco-2 Permeability
3.1.2. Protein Binding and Blood Partitioning
3.1.3. Estimation of Hepatic Clearance from In Vitro Metabolic Stability Assays
3.2. In Vivo PK Studies in Preclinical Species
3.2.1. PK Characteristics of IDP-73152 in Preclinical Species
3.2.2. Tissue Distribution of IDP-73152
3.3. PBPK Modeling
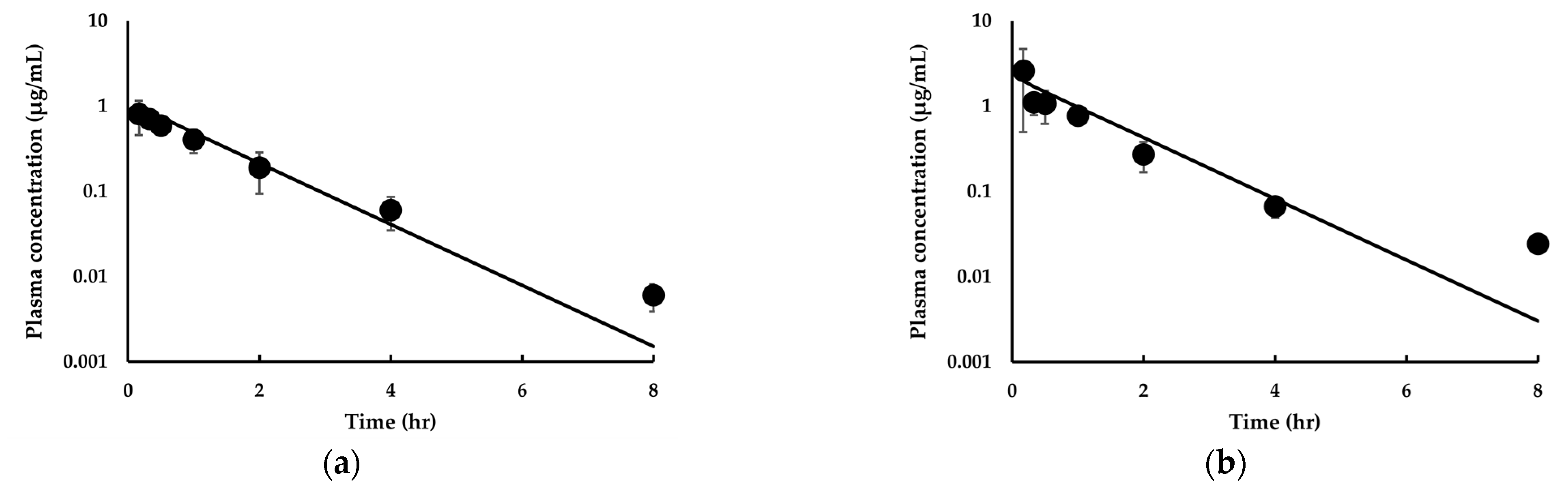
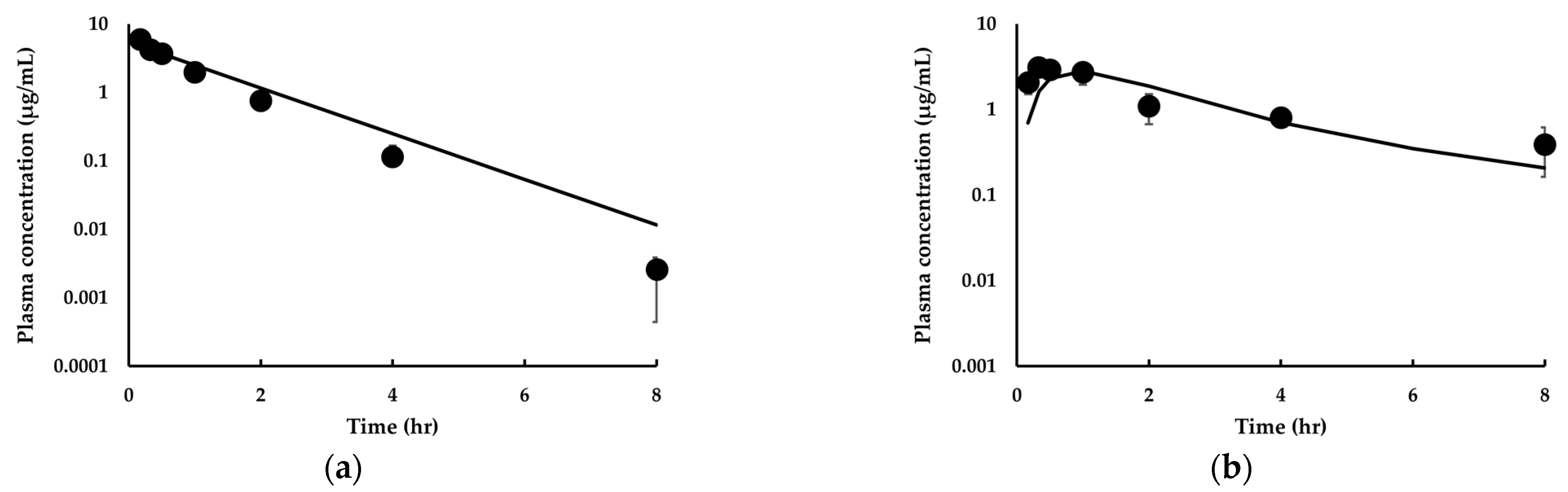
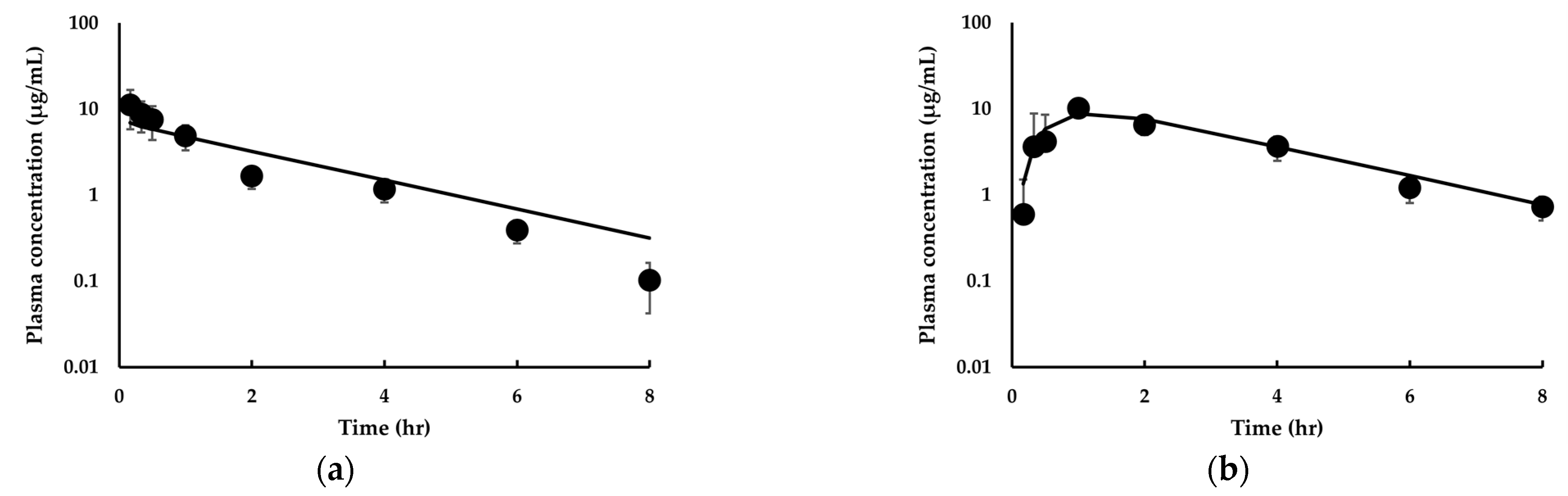
3.3.1. Model Development and Comparison with Experimental Data for IDP-73152
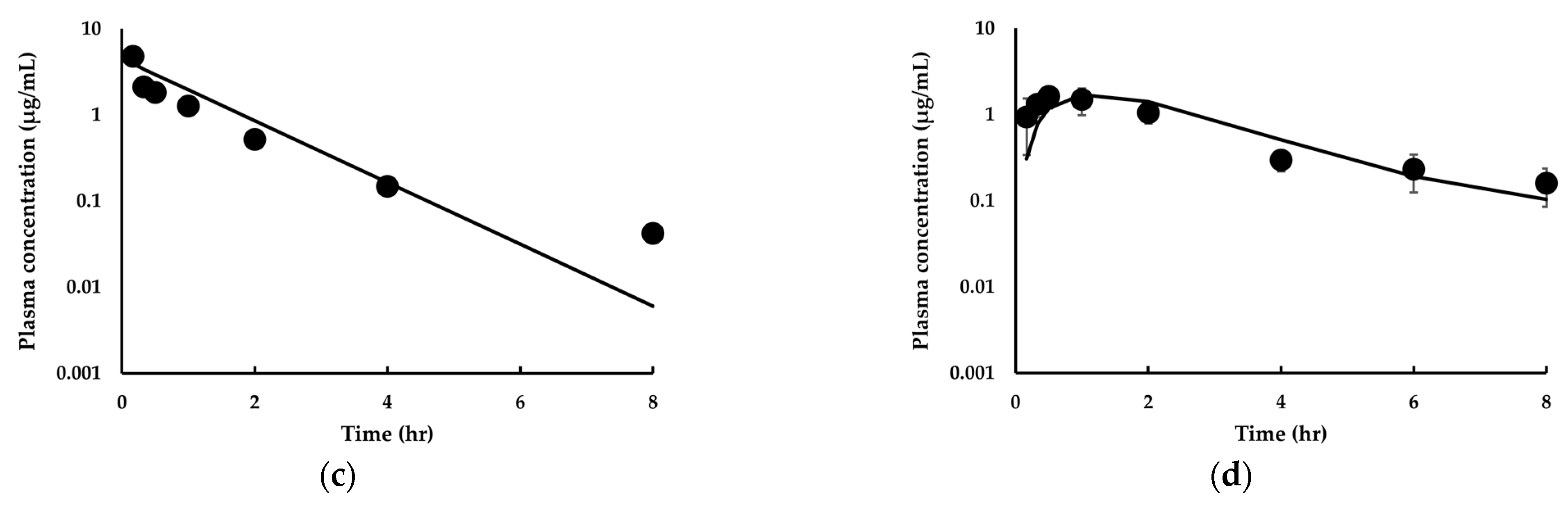
3.3.2. Model Extension to Mice and Dogs for IDP-73152
3.3.3. Estimation of Human PK for IDP-73152
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Adams, J.M. On the release of the formyl group from nascent protein. J. Mol. Biol. 1968, 33, 571–589. [Google Scholar] [CrossRef]
- Livingston, D.M.; Leder, P. Deformylation and protein biosynthesis. Biochemistry 1969, 8, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Ball, L.A.; Kaesberg, P. Cleavage of the N-terminal formylmethionine residue from a bacteriophage coat protein in vitro. J. Mol. Biol. 1973, 79, 531–537. [Google Scholar] [CrossRef]
- Jain, R.; Chen, D.; White, R.J.; Patel, D.V.; Yuan, Z. Bacterial Peptide deformylase inhibitors: A new class of antibacterial agents. Curr. Med. Chem. 2005, 12, 1607–1621. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Dahlgren, C.; Bylund, J. Subinhibitory concentrations of the deformylase inhibitor actinonin increase bacterial release of neutrophil-activating peptides: A new approach to antimicrobial chemotherapy. Antimicrob. Agents Chemother. 2003, 47, 2545–2550. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.J.; Kelly, B.K.; Miller, G.A. Actinonin: An antibiotic substance produced by an actinomycete. Nature 1962, 195, 701–702. [Google Scholar] [CrossRef]
- Clements, J.M.; Beckett, R.P.; Brown, A.; Catlin, G.; Lobell, M.; Palan, S.; Thomas, W.; Whittaker, M.; Wood, S.; Salama, S.; et al. Antibiotic activity and characterization of BB-3497, a novel peptide deformylase inhibitor. Antimicrob. Agents Chemother. 2001, 45, 563–570. [Google Scholar] [CrossRef]
- Ramanathan-Girish, S.; McColm, J.; Clements, J.M.; Taupin, P.; Barrowcliffe, S.; Hevizi, J.; Safrin, S.; Moore, C.; Patou, G.; Moser, H.; et al. Pharmacokinetics in animals and humans of a first-in-class peptide deformylase inhibitor. Antimicrob. Agents Chemother. 2004, 48, 4835–4842. [Google Scholar] [CrossRef]
- Lee, H.Y.; An, K.M.; Jung, J.; Koo, J.M.; Kim, J.G.; Yoon, J.M.; Lee, M.J.; Jang, H.; Lee, H.S.; Park, S.; et al. Identification of novel aminopiperidine derivatives for antibacterial activity against Gram-positive bacteria. Bioorg. Med. Chem. Lett. 2016, 26, 3148–3152. [Google Scholar] [CrossRef]
- Lee, M.; Kim, D.; Shin, J.; Lee, H.Y.; Park, S.; Lee, H.S.; Kang, J.H.; Chung, S.J. Quantification of IDP-73152, a novel antibiotic, in plasma from mice, rats and humans using an ultra-high performance liquid chromatography/tandem mass spectrometry method for use in pharmacokinetic studies. J. Pharm. Biomed. Anal. 2017, 145, 364–371. [Google Scholar] [CrossRef]
- Shin, D.; Park, S.I.; Lee, H.S.; An, K.M.; Jung, J.; Lee, M.; Yu, K.S. Pharmacokinetics and tolerability of IDP-73152 mesylate after a single oral administration under fasted and fed conditions in healthy volunteers. Drug Des. Devel. Ther. 2019, 13, 2483–2490. [Google Scholar] [CrossRef] [PubMed]
- Ina Hubatsch, E.G.E.R.P.A. Determination of drug permeability and prediction of drug absorption in Caco-2 monolayers. Nat. Protoc. 2007, 2, 2111–2119. [Google Scholar] [CrossRef] [PubMed]
- Waters, N.J.; Jones, R.; Williams, G.; Sohal, B. Validation of a rapid equilibrium dialysis approach for the measurement of plasma protein binding. J. Pharm. Sci. 2008, 97, 4586–4595. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Li, S.; Yang, H.; Lee, F.; Wu, J.T.; Qian, M.G. A novel liquid chromatography/tandem mass spectrometry based depletion method for measuring red blood cell partitioning of pharmaceutical compounds in drug discovery. Rapid Commun. Mass Spectrom. 2005, 19, 250–254. [Google Scholar] [CrossRef]
- De Smet, K.; Beken, S.; Vanhaecke, T.; Pauwels, M.; Vercruysse, A.; Rogiers, V. Isolation of rat hepatocytes. Methods Mol. Biol. 1998, 107, 295–301. [Google Scholar] [CrossRef]
- Papeleu, P.; Vanhaecke, T.; Henkens, T.; Elaut, G.; Vinken, M.; Snykers, S.; Rogiers, V. Isolation of rat hepatocytes. Methods Mol. Biol. 2006, 320, 229–237. [Google Scholar] [CrossRef]
- Yim, C.S.; Jeong, Y.S.; Lee, S.Y.; Pyeon, W.; Ryu, H.M.; Lee, J.H.; Lee, K.R.; Maeng, H.J.; Chung, S.J. Specific Inhibition of the Distribution of Lobeglitazone to the Liver by Atorvastatin in Rats: Evidence for a Rat Organic Anion Transporting Polypeptide 1B2-Mediated Interaction in Hepatic Transport. Drug Metab. Dispos. 2017, 45, 246–259. [Google Scholar] [CrossRef]
- Niehues, S.M.; Unger, J.K.; Malinowski, M.; Neymeyer, J.; Hamm, B.; Stockmann, M. Liver volume measurement: Reason of the difference between in vivo CT-volumetry and intraoperative ex vivo determination and how to cope it. Eur. J. Med. Res. 2010, 15, 345–350. [Google Scholar] [CrossRef]
- Musther, H.; Harwood, M.D.; Yang, J.; Turner, D.B.; Rostami-Hodjegan, A.; Jamei, M. The Constraints, Construction, and Verification of a Strain-Specific Physiologically Based Pharmacokinetic Rat Model. J. Pharm. Sci. 2017, 106, 2826–2838. [Google Scholar] [CrossRef]
- Barter, Z.E.; Chowdry, J.E.; Harlow, J.R.; Snawder, J.E.; Lipscomb, J.C.; Rostami-Hodjegan, A. Covariation of human microsomal protein per gram of liver with age: Absence of influence of operator and sample storage may justify interlaboratory data pooling. Drug Metab. Dispos. 2008, 36, 2405–2409. [Google Scholar] [CrossRef]
- Ito, K.; Iwatsubo, T.; Kanamitsu, S.; Nakajima, Y.; Sugiyama, Y. Quantitative prediction of in vivo drug clearance and drug interactions from in vitro data on metabolism, together with binding and transport. Annu. Rev. Pharmacol. Toxicol. 1998, 38, 461–499. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Noh, C.K.; Yim, C.S.; Jeong, Y.S.; Ahn, S.H.; Lee, W.; Kim, D.D.; Chung, S.J. Kinetics of the Absorption, Distribution, Metabolism, and Excretion of Lobeglitazone, a Novel Activator of Peroxisome Proliferator-Activated Receptor Gamma in Rats. J. Pharm. Sci. 2015, 104, 3049–3059. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.S.; Gross, J.F. Estimation of tissue-to-plasma partition coefficients used in physiological pharmacokinetic models. J. Pharm. Biopharm. 1979, 7, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Sugiyama, Y.; Awazu, S.; Hanano, M. In vitro and in vivo evaluation of the tissue-to-blood partition coefficient for physiological pharmacokinetic models. J. Pharm. Biopharm. 1982, 10, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.R.; Chae, Y.J.; Maeng, H.J.; Lee, J.; Kim, D.D.; Chong, S.; Shim, C.K.; Chung, S.J. Physiologically based pharmacokinetic modeling of SNU-0039, an anti-Alzheimer’s agent, in rats. J. Pharm. Pharm. 2011, 38, 637–651. [Google Scholar] [CrossRef]
- Davies, B.; Morris, T. Physiological parameters in laboratory animals and humans. Pharm. Res. 1993, 10, 1093–1095. [Google Scholar] [CrossRef]
- Brown, R.P.; Delp, M.D.; Lindstedt, S.L.; Rhomberg, L.R.; Beliles, R.P. Physiological parameter values for physiologically based pharmacokinetic models. Toxicol. Ind. Health 1997, 13, 407–484. [Google Scholar] [CrossRef]
- National Health and Nutrition Examination Survey. Available online: https://wwwn.cdc.gov/nchs/nhanes/Default.aspx (accessed on 6 May 2022).
- Yang, J.; Jamei, M.; Yeo, K.R.; Rostami-Hodjegan, A.; Tucker, G.T. Misuse of the well-stirred model of hepatic drug clearance. Drug Metab. Dispos. 2007, 35, 501–502. [Google Scholar] [CrossRef]
- Oie, S.; Tozer, T.N. Effect of altered plasma protein binding on apparent volume of distribution. J. Pharm. Sci. 1979, 68, 1203–1205. [Google Scholar] [CrossRef]
- Berezhkovskiy, L.M. A valid equation for the well-stirred perfusion limited physiologically based pharmacokinetic model that consistently accounts for the blood-tissue drug distribution in the organ and the corresponding valid equation for the steady state volume of distribution. J. Pharm. Sci. 2010, 99, 475–485. [Google Scholar] [CrossRef]
- Yu, L.X.; Amidon, G.L. A compartmental absorption and transit model for estimating oral drug absorption. Int. J. Pharm. 1999, 186, 119–125. [Google Scholar] [CrossRef]
- Clemens, E.T.; Stevens, C.E. A comparison of gastrointestinal transit time in ten species of mammal. J. Agric. Sci. 1980, 94, 735–737. [Google Scholar] [CrossRef]
- Sjögren, E.; Abrahamsson, B.; Augustijns, P.; Becker, D.; Bolger, M.B.; Brewster, M.; Brouwers, J.; Flanagan, T.; Harwood, M.; Heinen, C.; et al. In vivo methods for drug absorption—Comparative physiologies, model selection, correlations with in vitro methods (IVIVC), and applications for formulation/API/excipient characterization including food effects. Eur. J. Pharm. Sci. 2014, 57, 99–151. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Jiang, L.; Chen, T.M.; Hwang, K.K. A comparative study of artificial membrane permeability assay for high throughput profiling of drug absorption potential. Eur. J. Med. Chem. 2002, 37, 399–407. [Google Scholar] [CrossRef]
- Lennernas, H. Intestinal permeability and its relevance for absorption and elimination. Xenobiotica 2007, 37, 1015–1051. [Google Scholar] [CrossRef]
- Hu, Y.; Smith, D.E. In Silico Prediction of the Absorption and Disposition of Cefadroxil in Humans using an Intestinal Permeability Method Scaled from Humanized PepT1 Mice. Drug Metab. Dispos. 2019, 47, 173–183. [Google Scholar] [CrossRef]
- Agoram, B.; Woltosz, W.S.; Bolger, M.B. Predicting the impact of physiological and biochemical processes on oral drug bioavailability. Adv. Drug Deliv. Rev. 2001, 50 (Suppl. S1), S41–S67. [Google Scholar] [CrossRef]
- Hendriksen, B.A.; Felix, M.V.; Bolger, M.B. The composite solubility versus pH profile and its role in intestinal absorption prediction. AAPS Pharm. Sci. 2003, 5, E4. [Google Scholar] [CrossRef]
- Tang, H.; Hussain, A.; Leal, M.; Mayersohn, M.; Fluhler, E. Interspecies prediction of human drug clearance based on scaling data from one or two animal species. Drug Metab. Dispos. 2007, 35, 1886–1893. [Google Scholar] [CrossRef]
- Press, B. Optimization of the Caco-2 Permeability Assay to Screen Drug Compounds for Intestinal Absorption and Efflux. In Permeability Barrier: Methods and Protocols; Turksen, K., Ed.; Humana Press: Totowa, NJ, USA, 2011; pp. 139–154. [Google Scholar]
- Nassar, A.F.; Hollenberg, P.F.; Scatina, J. Drug Metabolism Handbook: Concepts and Applications; Wiley: Hoboken, NJ, USA, 2009. [Google Scholar]
- Wilkinson, G.R.; Shand, D.G. Commentary: A physiological approach to hepatic drug clearance. Clin. Pharmacol. Ther. 1975, 18, 377–390. [Google Scholar] [CrossRef]
- Antibiotic Resistance Threats in the United States. Available online: https://www.cdc.gov/drugresistance/pdf/threats-report/2019-ar-threats-report-508.pdf (accessed on 6 May 2022).
- Aslam, B.; Wang, W.; Arshad, M.I.; Khurshid, M.; Muzammil, S.; Rasool, M.H.; Nisar, M.A.; Alvi, R.F.; Aslam, M.A.; Qamar, M.U.; et al. Antibiotic resistance: A rundown of a global crisis. Infect. Drug Resist. 2018, 11, 1645–1658. [Google Scholar] [CrossRef] [PubMed]
- Fieulaine, S.; Alves de Sousa, R.; Maigre, L.; Hamiche, K.; Alimi, M.; Bolla, J.M.; Taleb, A.; Denis, A.; Pages, J.M.; Artaud, I.; et al. A unique peptide deformylase platform to rationally design and challenge novel active compounds. Sci. Rep. 2016, 6, 35429. [Google Scholar] [CrossRef] [PubMed]
- Butler, D.; Chen, D.; O’Dwyer, K.; Lewandowski, T.; Aubart, K.; Zalacain, M. Potent sub-MIC effect of GSK1322322 and other peptide deformylase inhibitors on in vitro growth of Staphylococcus aureus. Antimicrob. Agents Chemother. 2014, 58, 290–296. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Osborne, C.S.; Neckermann, G.; Fischer, E.; Pecanka, R.; Yu, D.; Manni, K.; Goldovitz, J.; Amaral, K.; Dzink-Fox, J.; Ryder, N.S. In vivo characterization of the peptide deformylase inhibitor LBM415 in murine infection models. Antimicrob. Agents Chemother. 2009, 53, 3777–3781. [Google Scholar] [CrossRef] [PubMed]
- Lofland, D.; Difuntorum, S.; Waller, A.; Clements, J.M.; Weaver, M.K.; Karlowsky, J.A.; Johnson, K. In vitro antibacterial activity of the peptide deformylase inhibitor BB-83698. J. Antimicrob. Chemother. 2004, 53, 664–668. [Google Scholar] [CrossRef] [PubMed]
- Broughton, B.J.; Chaplen, P.; Freeman, W.A.; Warren, P.J.; Wooldridge, K.R.; Wright, D.E. Studies concerning the antibiotic actinonin. Part VIII. Structure-activity relationships in the actinonin series. J. Chem. Soc. Perkin. 1975, 1, 857–860. [Google Scholar] [CrossRef]
- Rodgers, T.; Leahy, D.; Rowland, M. Physiologically based pharmacokinetic modeling 1: Predicting the tissue distribution of moderate-to-strong bases. J. Pharm. Sci. 2005, 94, 1259–1276. [Google Scholar] [CrossRef]
- Rodgers, T.; Rowland, M. Physiologically based pharmacokinetic modelling 2: Predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J. Pharm. Sci. 2006, 95, 1238–1257. [Google Scholar] [CrossRef]
- Rodgers, T.; Rowland, M. Mechanistic approaches to volume of distribution predictions: Understanding the processes. Pharm. Res. 2007, 24, 918–933. [Google Scholar] [CrossRef]
- Radford, A.J.; Rhodes, F.A. The association of jaundice with lobar pneumonia in the territory of Papua and New Guinea. Med. J. Aust. 1967, 2, 678–681. [Google Scholar] [CrossRef]
- Charlton, M.; Thompson, J.P. Pharmacokinetics in sepsis. BJA Educ. 2019, 19, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Evers, R.; Piquette-Miller, M.; Polli, J.W.; Russel, F.G.M.; Sprowl, J.A.; Tohyama, K.; Ware, J.A.; de Wildt, S.N.; Xie, W.; Brouwer, K.L.R.; et al. Disease-Associated Changes in Drug Transporters May Impact the Pharmacokinetics and/or Toxicity of Drugs: A White Paper From the International Transporter Consortium. Clin. Pharmacol. Ther. 2018, 104, 900–915. [Google Scholar] [CrossRef] [PubMed]
- Di, L.; Kerns, E.H. Drug-like Properties: Concepts, Structure Design and Methods from ADME to Toxicity Optimization, 2nd ed.; Academic Press: London, UK, 2016. [Google Scholar]
- Cheng, H.; Staubus, A.E.; Shum, L. An area function method for estimating the apparent absorption rate constant. Pharm. Res. 1988, 5, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Gascon, A.; Solinis, M.A.; Isla, A. The Role of PK/PD Analysis in the Development and Evaluation of Antimicrobials. Pharmaceutics 2021, 13, 833. [Google Scholar] [CrossRef] [PubMed]
- Landersdorfer, C.B.; Nation, R.L. Limitations of Antibiotic MIC-Based PK-PD Metrics: Looking Back to Move Forward. Front Pharmacol 2021, 12, 770518. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Mice 1 | Rats 2 | Dogs 2 | |
|---|---|---|---|---|
| Intravenous PK | ||||
| Dose (mg/kg) | 10 | 10 | 10 | |
| CLp (L/h/kg) | 1.52 | 2.00 ± 0.16 | 0.664 ± 0.259 | |
| Vss (L/kg) | 1.43 | 2.54 ± 0.26 | 1.15 ± 0.46 | |
| AUCinf 3 (μg·h/mL) | 6.59 | 5.03 ± 0.38 | 16.6 ± 5.9 | |
| t1/2 (h) | 0.731 | 1.43 ± 0.11 | 1.18 ± 0.14 | |
| Oral PK | ||||
| Dose (mg/kg) | 20 | 20 | 20 | |
| Cmax 4 (μg/mL) | 3.10 | 1.65 ± 0.44 | 10.5 ± 2.0 | |
| Tmax 5 (h) | 0.333 | 0.638 ± 0.29 | 0.777 ± 0.387 | |
| AUCinf (μg·h/mL) | 10.9 | 5.57 ± 1.59 | 31.6 ± 5.9 | |
| t1/2 6 (h) | 2.86 | 2.97 ± 1.40 | 1.81 ± 0.04 | |
| F 7 (%) | 78.1 | 55.3 | 95.5 | |
| Tissue | KP,ss1 |
|---|---|
| Adipose tissue | 0.853 ± 0.196 |
| Brain | 0.0729 ± 0.0167 |
| Heart | 2.15 ± 0.22 |
| Kidney | 8.68 ± 1.83 |
| Liver | 12.2 ± 3.3 |
| Lung | 7.13 ± 1.28 |
| Muscle | 1.37 ± 0.30 |
| Skin | 1.08 ± 0.38 |
| Spleen | 4.82 ± 1.30 |
| Testis | 0.338 ± 0.089 |
| Parameter | Value | |||
|---|---|---|---|---|
| Intra venous PK | ||||
| Dose (mg/kg) | 2.5 | 5 | 10 | |
| Observed AUCinf (μg·h/mL) | 1.32 ± 0.13 | 3.27 ± 1.82 | 5.03 ± 0.38 | |
| Predicted AUCinf (μg·h/mL) | 1.44 | 2.87 | 5.74 | |
| AUC ratio 1 | 1.09 | 0.826 | 1.14 | |
| Oral PK | ||||
| Dose (mg/kg) | 20 | |||
| Observed Cmax (μg/mL) | 1.65 ± 0.44 | |||
| Predicted Cmax (μg/mL) | 1.69 | |||
| Cmax ratio 2 | 1.02 | |||
| Observed AUCinf (μg·h/mL) | 5.57 ± 1.59 | |||
| Predicted AUCinf (μg·h/mL) | 5.71 | |||
| AUC ratio | 1.03 | |||
| Parameter | Value | |
|---|---|---|
| Intra venous PK | ||
| Dose (mg/kg) | 10 | |
| Observed AUCinf (μg·h/mL) | 6.59 | |
| Predicted AUCinf (μg·h/mL) | 7.58 | |
| AUC ratio | 1.15 | |
| Oral PK | ||
| Dose (mg/kg) | 20 | |
| Observed Cmax (μg/mL) | 3.10 | |
| Predicted Cmax (μg/mL) | 2.81 | |
| Cmax ratio | 0.906 | |
| Observed AUCinf (μg·h/mL) | 10.3 | |
| Predicted AUCinf (μg·h/mL) | 9.06 | |
| AUC ratio | 0.880 | |
| Parameter | Value | |
|---|---|---|
| Intra venous PK | ||
| Dose (mg/kg) | 10 | |
| Observed AUCinf (μg·h/mL) | 16.6 ± 5.9 | |
| Predicted AUCinf (μg·h/mL) | 18.7 | |
| AUC ratio | 1.13 | |
| Oral PK | ||
| Dose (mg/kg) | 20 | |
| Observed Cmax (μg/mL) | 10.5 ± 2.0 | |
| Predicted Cmax (μg/mL) | 8.74 | |
| Cmax ratio | 0.832 | |
| Observed AUCinf (μg·h/mL) | 31.6 ± 5.9 | |
| Predicted AUCinf (μg·h/mL) | 34.1 | |
| AUC ratio | 1.08 | |
| Parameters | Value | |
|---|---|---|
| Dose (mg) | 640 | 1280 |
| Observed Cmax (μg/mL) | 8.92 ± 1.17 | 13.2 ± 2.50 |
| Predicted Cmax (μg/mL) | 4.44 | 8.87 |
| Cmax ratio | 0.498 | 0.671 |
| Observed AUCinf (μg·h/mL) | 43.4 ± 4.47 | 77.4 ± 15.4 |
| Predicted AUCinf (μg·h/mL) | 43.4 | 86.9 |
| AUC ratio | 1.00 | 1.12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, M.; Jeong, Y.-S.; Kim, M.-S.; An, K.-M.; Chung, S.-J. Prediction of Pharmacokinetics of IDP-73152 in Humans Using Physiologically-Based Pharmacokinetics. Pharmaceutics 2022, 14, 1157. https://doi.org/10.3390/pharmaceutics14061157
Lee M, Jeong Y-S, Kim M-S, An K-M, Chung S-J. Prediction of Pharmacokinetics of IDP-73152 in Humans Using Physiologically-Based Pharmacokinetics. Pharmaceutics. 2022; 14(6):1157. https://doi.org/10.3390/pharmaceutics14061157
Chicago/Turabian StyleLee, Myongjae, Yoo-Seong Jeong, Min-Soo Kim, Kyung-Mi An, and Suk-Jae Chung. 2022. "Prediction of Pharmacokinetics of IDP-73152 in Humans Using Physiologically-Based Pharmacokinetics" Pharmaceutics 14, no. 6: 1157. https://doi.org/10.3390/pharmaceutics14061157
APA StyleLee, M., Jeong, Y.-S., Kim, M.-S., An, K.-M., & Chung, S.-J. (2022). Prediction of Pharmacokinetics of IDP-73152 in Humans Using Physiologically-Based Pharmacokinetics. Pharmaceutics, 14(6), 1157. https://doi.org/10.3390/pharmaceutics14061157