
The Influence of Short Motifs on the Anticancer Activity of HB43 Peptide

 , , ,
, , ,  , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sequence Alignment by ADAPTABLE Web Server
2.2. Peptide Synthesis
2.3. Cell Cultures
2.4. Cell Viability Assay
2.5. Gelatin Zymography
2.6. Flow Cytometry Analysis of Cell Cycle
2.7. Statistical Analysis
2.8. Sample Preparation
2.9. NMR Acquisition and Processing
2.10. CD Spectroscopy
2.11. Molecular Dynamics Simulations
3. Results and Discussion
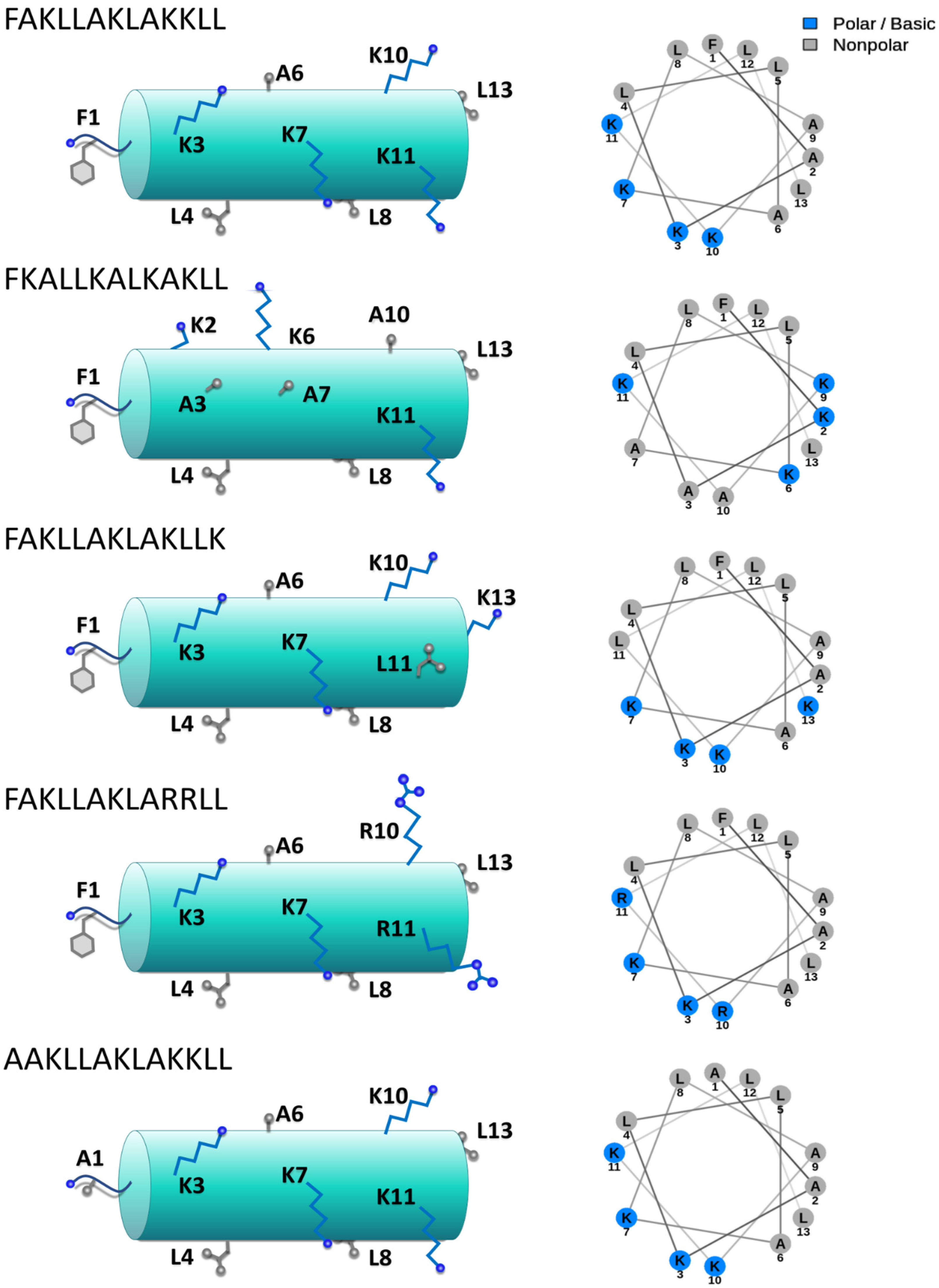
3.1. Design of Ad-Hoc Mutations in Conserved Motifs Found in the HB43-Related Family of Anticancer Peptides
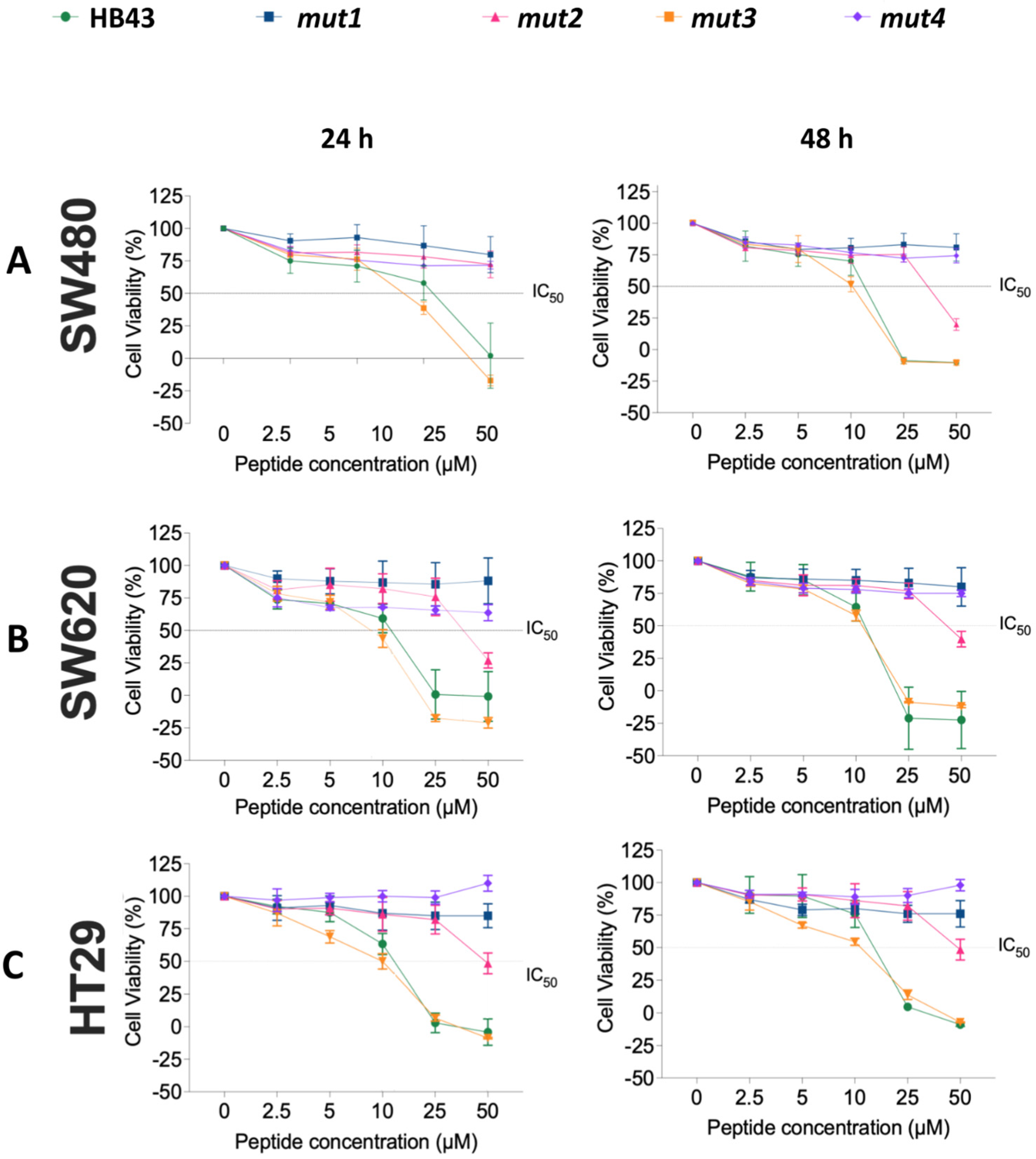
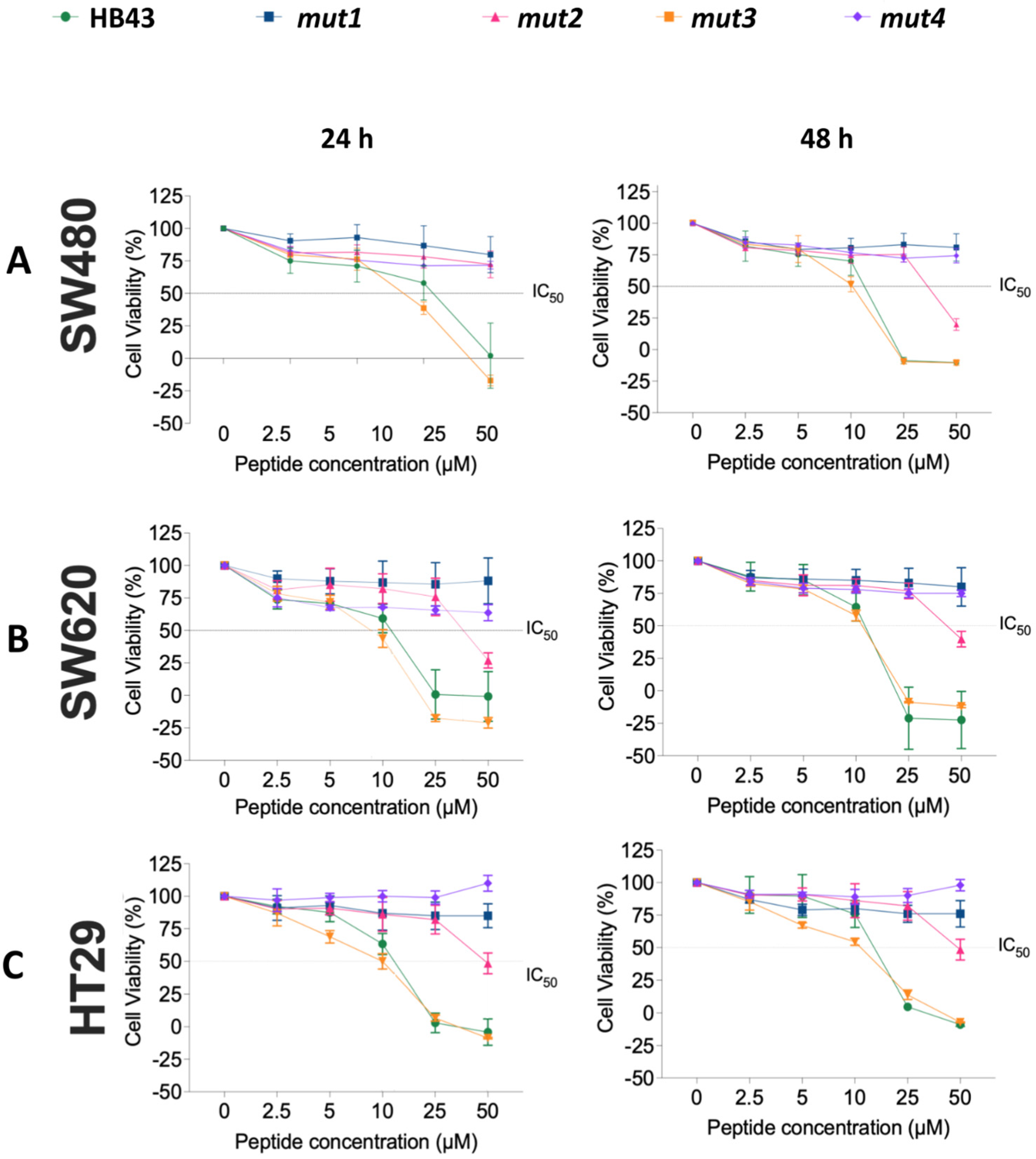
3.2. Effect of HB43 and the Designed Mutants on Cancer Cell Viability
3.3. HB43 and Mutants Do Not Alter Cell Cycle Distribution of Colon Cancer Cell Lines
3.4. HB43 and Mutants Do Not Have a Significant Impact on MMP Activity
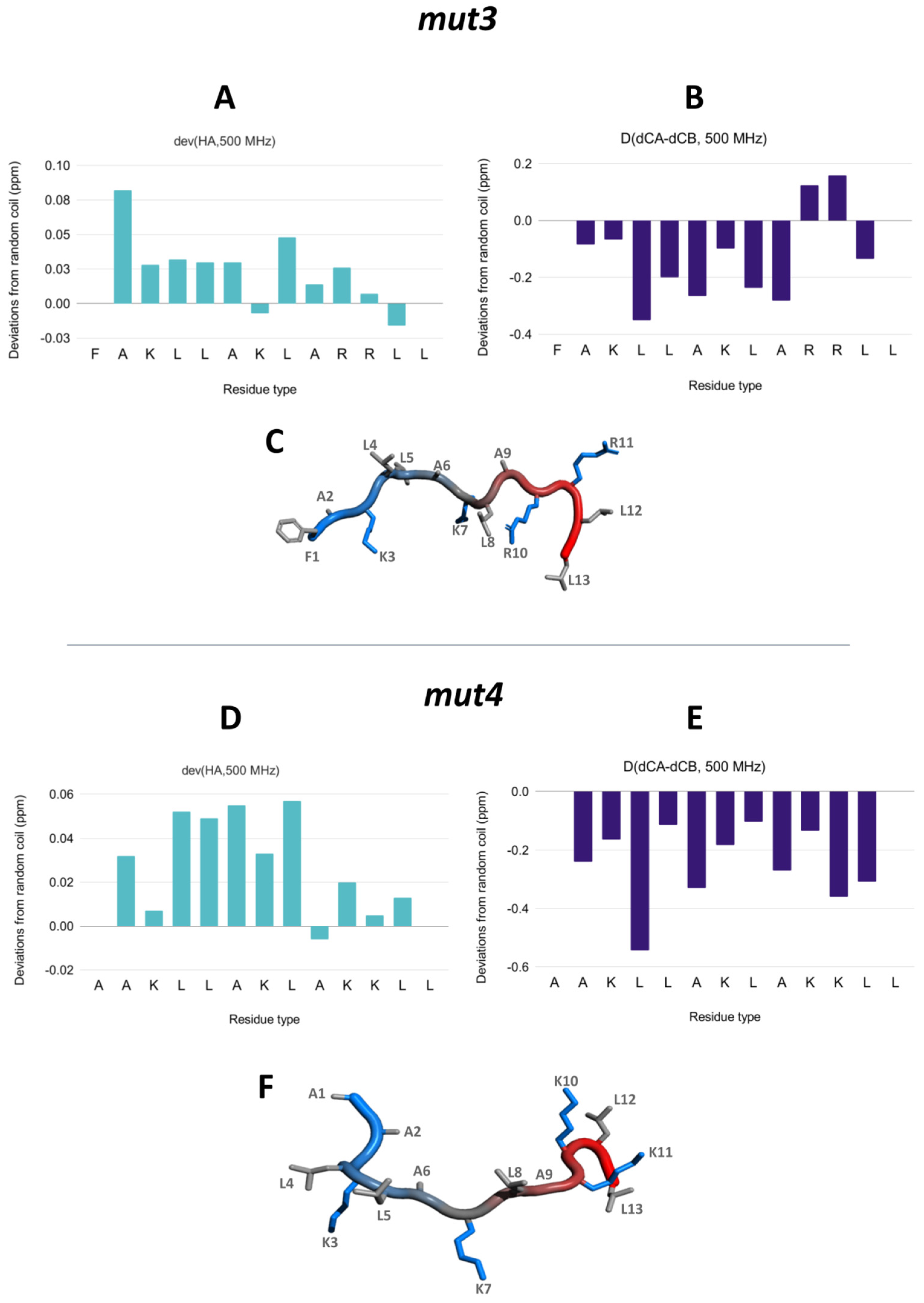
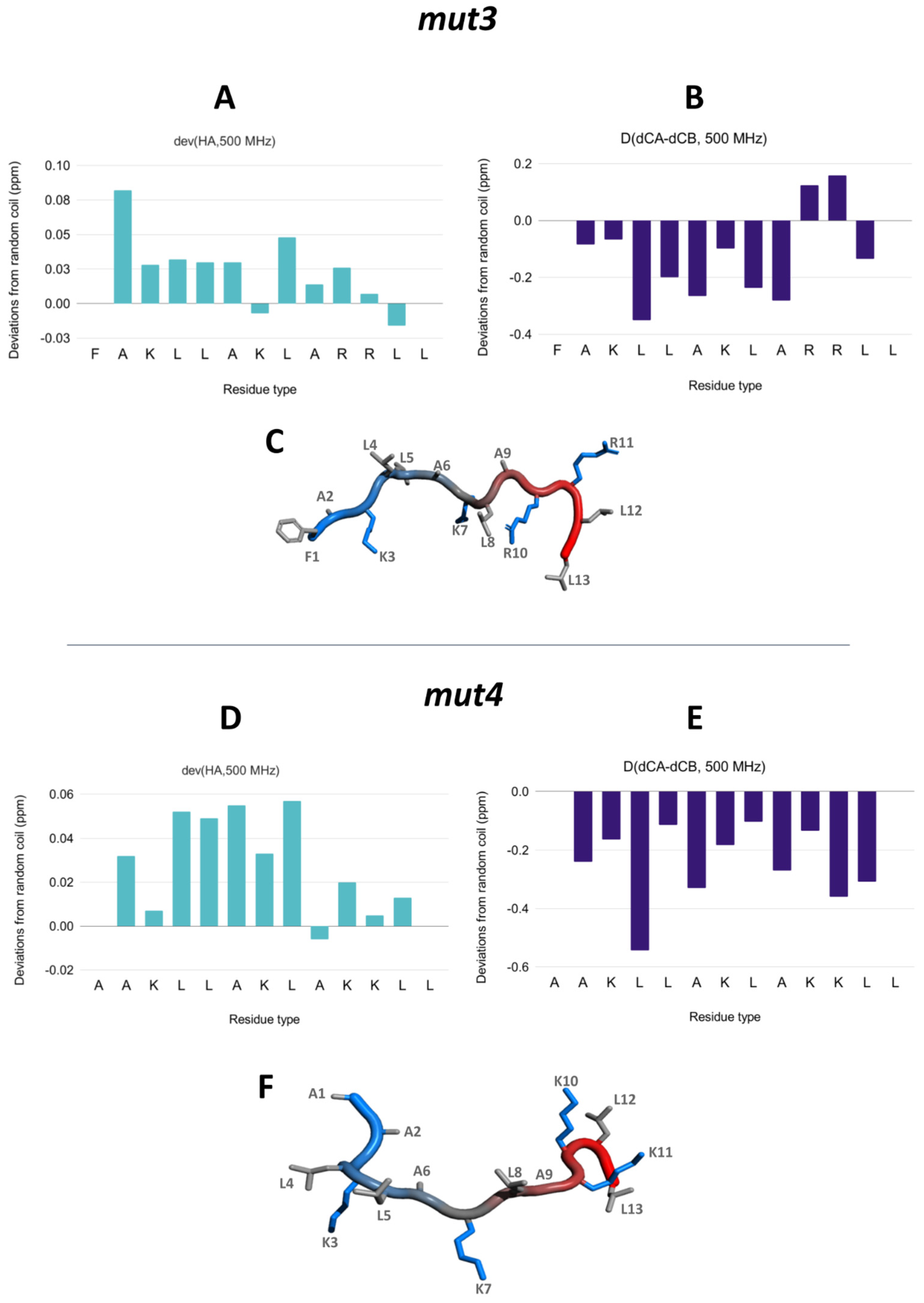
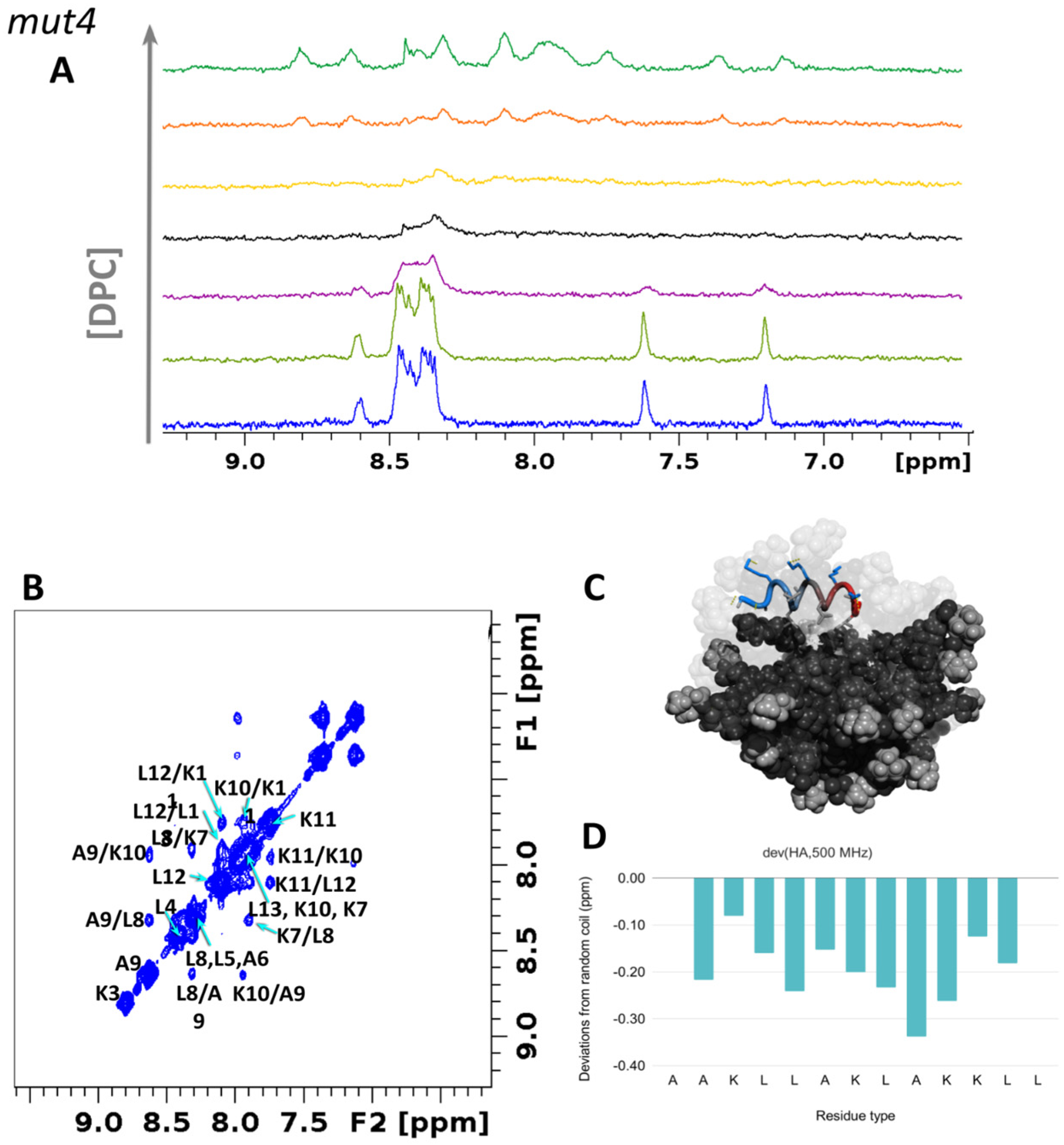
3.5. NMR Assignment and Structure Determination of Mutants in Solution
3.6. Interaction with Model Membranes
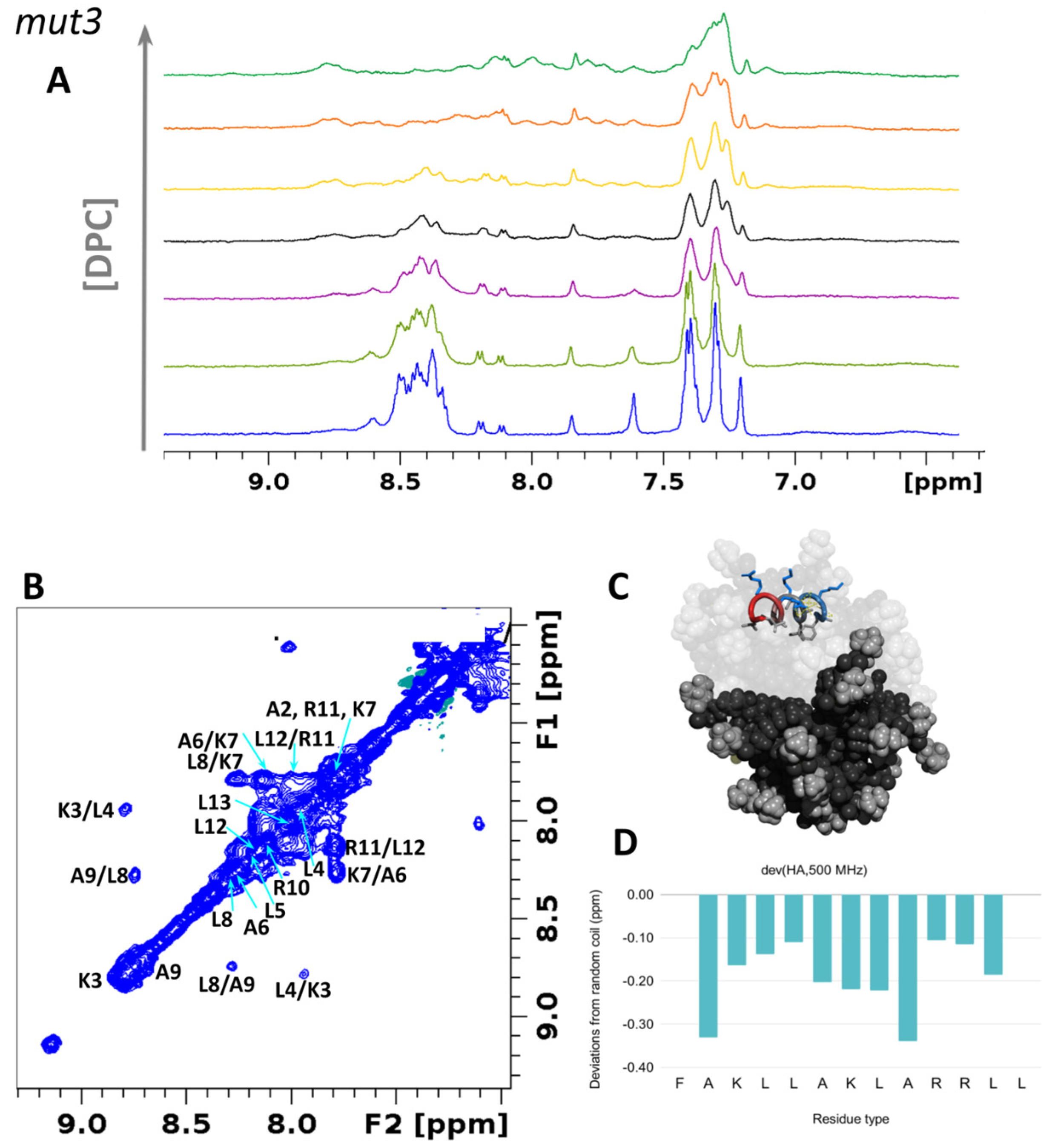
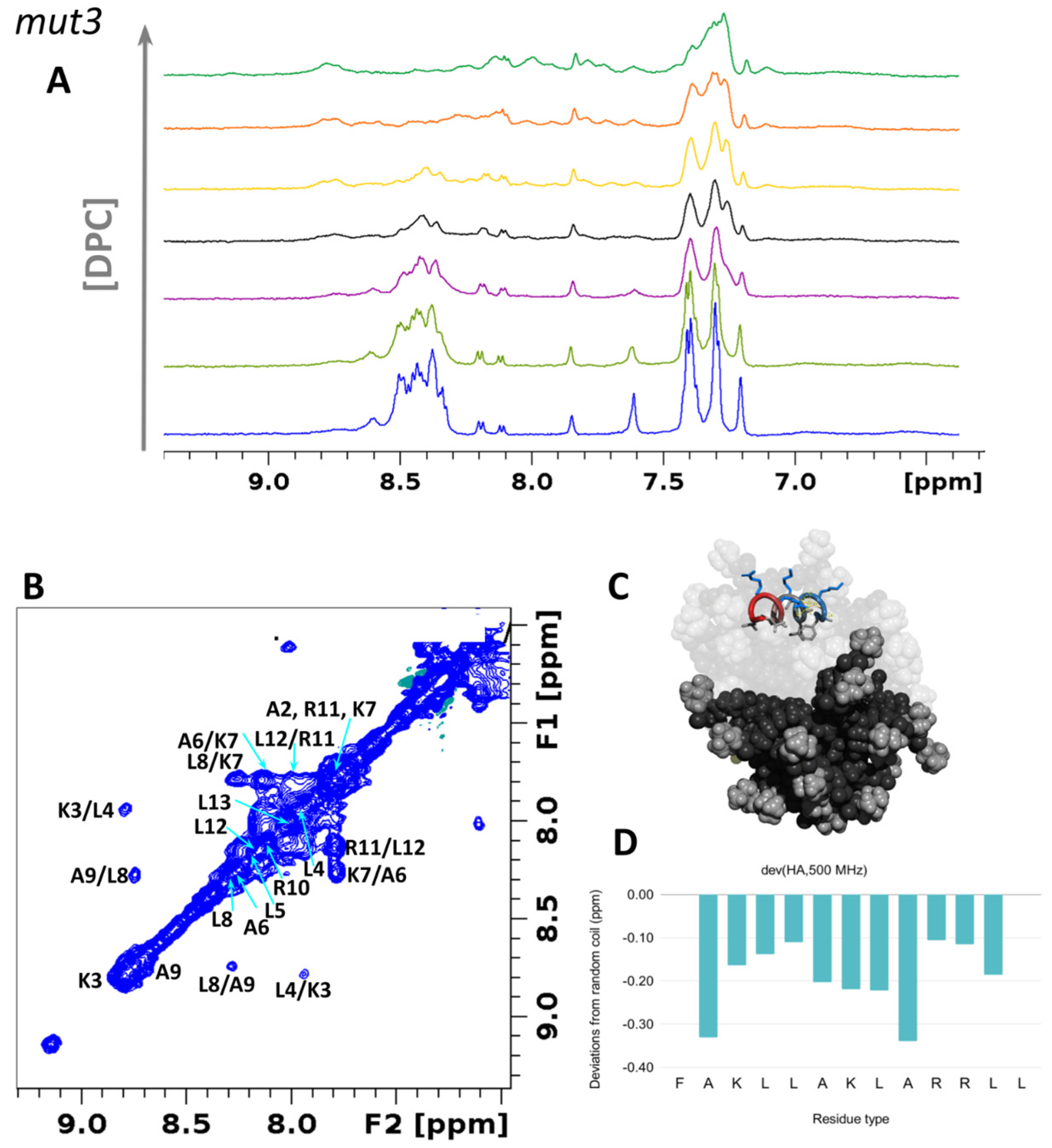
3.6.1. Structural Studies in Micelles
3.6.2. Interaction with DMPC/DHPC Bicelles
3.6.3. Interaction with SUVs
3.6.4. Interaction with MLVs
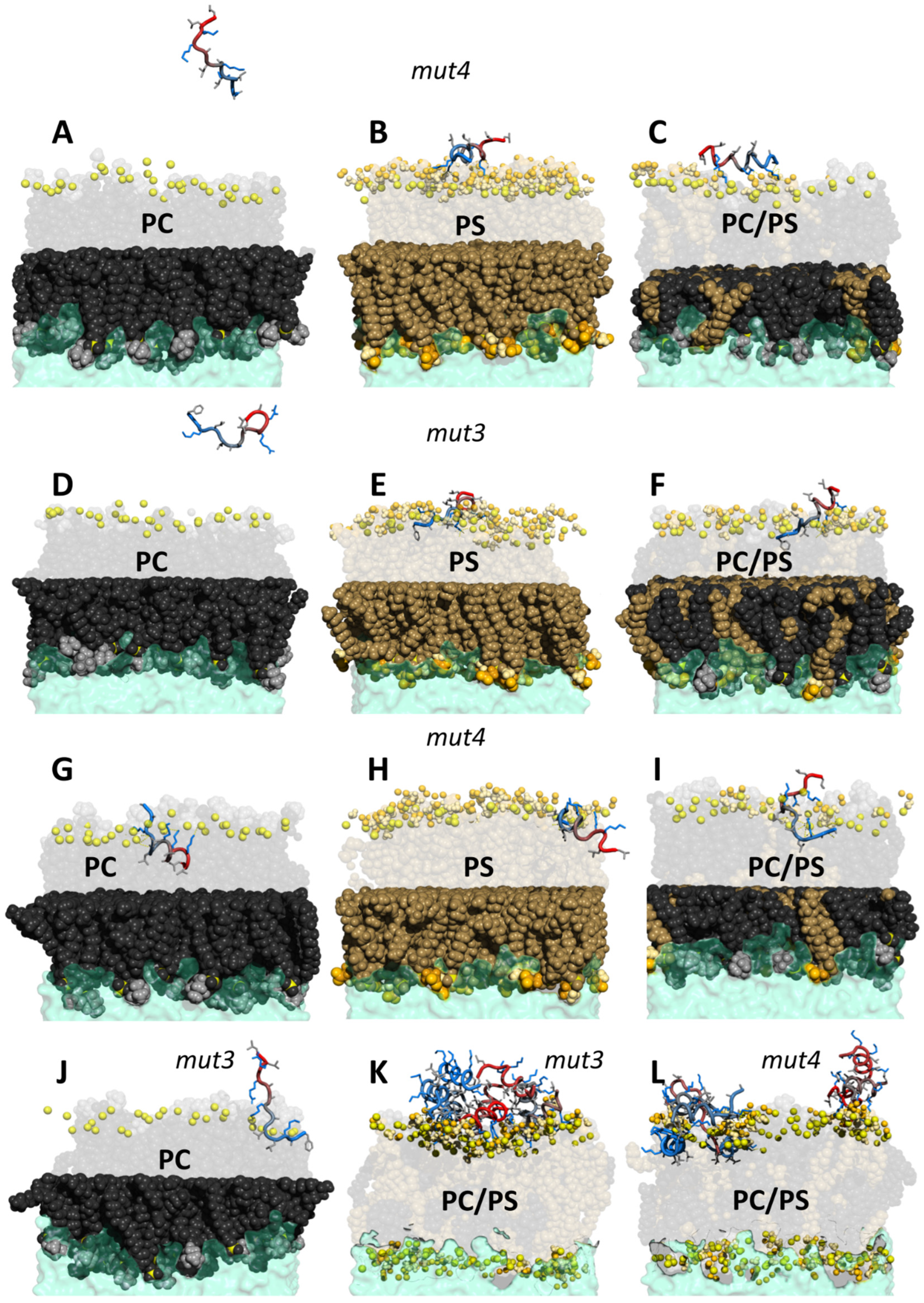
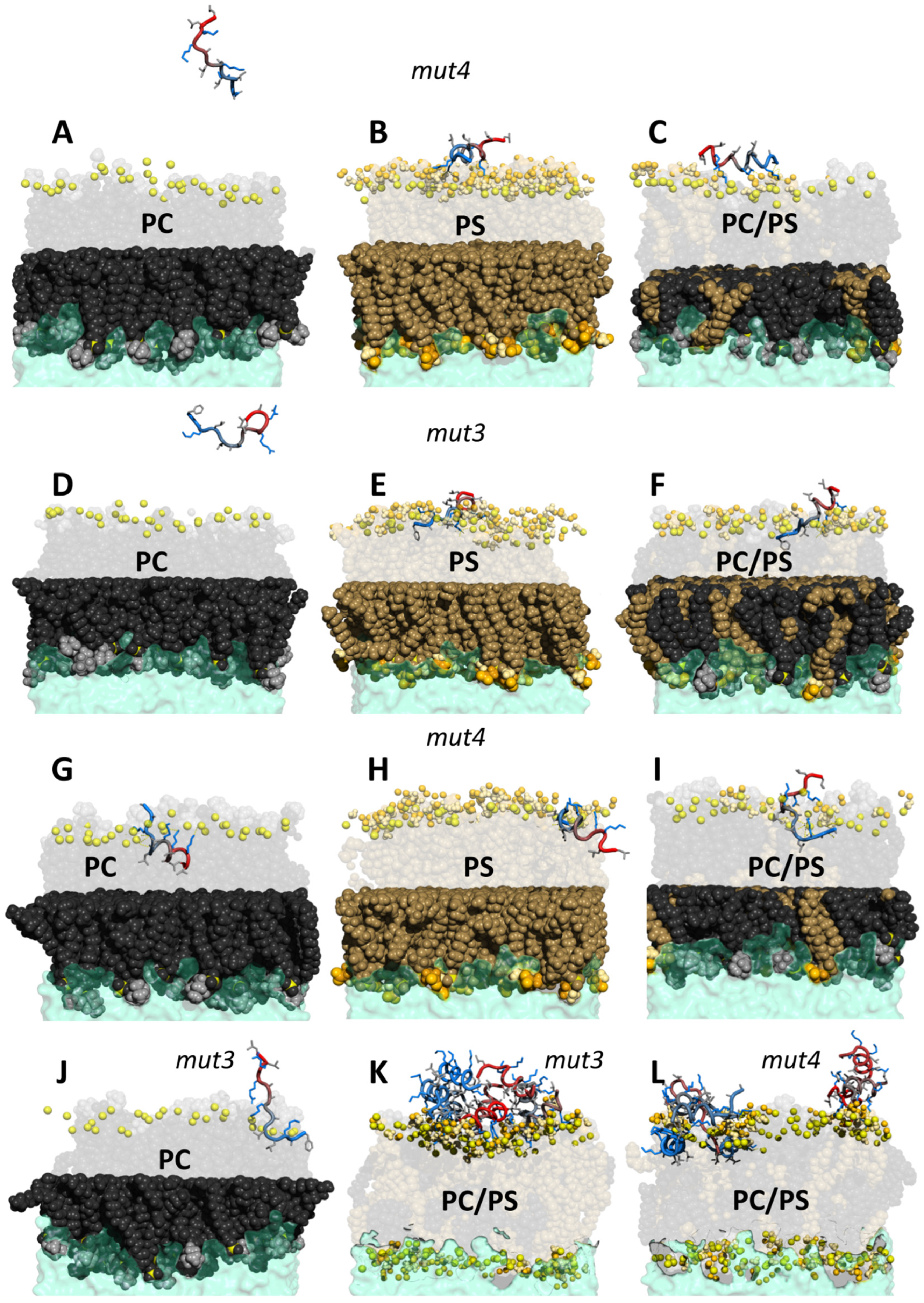
3.7. MD Simulations of PeptideLipid Interactions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Riedl, S.; Zweytick, D.; Lohner, K. Membrane-Active Host Defense Peptides--Challenges and Perspectives for the Development of Novel Anticancer Drugs. Chem. Phys. Lipids 2011, 164, 766–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoskin, D.W.; Ramamoorthy, A. Studies on Anticancer Activities of Antimicrobial Peptides. Biochim. Biophys. Acta 2008, 1778, 357–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulder, K.C.L.; Lima, L.A.; Miranda, V.J.; Dias, S.C.; Franco, O.L. Current Scenario of Peptide-Based Drugs: The Key Roles of Cationic Antitumor and Antiviral Peptides. Front. Microbiol. 2013, 4, 321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Feng, Q.; Yan, Q.; Hao, X.; Chen, Y. Alpha-Helical Cationic Anticancer Peptides: A Promising Candidate for Novel Anticancer Drugs. Mini Rev. Med. Chem. 2015, 15, 73–81. [Google Scholar] [CrossRef]
- Felício, M.R.; Silva, O.N.; Gonçalves, S.; Santos, N.C.; Franco, O.L. Peptides with Dual Antimicrobial and Anticancer Activities. Front. Chem. 2017, 5, 5. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Dong, C.; Li, X.; Han, W.; Su, X. Anticancer Potential of Bioactive Peptides from Animal Sources (Review). Oncol. Rep. 2017, 38, 637–651. [Google Scholar] [CrossRef] [Green Version]
- Teng, Q.-X.; Luo, X.; Lei, Z.-N.; Wang, J.-Q.; Wurpel, J.; Qin, Z.; Yang, D.-H. The Multidrug Resistance-Reversing Activity of a Novel Antimicrobial Peptide. Cancers 2020, 12, 1963. [Google Scholar] [CrossRef]
- Tyagi, A.; Tuknait, A.; Anand, P.; Gupta, S.; Sharma, M.; Mathur, D.; Joshi, A.; Singh, S.; Gautam, A.; Raghava, G.P. CancerPPD: A Database of Anticancer Peptides and Proteins. Nucleic Acids Res. 2015, 43, D837–D843. [Google Scholar] [CrossRef] [Green Version]
- Timur, S.S.; Gürsoy, R.N. The Role of Peptide-Based Therapeutics in Oncotherapy. J. Drug Target. 2021, 29, 1048–1062. [Google Scholar] [CrossRef]
- Grand View Research Peptide Therapeutics Market by Application (Cancer, Cardiovascular Disorder, Metabolic Disorder, Respiratory Disorder, Pain, Dermatology), by Type (Generic, Innovative) By Type of Manufacturers (In-House, Outsourced), and Segment Forecasts, 2018–2025. Available online: https://www.grandviewresearch.com/industry-analysis/peptide-therapeutics-market (accessed on 10 December 2021).
- Papo, N.; Shai, Y. Host Defense Peptides as New Weapons in Cancer Treatment. Cell. Mol. Life Sci. 2005, 62, 784–790. [Google Scholar] [CrossRef]
- Huang, Y.-B.; He, L.-Y.; Jiang, H.-Y.; Chen, Y.-X. Role of Helicity on the Anticancer Mechanism of Action of Cationic-Helical Peptides. Int. J. Mol. Sci. 2012, 13, 6849–6862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos-Martín, F.; D’Amelio, N. Molecular Basis of the Anticancer and Antibacterial Properties of CecropinXJ Peptide: An In Silico Study. Int. J. Mol. Sci. 2021, 22, 691. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Martín, F.; Herrera-León, C.; D’Amelio, N. Molecular Basis of the Anticancer, Apoptotic and Antibacterial Activities of Bombyx Mori Cecropin A. Arch. Biochem. Biophys. 2021, 715, 109095. [Google Scholar] [CrossRef] [PubMed]
- Trinidad-Calderón, P.A.; Varela-Chinchilla, C.D.; García-Lara, S. Natural Peptides Inducing Cancer Cell Death: Mechanisms and Properties of Specific Candidates for Cancer Therapeutics. Molecules 2021, 26, 7453. [Google Scholar] [CrossRef]
- Pinho, S.S.; Reis, C.A. Glycosylation in Cancer: Mechanisms and Clinical Implications. Nat. Rev. Cancer 2015, 15, 540–555. [Google Scholar] [CrossRef]
- Alves, A.C.; Ribeiro, D.; Nunes, C.; Reis, S. Biophysics in Cancer: The Relevance of Drug-Membrane Interaction Studies. Biochim. Biophys. Acta 2016, 1858, 2231–2244. [Google Scholar] [CrossRef]
- Bernardes, N.; Fialho, A.M. Perturbing the Dynamics and Organization of Cell Membrane Components: A New Paradigm for Cancer-Targeted Therapies. Int. J. Mol. Sci. 2018, 19, 3871. [Google Scholar] [CrossRef] [Green Version]
- Zalba, S.; Ten Hagen, T.L.M. Cell Membrane Modulation as Adjuvant in Cancer Therapy. Cancer Treat. Rev. 2017, 52, 48–57. [Google Scholar] [CrossRef] [Green Version]
- Sok, M.; Sentjurc, M.; Schara, M. Membrane Fluidity Characteristics of Human Lung Cancer. Cancer Lett. 1999, 139, 215–220. [Google Scholar] [CrossRef]
- Bray, B.L. Large-Scale Manufacture of Peptide Therapeutics by Chemical Synthesis. Nat. Rev. Drug Discov. 2003, 2, 587–593. [Google Scholar] [CrossRef]
- Vlieghe, P.; Lisowski, V.; Martinez, J.; Khrestchatisky, M. Synthetic Therapeutic Peptides: Science and Market. Drug Discov. Today 2010, 15, 40–56. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.; Shukla, P. Antimicrobial Peptides: Recent Insights on Biotechnological Interventions and Future Perspectives. Protein Pept. Lett. 2019, 26, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Yaghoubi, A.; Khazaei, M.; Hasanian, S.M.; Avan, A.; Cho, W.C.; Soleimanpour, S. Bacteriotherapy in Breast Cancer. Int. J. Mol. Sci. 2019, 20, 5880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neamtu, I.; Rusu, A.G.; Diaconu, A.; Nita, L.E.; Chiriac, A.P. Basic Concepts and Recent Advances in Nanogels as Carriers for Medical Applications. Drug Deliv. 2017, 24, 539–557. [Google Scholar] [CrossRef] [Green Version]
- Haupt, K.; Medina Rangel, P.X.; Bui, B.T.S. Molecularly Imprinted Polymers: Antibody Mimics for Bioimaging and Therapy. Chem. Rev. 2020, 120, 9554–9582. [Google Scholar] [CrossRef]
- Arnfast, L.; Madsen, C.G.; Jorgensen, L.; Baldursdottir, S. Design and Processing of Nanogels as Delivery Systems for Peptides and Proteins. Ther. Deliv. 2014, 5, 691–708. [Google Scholar] [CrossRef]
- Wagner, A.M.; Gran, M.P.; Peppas, N.A. Designing the New Generation of Intelligent Biocompatible Carriers for Protein and Peptide Delivery. Acta Pharm. Sin. B 2018, 8, 147–164. [Google Scholar] [CrossRef]
- Craik, D.J.; Fairlie, D.P.; Liras, S.; Price, D. The Future of Peptide-Based Drugs. Chem. Biol. Drug Des. 2013, 81, 136–147. [Google Scholar] [CrossRef]
- Grisoni, F.; Neuhaus, C.S.; Gabernet, G.; Müller, A.T.; Hiss, J.A.; Schneider, G. Designing Anticancer Peptides by Constructive Machine Learning. ChemMedChem 2018, 13, 1300–1302. [Google Scholar] [CrossRef]
- Kumar, P.; Kizhakkedathu, J.N.; Straus, S.K. Antimicrobial Peptides: Diversity, Mechanism of Action and Strategies to Improve the Activity and Biocompatibility In Vivo. Biomolecules 2018, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Jamasbi, E.; Lucky, S.S.; Li, W.; Hossain, M.A.; Gopalakrishnakone, P.; Separovic, F. Effect of Dimerized Melittin on Gastric Cancer Cells and Antibacterial Activity. Amino Acids 2018, 50, 1101–1110. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Yang, H.; Wan, L.; Cai, H.-W.; Li, S.-F.; Li, Y.-P.; Cheng, J.-Q.; Lu, X.-F. Enhancement of Cytotoxicity of Antimicrobial Peptide Magainin II in Tumor Cells by Bombesin-Targeted Delivery. Acta Pharmacol. Sin. 2011, 32, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Papo, N.; Seger, D.; Makovitzki, A.; Kalchenko, V.; Eshhar, Z.; Degani, H.; Shai, Y. Inhibition of Tumor Growth and Elimination of Multiple Metastases in Human Prostate and Breast Xenografts by Systemic Inoculation of a Host Defense—Like Lytic Peptide. Cancer Res. 2006, 66, 5371–5378. [Google Scholar] [CrossRef] [Green Version]
- Owen, D.R. Short Bioactive Peptides. U.S. Patent US6875744B2, 5 April 2005. [Google Scholar]
- Hamdan, F.; Bigdeli, Z.; Asghari, S.M.; Sadremomtaz, A.; Balalaie, S. Synthesis of Modified RGD-Based Peptides and Their in Vitro Activity. ChemMedChem 2019, 14, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, S.S.; Bang, Y.J.; Kim, S.J.; Lee, B.J. In Vitro Activities of Native and Designed Peptide Antibiotics against Drug Sensitive and Resistant Tumor Cell Lines. Peptides 2003, 24, 945–953. [Google Scholar] [CrossRef]
- Henk, W.G.; Todd, W.J.; Enright, F.M.; Mitchell, P.S. The Morphological Effects of Two Antimicrobial Peptides, Hecate-1 and Melittin, on Escherichia Coli. Scanning Microsc. 1995, 9, 19. [Google Scholar]
- Slaninová, J.; Mlsová, V.; Kroupová, H.; Alán, L.; Tůmová, T.; Monincová, L.; Borovičková, L.; Fučík, V.; Ceřovský, V. Toxicity Study of Antimicrobial Peptides from Wild Bee Venom and Their Analogs toward Mammalian Normal and Cancer Cells. Peptides 2012, 33, 18–26. [Google Scholar] [CrossRef]
- Zhang, L.; Parente, J.; Harris, S.M.; Woods, D.E.; Hancock, R.E.W.; Falla, T.J. Antimicrobial Peptide Therapeutics for Cystic Fibrosis. Antimicrob. Agents Chemother. 2005, 49, 2921–2927. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Kennedy-Stoskopf, S.; Jaynes, J.M.; Thurmond, L.M.; Tompkins, W.A. Inhibitory Activity of Synthetic Peptide Antibiotics on Feline Immunodeficiency Virus Infectivity in Vitro. J. Virol. 2002, 76, 9952–9961. [Google Scholar] [CrossRef] [Green Version]
- Herrera-León, C.; Ramos-Martín, F.; Antonietti, V.; Sonnet, P.; D’Amelio, N. The Impact of Phosphatidylserine Exposure on Cancer Cell Membranes on the Activity of the Anticancer Peptide HB43. FEBS J. 2021, 289, 1984–2003. [Google Scholar] [CrossRef]
- Forde, E.; Humphreys, H.; Greene, C.M.; Fitzgerald-Hughes, D.; Devocelle, M. Potential of Host Defense Peptide Prodrugs as Neutrophil Elastase-Dependent Anti-Infective Agents for Cystic Fibrosis. Antimicrob. Agents Chemother. 2014, 58, 978–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zapotoczna, M.; Forde, É.; Hogan, S.; Humphreys, H.; O’Gara, J.P.; Fitzgerald-Hughes, D.; Devocelle, M.; O’Neill, E. Eradication of Staphylococcus Aureus Biofilm Infections Using Synthetic Antimicrobial Peptides. J. Infect. Dis. 2017, 215, 975–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinthuvanich, C.; Veiga, A.S.; Gupta, K.; Gaspar, D.; Blumenthal, R.; Schneider, J.P. Anticancer β-Hairpin Peptides: Membrane-Induced Folding Triggers Activity. J. Am. Chem. Soc. 2012, 134, 6210–6217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos-Martín, F.; Annaval, T.; Buchoux, S.; Sarazin, C.; D’Amelio, N. ADAPTABLE: A Comprehensive Web Platform of Antimicrobial Peptides Tailored to the User’s Research. Life Sci. Alliance 2019, 2, e201900512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monnier, N.; Furlan, A.L.; Buchoux, S.; Deleu, M.; Dauchez, M.; Rippa, S.; Sarazin, C. Exploring the Dual Interaction of Natural Rhamnolipids with Plant and Fungal Biomimetic Plasma Membranes through Biophysical Studies. Int. J. Mol. Sci. 2019, 20, 1009. [Google Scholar] [CrossRef] [Green Version]
- Furlan, A.L.; Castets, A.; Nallet, F.; Pianet, I.; Grélard, A.; Dufourc, E.J.; Géan, J. Red Wine Tannins Fluidify and Precipitate Lipid Liposomes and Bicelles. A Role for Lipids in Wine Tasting? Langmuir 2014, 30, 5518–5526. [Google Scholar] [CrossRef] [PubMed]
- Furlan, A.L.; Jobin, M.-L.; Pianet, I.; Dufourc, E.J.; Géan, J. Flavanol/Lipid Interaction: A Novel Molecular Perspective in the Description of Wine Astringency & Bitterness and Antioxidant Action. Tetrahedron 2015, 71, 3143–3147. [Google Scholar]
- Grélard, A.; Guichard, P.; Bonnafous, P.; Marco, S.; Lambert, O.; Manin, C.; Ronzon, F.; Dufourc, E.J. Hepatitis B Subvirus Particles Display Both a Fluid Bilayer Membrane and a Strong Resistance to Freeze Drying: A Study by Solid-state NMR, Light Scattering, and Cryo-electron Microscopy/Tomography. FASEB J. 2013, 27, 4316–4326. [Google Scholar] [CrossRef]
- Wishart, D.S.; Bigam, C.G.; Yao, J.; Abildgaard, F.; Dyson, H.J.; Oldfield, E.; Markley, J.L.; Sykes, B.D. 1H, 13C and 15N Chemical Shift Referencing in Biomolecular NMR. J. Biomol. NMR 1995, 6, 135–140. [Google Scholar] [CrossRef]
- Nielsen, J.T.; Mulder, F.A.A. POTENCI: Prediction of Temperature, Neighbor and PH-Corrected Chemical Shifts for Intrinsically Disordered Proteins. J. Biomol. NMR 2018, 70, 141–165. [Google Scholar] [CrossRef]
- Davis, J.H.; Jeffrey, K.R.; Bloom, M.; Valic, M.I.; Higgs, T.P. Quadrupolar Echo Deuteron Magnetic Resonance Spectroscopy in Ordered Hydrocarbon Chains. Chem. Phys. Lett. 1976, 42, 390–394. [Google Scholar] [CrossRef]
- Jo, S.; Lim, J.B.; Klauda, J.B.; Im, W. CHARMM-GUI Membrane Builder for Mixed Bilayers and Its Application to Yeast Membranes. Biophys. J. 2009, 97, 50–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Wu, E.L.; Cheng, X.; Jo, S.; Rui, H.; Song, K.C.; Dávila-Contreras, E.M.; Qi, Y.; Lee, J.; Monje-Galvan, V.; Venable, R.M.; et al. CHARMM-GUI Membrane Builder toward Realistic Biological Membrane Simulations. J. Comput. Chem. 2014, 35, 1997–2004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A Unified Platform for Automated Protein Structure and Function Prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Zhang, Y. I-TASSER Server: New Development for Protein Structure and Function Predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein Structure and Function Prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2016, 14, 71–73. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.; Kim, T.; Im, W. Automated Builder and Database of Protein/Membrane Complexes for Molecular Dynamics Simulations. PLoS ONE 2007, 2, e880. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Jo, S.; Lee, H.S.; Klauda, J.B.; Im, W. CHARMM-GUI Micelle Builder for Pure/Mixed Micelle and Protein/Micelle Complex Systems. J. Chem. Inf. Model. 2013, 53, 2171–2180. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; Hermans, J. Interaction Models for Water in Relation to Protein Hydration. Jerus. Symp. Quantum Chem. Biochem. 1981, 14, 331–342. [Google Scholar]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Nosé, S.; Klein, M.L. Constant Pressure Molecular Dynamics for Molecular Systems. Mol. Phys. 1983, 50, 1055–1076. [Google Scholar] [CrossRef]
- Nosé, S. A Unified Formulation of the Constant Temperature Molecular Dynamics Methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef] [Green Version]
- Hoover, W.G. Canonical Dynamics: Equilibrium Phase-Space Distributions. Phys. Rev. A Gen. Phys. 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.J.; Klauda, J.B.; Sodt, A.J. Simulation Best Practices for Lipid Membranes [Article v1.0]. Living J. Comput. Mol. Sci. 2019, 1, 5966. [Google Scholar] [CrossRef]
- Lemkul, J. From Proteins to Perturbed Hamiltonians: A Suite of Tutorials for the GROMACS-2018 Molecular Simulation Package [Article v1.0]. Living J. Comput. Mol. Sci. 2019, 1, 5068. [Google Scholar] [CrossRef]
- Koradi, R.; Billeter, M.; Wüthrich, K. MOLMOL: A Program for Display and Analysis of Macromolecular Structures. J. Mol. Graph. 1996, 14, 51–55. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Janert, P.K. Gnuplot in Action: Understanding Data with Graphs; Manning Publications: New York, NY, USA, 2010; ISBN 9781933988399. [Google Scholar]
- DeLano, W.L. Pymol: An Open-Source Molecular Graphics Tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Shahmiri, M.; Enciso, M.; Mechler, A. Controls and Constrains of the Membrane Disrupting Action of Aurein 1.2. Sci. Rep. 2015, 5, 16378. [Google Scholar] [CrossRef] [Green Version]
- Dennison, S.R.; Harris, F.; Phoenix, D.A. A Study on the Importance of Phenylalanine for Aurein Functionality. Protein Pept. Lett. 2009, 16, 1455–1458. [Google Scholar] [CrossRef]
- Arora, A.; Majhi, S.; Mishra, A. Designing a Short, Potent, Pore-Forming Antimicrobial Peptide. Mater. Today Proc. 2022, 49, 2392–2396. [Google Scholar] [CrossRef]
- Liu, P.; Zeng, X.; Wen, X. Design and Synthesis of New Cationic Antimicrobial Peptides with Low Cytotoxicity. Int. J. Pept. Res. Ther. 2021, 27, 831–840. [Google Scholar] [CrossRef]
- Rice, A.; Wereszczynski, J. Probing the Disparate Effects of Arginine and Lysine Residues on Antimicrobial Peptide/Bilayer Association. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1941–1950. [Google Scholar] [CrossRef]
- Mól, A.R.; Castro, M.S.; Fontes, W. NetWheels: A Web Application to Create High Quality Peptide Helical Wheel and Net Projections. BioRxiv 2018, 416347. [Google Scholar] [CrossRef] [Green Version]
- Gaspar, D.; Veiga, A.S.; Castanho, M.A.R.B. From Antimicrobial to Anticancer Peptides. A Review. Front. Microbiol. 2013, 4, 294. [Google Scholar] [CrossRef] [Green Version]
- Thundimadathil, J. Cancer Treatment Using Peptides: Current Therapies and Future Prospects. J. Amino Acids 2012, 2012, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zucker, S.; Vacirca, J. Role of Matrix Metalloproteinases (MMPs) in Colorectal Cancer. Cancer Metastasis Rev. 2004, 23, 101–117. [Google Scholar] [CrossRef] [PubMed]
- Visse, R.; Nagase, H. Matrix Metalloproteinases and Tissue Inhibitors of Metalloproteinases: Structure, Function, and Biochemistry. Circ. Res. 2003, 92, 827–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darzynkiewicz, Z.; Huang, X.; Zhao, H. Analysis of Cellular DNA Content by Flow Cytometry. Curr. Protoc. Cytom. 2017, 82, 7.5.1–7.5.20. [Google Scholar] [PubMed]
- Zhou, W.; Pan, T.; Cui, H.; Zhao, Z.; Chu, P.K.; Yu, X.-F. Black Phosphorus: Bioactive Nanomaterials with Inherent and Selective Chemotherapeutic Effects. Angew. Chem. Int. Ed. Engl. 2019, 58, 769–774. [Google Scholar] [CrossRef]
- Roca-Lema, D.; Martinez-Iglesias, O.; Fernández de Ana Portela, C.; Rodríguez-Blanco, A.; Valladares-Ayerbes, M.; Díaz-Díaz, A.; Casas-Pais, A.; Prego, C.; Figueroa, A. In Vitro Anti-Proliferative and Anti-Invasive Effect of Polysaccharide-Rich Extracts from Trametes Versicolor and Grifola Frondosa in Colon Cancer Cells. Int. J. Med. Sci. 2019, 16, 231–240. [Google Scholar] [CrossRef] [Green Version]
- PodgÓrska, M.; Pietraszek-Gremplewicz, K.; OlszaŃska, J.; Nowak, D. The Role of Apelin and Apelin Receptor Expression in Migration and Invasiveness of Colon Cancer Cells. Anticancer Res. 2021, 41, 151–161. [Google Scholar] [CrossRef]
- Kallick, D.A.; Tessmer, M.R.; Watts, C.R.; Li, C.Y. The Use of Dodecylphosphocholine Micelles in Solution NMR. J. Magn. Reson. Ser. B 1995, 109, 60–65. [Google Scholar] [CrossRef]
- Roumestand, C.; Louis, V.; Aumelas, A.; Grassy, G.; Calas, B.; Chavanieu, A. Oligomerization of Protegrin-1 in the Presence of DPC Micelles. A Proton High-Resolution NMR Study. FEBS Lett. 1998, 421, 263–267. [Google Scholar] [CrossRef] [Green Version]
- Beswick, V.; Guerois, R.; Cordier-Ochsenbein, F.; Coïc, Y.M.; Tam, H.D.; Tostain, J.; Noël, J.P.; Sanson, A.; Neumann, J.M. Dodecylphosphocholine Micelles as a Membrane-like Environment: New Results from NMR Relaxation and Paramagnetic Relaxation Enhancement Analysis. Eur. Biophys. J. 1999, 28, 48–58. [Google Scholar] [CrossRef]
- Legrand, B.; Laurencin, M.; Sarkis, J.; Duval, E.; Mouret, L.; Hubert, J.-F.; Collen, M.; Vié, V.; Zatylny-Gaudin, C.; Henry, J.; et al. Structure and Mechanism of Action of a de Novo Antimicrobial Detergent-like Peptide. Biochim. Biophys. Acta 2011, 1808, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Sykes, B.D.; Richards, F.M. The Chemical Shift Index: A Fast and Simple Method for the Assignment of Protein Secondary Structure through NMR Spectroscopy. Biochemistry 1992, 31, 1647–1651. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Sykes, B.D. The 13C Chemical-Shift Index: A Simple Method for the Identification of Protein Secondary Structure Using 13C Chemical-Shift Data. J. Biomol. NMR 1994, 4, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S. Interpreting Protein Chemical Shift Data. Prog. Nucl. Magn. Reson. Spectrosc. 2011, 58, 62–87. [Google Scholar] [CrossRef]
- de Planque, M.R.R.; Bonev, B.B.; Demmers, J.A.A.; Greathouse, D.V.; Koeppe, R.E., 2nd; Separovic, F.; Watts, A.; Killian, J.A. Interfacial Anchor Properties of Tryptophan Residues in Transmembrane Peptides Can Dominate over Hydrophobic Matching Effects in Peptide-Lipid Interactions. Biochemistry 2003, 42, 5341–5348. [Google Scholar] [CrossRef]
- Petersen, F.N.R.; Jensen, M.Ø.; Nielsen, C.H. Interfacial Tryptophan Residues: A Role for the Cation-Pi Effect? Biophys. J. 2005, 89, 3985–3996. [Google Scholar] [CrossRef] [Green Version]
- Sparks, K.A.; Gleason, N.J.; Gist, R.; Langston, R.; Greathouse, D.V.; Koeppe, R.E., 2nd. Comparisons of Interfacial Phe, Tyr, and Trp Residues as Determinants of Orientation and Dynamics for GWALP Transmembrane Peptides. Biochemistry 2014, 53, 3637–3645. [Google Scholar] [CrossRef]
- Shahmiri, M.; Cornell, B.; Mechler, A. Phenylalanine Residues Act as Membrane Anchors in the Antimicrobial Action of Aurein 1.2. Biointerphases 2017, 12, 05G605. [Google Scholar] [CrossRef]
- Da Costa, G.; Mouret, L.; Chevance, S.; Le Rumeur, E.; Bondon, A. NMR of Molecules Interacting with Lipids in Small Unilamellar Vesicles. Eur. Biophys. J. 2007, 36, 933–942. [Google Scholar] [CrossRef]
- Bevers, E.M.; Williamson, P.L. Getting to the Outer Leaflet: Physiology of Phosphatidylserine Exposure at the Plasma Membrane. Physiol. Rev. 2016, 96, 605–645. [Google Scholar] [CrossRef]
- Henderson, T.O.; Glonek, T.; Myers, T.C. Phosphorus-31 Nuclear Magnetic Resonance Spectroscopy of Phospholipids. Biochemistry 1974, 13, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Abu-Baker, S.; Qi, X.; Lorigan, G.A. Investigating the Interaction of Saposin C with POPS and POPC Phospholipids: A Solid-State NMR Spectroscopic Study. Biophys. J. 2007, 93, 3480–3490. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 24 h | 48 h | |||||
|---|---|---|---|---|---|---|
| Peptide | SW480 | SW620 | HT29 | SW480 | SW620 | HT29 |
| HB43 | 12 ± 4 | 10 ± 1 | 11 ± 1 | 11.4 ± 0.3 | 10.9 ± 0.7 | 13.3 ± 0.4 |
| mut2 | 34 ± 2 | 40 ± 3 | 50 ± 3 | 39 ± 3 | 47 ± 3 | 50 ± 3 |
| mut3 | 8 ± 1 | 9 ± 1 | 9 ± 1 | 10.0 ± 0.4 | 10.94 ± 0.07 | 9.5 ± 0.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herrera-León, C.; Ramos-Martín, F.; El Btaouri, H.; Antonietti, V.; Sonnet, P.; Martiny, L.; Zevolini, F.; Falciani, C.; Sarazin, C.; D’Amelio, N. The Influence of Short Motifs on the Anticancer Activity of HB43 Peptide. Pharmaceutics 2022, 14, 1089. https://doi.org/10.3390/pharmaceutics14051089
Herrera-León C, Ramos-Martín F, El Btaouri H, Antonietti V, Sonnet P, Martiny L, Zevolini F, Falciani C, Sarazin C, D’Amelio N. The Influence of Short Motifs on the Anticancer Activity of HB43 Peptide. Pharmaceutics. 2022; 14(5):1089. https://doi.org/10.3390/pharmaceutics14051089
Chicago/Turabian StyleHerrera-León, Claudia, Francisco Ramos-Martín, Hassan El Btaouri, Viviane Antonietti, Pascal Sonnet, Laurent Martiny, Fabrizia Zevolini, Chiara Falciani, Catherine Sarazin, and Nicola D’Amelio. 2022. "The Influence of Short Motifs on the Anticancer Activity of HB43 Peptide" Pharmaceutics 14, no. 5: 1089. https://doi.org/10.3390/pharmaceutics14051089
APA StyleHerrera-León, C., Ramos-Martín, F., El Btaouri, H., Antonietti, V., Sonnet, P., Martiny, L., Zevolini, F., Falciani, C., Sarazin, C., & D’Amelio, N. (2022). The Influence of Short Motifs on the Anticancer Activity of HB43 Peptide. Pharmaceutics, 14(5), 1089. https://doi.org/10.3390/pharmaceutics14051089