
Developing Clinically Relevant Dissolution Specifications (CRDSs) for Oral Drug Products: Virtual Webinar Series




 , , , ,
, , , ,  , ,
, ,
Abstract
:1. Introduction
- Introduction to CRDSs—the ‘what’ and the ‘why’;
- Introduction to PBPK/PBBM Modelling—the ‘when’ and the ‘how’;
- How to develop CRDSs, including case studies from industry;
- Overview of global regulatory trends within CRDSs, including progress, challenges, and emerging opportunities;
- Future developments with PBBM/PBPK software packages;
- Emerging opportunities within PBPK/PBBM modelling to support CRDSs, including new research areas.
2. Summary of Webinars
2.1. Webinar 1: Clinically Relevant Dissolution Specifications: Why, What, How, and When? (Paul A. Dickinson and Andreas Abend)
2.1.1. The Why and What (Paul Dickinson)
- Identity (safety and efficacy);
- Assay (safety and efficacy);
- Related substances/degradants (safety).
- An important surrogate of clinical performance;
- A routine test of product quality.
‘Drug absorption from a solid dosage form after oral administration depends on the release of the drug substance from the drug product, the dissolution or solubilization of the drug under physiological conditions, and the permeability across the gastrointestinal tract. Because of the critical nature of the first two of these steps, in vitro dissolution may be relevant to the prediction of in vivo performance. Based on this general consideration, in vitro dissolution tests for immediate release solid oral dosage forms, such as tablets and capsules, are used to (1) assess the lot-to-lot quality of a drug product; (2) guide development of new formulations; and (3) ensure continuing product quality and performance after certain changes, such as changes in the formulation, the manufacturing process, the site of manufacture, and the scale-up of the manufacturing process’[12]
‘A dissolution procedure intended to be used as a routine control test for immediate release drug products should be robust, reproducible and discriminatory in order to assure a consistent product quality and to detect product quality attributes, which, if altered, may affect the in vivo performance’[13].
2.1.2. The How and When (Andreas Abend)
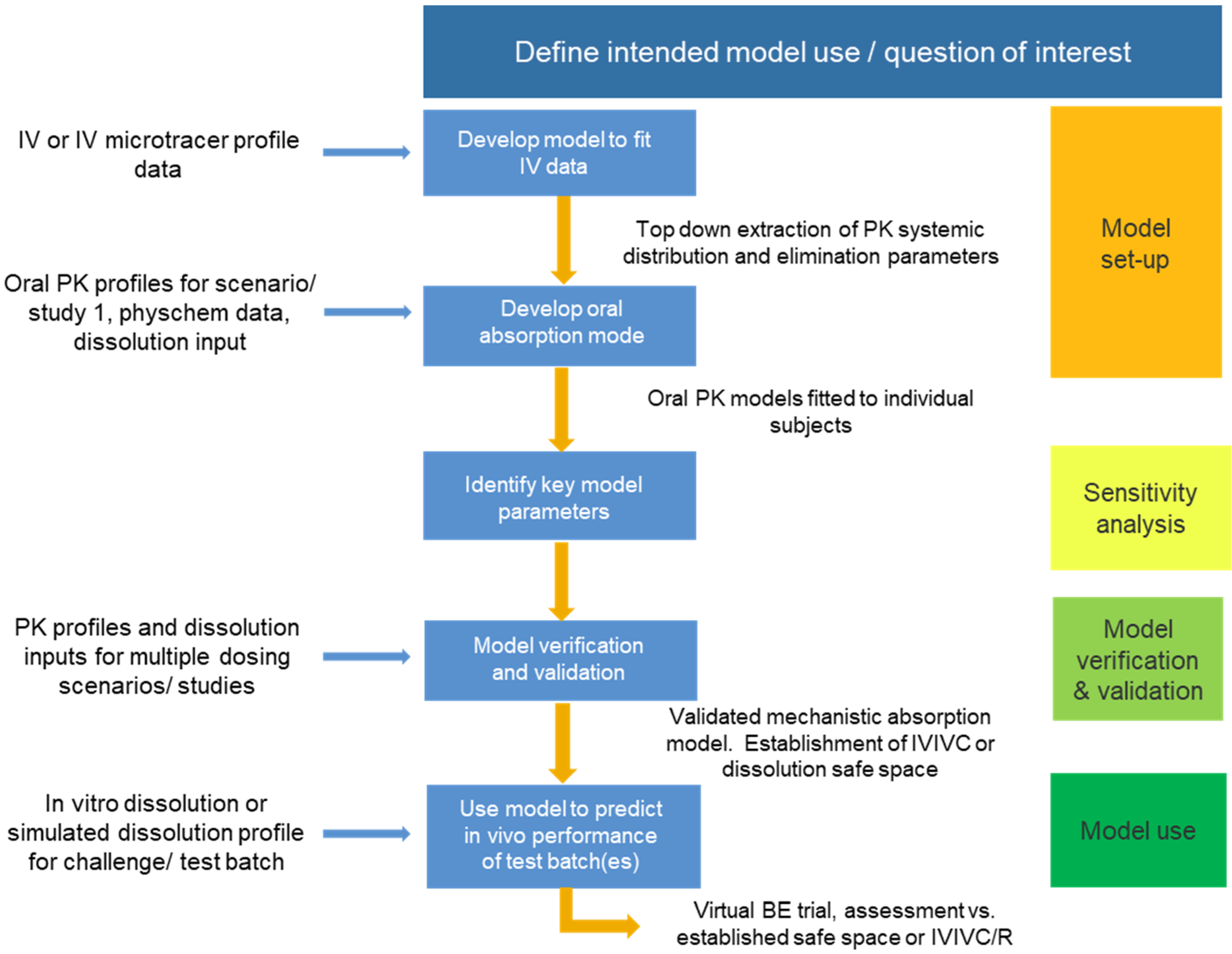
2.2. Webinar 2: Introduction to PBPK/PBBM: How to Build a PBBM Model and Why? (Andrea Moir and Sue Cole)
- Guideline on the pharmacokinetic and clinical evaluation, modified release dosage forms 2014 [22];
- Guideline on the reporting of physiologically based pharmacokinetic (PBPK) modelling and simulation 2018 [23];
- FDA Guidance on ‘Physiologically Based Pharmacokinetic Analyses—Format and Content Guidance for Industry’ [24];
- The use of physiologically based pharmacokinetic analyses—biopharmaceutics applications for oral drug product development, manufacturing changes, and controls [10].
- Question of interest;
- Context of use—how is the model used to answer the question?;
- Assess model risk—what weight does the model bring to the decision? Additionally, what would be the consequence of a wrong decision?;
- Establish risk-informed credibility assessment—this needs to be commensurate with the risk and requires validation and verification activities;
- Model credibility assessment—considered against the context of use. This approach is consistent with that recommended by EMA Guidance. To date, there are no examples in the literature of the application of this framework to PBBM, but it is referred to in the recent draft FDA guideline ‘The Use of Physiologically Based Pharmacokinetic Analyses’ [10].
2.3. Webinar 3: How to Develop CRDSs including Case Studies from the Industry (Xavier Pepin, Diansong Zhou, and Christophe Tistaert)
2.3.1. Case Study 1—PBPK Application to Evaluate Acalabrutinib Absorption and Drug Substance PSD Safe Space Size (Xavier Pepin and Diansong Zhou)
2.3.2. Case Study 2: Establishing Clinically Relevant Specifications in Pre-Approval and Post-Approval Environment for an Orally Administered Compound Formulated in Several Immediate release Drug Products (Christophe Tistaert)
2.4. Webinar 4: Overview of Global Regulatory Trends within CRDS including Progress, Challenges, and Emerging Opportunities (Aris Dokoumetzidis and Om Anand)
2.4.1. Absorption PBPK Models in Regulatory Applications: The EMA Experience (Aris Dokoumetzidis)
2.4.2. Clinically Relevant Dissolution Specifications: A Biopharmaceutics Risk-Based Approach: An FDA Perspective (Om Anand)
2.5. Webinar 5: Future Developments with PBBM/PBPK Software Packages (David Turner and Maxime Le Merdy)
2.5.1. Virtual Bioequivalence, Mechanistic Models, and Advanced Models (David Turner)
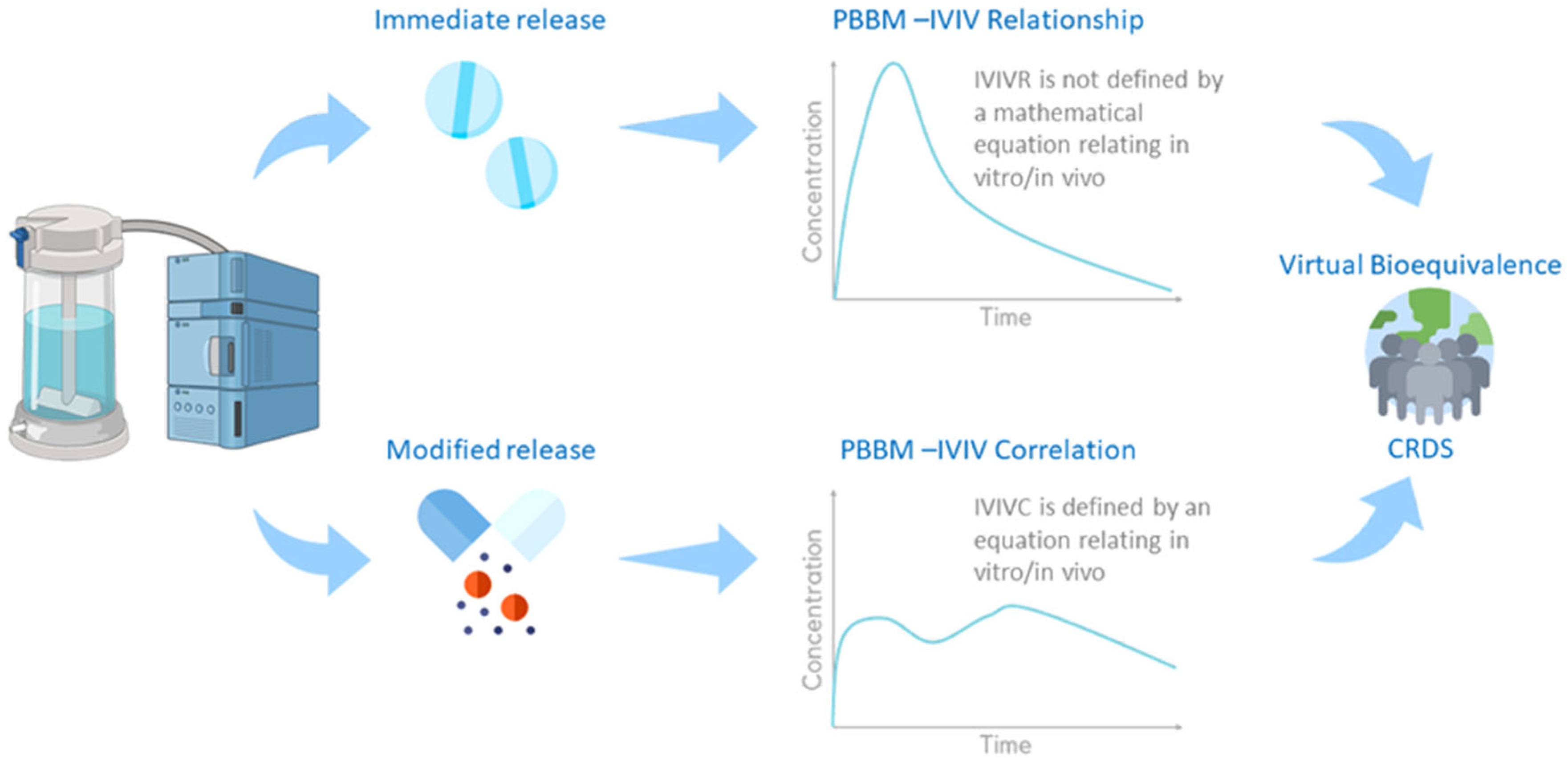
2.5.2. How to Use Modelling and Simulation to Link In Vitro Dissolution to Drugs In Vivo Behaviour (Maxime Le Merdy)
- (1)
- The Z-factor method has been used by the generic industry to (1) establish an IVIVR; (2) predict in vivo PK profiles of test and reference products based on their in vitro dissolution profiles and the validated IVIVR; (3) use virtual BE trials to predict the BE between the test and reference formulations for a BCS Class 2 compound [58].
- (2)
- The US FDA used an IVIVR to investigate the clinical impact of potential changes in warfarin crystalline form upon storage in different conditions. The IVIVR predicted PK profiles could be perfectly overlaid with BE clinical trial results, demonstrating the great predictive ability of this M&S virtual BE method [59].
2.6. Webinar 6: Emerging Opportunities within PBPK/PBBM Modelling to Support CRDSs, including New Research Areas (Adam Darwich, Brendan Griffin, and Jennifer Dressman)
2.6.1. Absorption Modelling: A Brief History, Emerging Trends, and Path Forward (Adam Darwich)
2.6.2. Predicting Preclinical Outcomes: An In Vitro–In Silico Approach to Guide Oral Formulation Design (Brendan Griffin)
2.6.3. An In Vitro–In Silico Approach to Predicting Oral Absorption of Drugs When Co-Administered with PPIs (Jennifer Dressman)
3. Q&A Sessions—Key Themes
4. Conclusions
- PBBM modelling approaches and their utility, including model verification and validation. Further dialogue is required to clearly understand requirements and manage expectations both for the industry and regulators;
- Role of dissolution data (QC or more biorelevant media) as appropriate input into PBBM models;
- Clinical study design to support the setting of clinically relevant dissolution specifications;
- Regulators and the industry should develop a CRDS roadmap and framework for implementing CRDSs;
- Opportunity to engage and set up an EMA–Industry workgroup on CRDSs and PBBM in pharmaceutical applications.
Author Contributions
Funding
Informed Consent Statement
Conflicts of Interest
Abbreviations
| ADAM | Advanced dissolution absorption and metabolism |
| ADME | Absorption distribution metabolism excretion |
| ADBSM | Advanced dynamic bile salt model |
| API | Active pharmaceutical ingredient |
| APS | Academy of Pharmaceutical Science |
| AUC | Area under the curve |
| BA | Bioavailability |
| BE | Bioequivalent |
| BCS | Biopharmaceutical Classification System |
| BioRAM | Biopharmaceutics risk assessment roadmap |
| BMI | Body mass index |
| BS | Bile salts |
| BSV | Between subject variability |
| CBA | Critical bioavailability attribute |
| CI | Confidence interval |
| CMC | Chemical and Manufacturing Controls |
| CMA | Critical material attributes |
| Cmax | Maximum concentration |
| Cp | Plasma concentration time profile |
| CPP | Critical process parameters |
| CQA | Critical quality attribute |
| CRDS | Clinically relevant dissolution specifications |
| CR | Controlled release |
| CV | Coefficient of Variation |
| DCS | Development classification system |
| DDI | Drug–drug interaction |
| DLM | Diffusion layer model |
| DP | Drug product |
| DS | Drug substance |
| EMA | European Medicines Agency |
| EU | European Union |
| FaSSIF | Fasted state simulated intestinal fluid |
| FaSSIPp | Porcine fasted state simulated intestinal fluid |
| FDA | Food and Drug Administration |
| FDCPD | Fixed-dose combination drug product |
| GI | Gastrointestinal |
| GIT | Gastrointestinal tract |
| GMR | Geometric mean ratio |
| HCl | Hydrochloric acid |
| HPLC | High-pressure liquid chromatography |
| ICH | International Committee for Harmonisation |
| IMMC | Interdigestive migrating motor complex |
| IMPD | Investigational medicinal products |
| IVO | Inter occasion variability |
| IR | Immediate release |
| IV | Intravenous |
| IVIVE | In vitro–in vivo extrapolation |
| IVIVC | In vitro–in vivo correlation |
| IVIVR | In vitro–in vivo relationship |
| MR | Modified release |
| MRI | Magnetic resonance imaging |
| M&S | Modelling and simulation |
| NDA | New drug application |
| PhEUR | European Pharmacopeia |
| PBDT | Physiologically based dissolution testing |
| PBPK | Physiologically based pharmacokinetics |
| PBBM | Physiologically based biopharmaceutics modelling |
| PK | Pharmacokinetics |
| PPI | Proton pump inhibitor |
| PPK | Population pharmacokinetics |
| P-PSD | Product particle size distribution |
| PSD | Particle size distribution |
| QC | Quality control |
| RWD | Real-world data |
| SIF | Simulated intestinal fluid |
| SGF | Simulated gastric fluid |
| SUPAC | Scale-up and post-approval changes |
| USP | US Pharmacopeia |
| VBE | Virtual bioequivalence |
| WSV | Within-subject variability |
References
- McAllister, M.; Flanagan, T.; Boon, K.; Pepin, X.; Tistaert, C.; Jamei, M.; Abend, A.; Kotzagiorgis, E.; Mackie, C. Meeting Report: Developing Clinically Relevant Dissolution Specifications for Oral Drug Products—Industrial and Regulatory Perspectives. Pharmaceutics 2020, 12, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heimbach, T.; Kesisoglou, F.; Novakovic, J.; Tistaert, C.; Mueller-Zsigmondy, M.; Kollipara, S.; Ahmed, T.; Mitra, A.; Suarez-Sharp, S. Establishing the Bioequivalence Safe Space for Immediate-Release Oral Dosage Forms using Physiologically Based Biopharmaceutics Modeling (PBBM): Case Studies. J. Pharm. Sci. 2021, 110, 3896–3906. [Google Scholar] [CrossRef] [PubMed]
- Loisios-Konstantinidis, I.; Dressman, J. Physiologically Based Pharmacokinetic/Pharmacodynamic Modeling to Support Waivers of In Vivo Clinical Studies: Current Status, Challenges, and Opportunities. Mol. Pharm. 2021, 18, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Jereb, R.; Kristl, A.; Mitra, A. Prediction of fasted and fed bioequivalence for immediate release drug products using physiologically based biopharmaceutics modeling (PBBM). Eur. J. Pharm. Sci. 2020, 155, 105554. [Google Scholar] [CrossRef] [PubMed]
- Bermejo, M.; Hens, B.; Mudie, D.; Paixao, P.; Tsume, Y.; Amidon, G.L.; Bermejo, M.; Hens, B.; Dickens, J.; Shedden, K.; et al. A Mechanistic Physiologically-Based Biopharmaceutics Modeling (PBBM) Approach to Assess the In Vivo Performance of an Orally Administered Drug Product: From IVIVC to IVIVP. Pharmaceutics 2020, 12, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamei, M.; Abrahamsson, B.; Karlsson, E.; Brown, J.; Bevernage, J.; Tistaert, C.; Bolger, M.B.; Mullin, J.M.; Heimbach, T.; Kesisoglou, F.; et al. Current status and future opportunities for incorporation of dissolution data in PBPK modeling for pharmaceutical development and regulatory applications: OrBiTo consortium commentary. Eur. J. Pharm. Biopharm. 2020, 155, 55–68. [Google Scholar] [CrossRef]
- Pepin, X.J.H.; Dressman, J.; Parrott, N.; Delvadia, P.; Mitra, A.; Zhang, X.; Babiskin, A.; Kolhatkar, V.; Seo, P.; Taylor, L.S.; et al. In Vitro Biopredictive Methods: A Workshop Summary Report. J. Pharm. Sci. 2021, 110, 567–583. [Google Scholar] [CrossRef]
- Pepin, X.J.H.; Parrott, N.; Dressman, J.; Delvadia, P.; Mitra, A.; Zhang, X.; Babiskin, A.; Kolhatkar, V.; Suarez-Sharp, S. Current State and Future Expectations of Translational Modeling Strategies to Support Drug Product Development, Manufacturing Changes and Controls: A Workshop Summary Report. J. Pharm. Sci. 2021, 110, 555–566. [Google Scholar] [CrossRef]
- Mitra, A.; Suarez-Sharp, S.; Pepin, X.J.H.; Flanagan, T.; Zhao, Y.; Kotzagiorgis, E.; Parrott, N.; Sharan, S.; Tistaert, C.; Heimbach, T.; et al. Applications of Physiologically Based Biopharmaceutics Modeling (PBBM) to Support Drug Product Quality: A Workshop Summary Report. J. Pharm. Sci. 2021, 110, 594–609. [Google Scholar] [CrossRef]
- FDA. The Use of Physiologically Based Pharmacokinetic Analyses—Biopharmaceutics Applications for Oral Drug Product Development, Manufacturing Changes, and Controls. Guidance for Industry; Center for Drug Evaluation and Research (CDER), Ed.; F.A.D.A. USA Department of Health and Human Services: Washington, DC, USA, 2020.
- Dickinson, P.A.; Stott, P.W.; Townsend, A.I.; Smart, J.P.; Ghahramani, P.; Hammett, T.; Billett, L.; Behn, S.; Gibb, R.C.; Abrahamsson, B. Clinical Relevance of Dissolution Testing in Quality by Design. AAPS J. 2008, 10, 380. [Google Scholar] [CrossRef] [Green Version]
- U.S. Food and Drug Administration. Guidance for Industry: Dissolution Testing of Immediate Release Solid Oral Dosage Forms. 1997. Available online: https://www.fda.gov/media/70936/download (accessed on 15 December 2021).
- EMA, European Medicines Agency (EMA). Reflection Paper on the Dissolution Specification for Generic Solid Oral Immediate Release Products with Systemic Action (EMA/CHMP/CVMP/QWP/336031/2017); EMA: London, UK, 2017. [Google Scholar]
- Selen, A.; Dickinson, P.A.; Müllertz, A.; Crison, J.R.; Mistry, H.B.; Cruañes, M.T.; Martinez, M.N.; Lennernäs, H.; Wigal, T.L.; Swinney, D.C.; et al. The Biopharmaceutics Risk Assessment Roadmap for Optimizing Clinical Drug Product Performance. J. Pharm. Sci. 2014, 103, 3377–3397. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, P.A.; Kesisoglou, F.; Flanagan, T.; Martinez, M.N.; Mistry, H.B.; Crison, J.R.; Polli, J.E.; Cruañes, M.T.; Serajuddin, A.T.; Müllertz, A.; et al. Optimizing Clinical Drug Product Performance: Applying Biopharmaceutics Risk Assessment Roadmap (BioRAM) and the BioRAM Scoring Grid. J. Pharm. Sci. 2016, 105, 3243–3255. [Google Scholar] [CrossRef] [PubMed]
- Selen, A.A.M.; Kesisoglou, F.; Ho, R.J.Y.; Cook, J.A.; Dickinson, P.A.; Flanagan, T. Integrated Multi-stakeholder Systems Thinking Strategy: Decision-making with Biopharmaceutics Risk Assessment Roadmap (BioRAM) to Optimize Clinical Performance of Drug Products. AAPS J. 2020, 22, 97–117. [Google Scholar] [CrossRef] [PubMed]
- Gray, V.A.; Mann, J.C.; Barker, R.; Pepin, X.J.H. The Case for Physiologically Based Biopharmaceutics Modelling (PBBM): What do Dissolution Scientists Need to Know? Dissolution Technol. 2020, 27, 6–19. [Google Scholar] [CrossRef]
- Abend, A.H.T.; Cohen, M.; Kesisoglou, F.; Pepin, X.; Suarez-Sharp, S. Dissolution and Translational Modeling Strategies Enabling Patient-Centric Drug Product Development: The M-CERSI Workshop Summary Report. AAPS J. 2018, 20, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, F.C.R.; Zhao, L.; Rostami-Hodjegan, A. Scientific considerations to move towards biowaiver for biopharmaceutical classification system class III drugs: How modeling and simulation can help. Biopharm. Drug Dispos. 2021, 42, 118–127. [Google Scholar] [CrossRef]
- Shebley, M.; Sandhu, P.; Emami Riedmaier, A.; Jamei, M.; Narayanan, R.; Patel, A.; Peters, S.A.; Reddy, V.P.; Zheng, M.; de Zwart, L.; et al. Physiologically Based Pharmacokinetic Model Qualification and Reporting Procedures for Regulatory Submissions: A Consortium Perspective. Clin. Pharmacol. Ther. 2018, 104, 88–110. [Google Scholar] [CrossRef]
- Parrott, N.; Suarez-Sharp, S.; Kesisoglou, F.; Pathak, S.M.; Good, D.; Wagner, C.; Dallmann, A.; Mullin, J.; Patel, N.; Riedmaier, A.E.; et al. Best Practices in the Development and Validation of Physiologically Based Biopharmaceutics Modeling. A Workshop Summary Report. J. Pharm. Sci. 2021, 110, 584–593. [Google Scholar] [CrossRef]
- EMA. Guideline on the Pharmacokinetic and Clinical Evaluation of Modified Release Dosage Forms, in EMA/CHMP/EWP/280/96 Rev1; CHMP EMA, Ed.; EMA (European Medicines Agency): Amsterdam, The Netherlands, 2014. [Google Scholar]
- EMA. Guideline on the Reporting of Physiologically based Pharmacokinetic (PBPK) Modelling and Simulation, in EMA/CHMP/458101/2016; CHMP EMA, Ed.; EMA (European Medicines Agency): Amsterdam, The Netherlands, 2018. [Google Scholar]
- U.S. Food and Drug Administration. Guidance for Industry: Physiologically based Pharmacokinetic Analyses—Format and Content Guidance for Industry. 2018. Available online: https://www.fda.gov/media/101469/download (accessed on 15 December 2021).
- Manolis, E.; Rohou, S.; Hemmings, R.; Salmonson, T.; Karlsson, M.; Milligan, P.A. The Role of Modeling and Simulation in Development and Registration of Medicinal Products: Output From the EFPIA/EMA Modeling and Simulation Workshop. CPT Pharmacomet. Syst. Pharmacol. 2013, 2, 1–4. [Google Scholar] [CrossRef]
- FDA. Development of Best Practices in Physiologically Based Pharmacokinetic Modeling to Support Clinical Pharmacology Regulatory Decision-Making. 2019. Available online: https://www.fda.gov/drugs/news-events-human-drugs/development-best-practices-physiologically-based-pharmacokinetic-modeling-support-clinical (accessed on 1 February 2021).
- Kuemmel, C.; Yang, Y.; Zhang, X.; Florian, J.; Zhu, H.; Tegenge, M.; Huang, S.M.; Wang, Y.; Morrison, T.; Zineh, I. Consideration of a Credibility Assessment Framework in Model-Informed Drug Development: Potential Application to Physiologically-Based Pharmacokinetic Modeling and Simulation. CPT Pharmacomet. Syst. Pharmacol. 2020, 9, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Sanaka, M.K.Y.; Nishinakagawa, S.; Mineshita, S. Use of salivary acetaminophen concentration to assess gastric emptying rate of liquids. J. Gastroenterol. 2000, 35, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Magbool, S.P.; Parkman, H.P.; Friedenberg, F.K. Wireless capsule motility: Comparison of the SmartPill GI monitoring system with scintigraphy for measuring whole gut transit. Dig. Dis. Sci. 2009, 54, 2167–2174. [Google Scholar] [CrossRef] [PubMed]
- Pepin, X.J.H. Justification of Drug Product Dissolution Rate and Drug Substance Particle Size Specifications Based on Absorption PBPK Modeling for Lesinurad Immediate Release Tablets. Mol. Pharm. 2016, 13, 3256–3269. [Google Scholar] [CrossRef] [PubMed]
- Anand, O. Clinically Releavnt Dissolution Specifications-an FDA Perspective. 2021. Available online: https://www.apsgb.co.uk/wp-content/uploads/2021/05/Clinically-Relevant-Dissolution-Specifications-an-FDA-Perspective-__Om-Anand.pdf (accessed on 2 April 2022).
- FDA. Report on the State of Pharmaceutical Quality-Assuring Quality Medicines are Available for the American Public; Center for Drug Evaluation and Research Office of Pharmaceutical Quality; FDA (Food and Drug Administration): Silver Spring, MD, USA, 2018.
- Abdus-Samad, J. Drug Product Nomenclature. In Proceedings of the CDER Prescription Drug Labeling Conference; College Park, MD, USA, 4–5 December 2019. Available online: https://www.fda.gov/media/134015/download (accessed on 15 October 2021).
- FDA. CDER Patient-Focused Drug Development. 2021. Available online: https://www.fda.gov/drugs/development-approval-process-drugs/cder-patient-focused-drug-development (accessed on 1 February 2021).
- Peng, D.; Bercu, J.; Subashi, A.K.; Lawrence, X. Yu Patient-Centric Specification: Regulatory & Pharma Industry Progress. Pharm. Eng. 2019. Available online: https://ispe.org/pharmaceutical-engineering/patient-centric-specification-regulatory-industry-progress# (accessed on 2 April 2022).
- Abdou, E.A. Dissolution Chapter 35. In Remington: The Science and Practice of Pharmacy, 20th ed.; Gennaro, A.R., Remington, J.P., Eds.; Lippincott Williams: Philadelphia, PA, USA, 2000. [Google Scholar]
- U.S. Food and Drug Administration. Guidance for Industry: Extended Release Oral Dosage Forms: Development, Evaluation, and Application of In Vitro/In Vivo Correlations. 1997. Available online: https://www.fda.gov/media/70939/download (accessed on 15 December 2021).
- U.S. Food and Drug Administration. Guidance for Industry: Waiver of In Vivo Bioavailability and Bioequivalence Studies for Immediate Release Solid Oral Dosage Forms based on A Biopharmaceutics Classification System. 2017. Available online: https://www.gmp-compliance.org/files/guidemgr/UCM070246.pdf (accessed on 15 December 2021).
- U.S. Food and Drug Administration. Guidance for Industry: Dissolution Testing and Acceptance Criteria for Immediate-Release Solid Oral Dosage Form Drug Products Containing High Solubility Drug Substances-Guidance for Industry. 2018. Available online: https://www.fda.gov/media/92988/download (accessed on 15 December 2021).
- Zhao, Y.A.S.; Sharp, S.S. FDA expectations in building a safe space to gain regulatory flexibility based on PBBM. In Proceedings of the Current State and Future Expectations of Translational Modeling Strategies to Support Drug Product Development, Manufacturing Changes and Controls Workshop, College Park, MD, USA, 25 September 2019; Available online: https://cersi.umd.edu/sites/cersi.umd.edu/files/Day%203-1%20Zhao%20Suarez%20LM.pdf (accessed on 15 December 2021).
- Vinarov, Z.A.M.; Agundez, J.A.G. Impact of gastrointestinal tract variability on oral drug absorption and pharmacokinetics: An UNGAP review. Eur. J. Pharm. Sci. 2021, 162, 105812. [Google Scholar] [CrossRef]
- Loisios-Konstantinidis, I.; Hens, B.; Mitra, A.; Kim, S.; Chiann, C.; Cristofoletti, R. Using Physiologically Based Pharmacokinetic Modeling to Assess the Risks of Failing Bioequivalence Criteria: A Tale of Two Ibuprofen Products. AAPS J. 2020, 22, 113. [Google Scholar] [CrossRef]
- Bego, M.; Patel, N.; Cristofoletti, R.; Rostami-Hodjegan, A. Proof of Concept in Assignment of Within-Subject Variability During Virtual Bioequivalence Studies: Propagation of Intra-Subject Variation in Gastrointestinal Physiology Using Physiologically Based Pharmacokinetic Modeling. AAPS J. 2021, 24, 21. [Google Scholar] [CrossRef]
- Stamatopoulos, K.; Pathak, S.M.; Marciani, L.; Turner, D.B. A population-based PBPK model for prediction of time-variant bile salt disposition within GI luminal fluids. Mol. Pharm. 2020, 17, 1310–1323. [Google Scholar] [CrossRef]
- Frank, K.J.; Locher, K.; Zecevic, D.E. In vivo predictive mini-scale dissolution for weak bases: Advantages of pH-shift in combination with an absorptive compartment. Eur. J. Pharm. Sci. 2014, 61, 9. [Google Scholar] [CrossRef]
- Purohit, H.S.; Trasi, N.S.; Sun, D.D.; Chow, E.C.Y.; Wen, H.; Zhang, X.; Gao, Y.; Taylor, L.S. Investigating the Impact of Drug Crystallinity in Amorphous Tacrolimus Capsules on Pharmacokinetics and Bioequivalence Using Discriminatory In Vitro Dissolution Testing and Physiologically Based Pharmacokinetic Modeling and Simulation. J. Pharm. Sci. 2018, 107, 1330–1341. [Google Scholar] [CrossRef]
- Mudie, D.M.; Murray, K.; Hoad, C.L.; Pritchard, S.E.; Garnett, M.C.; Amidon, G.L.; Gowland, P.A.; Spiller, R.C.; Amidon, G.E.; Marciani, L. Quantification of gastrointestinal liquid volumes and distribution following a 240 mL dose of water in the fasted state. Mol. Pharm. 2014, 11, 3039–3047. [Google Scholar] [CrossRef]
- Pathak, S.M.; Ruff, A.; Kostewicz, E.S.; Patel, N.; Turner, D.B.; Jamei, M. Model-Based Analysis of Biopharmaceutic Experiments to Improve Mechanistic Oral Absorption Modeling: An Integrated in Vitro in Vivo Extrapolation Perspective Using Ketoconazole as a Model Drug. Mol. Pharm. 2017, 14, 4305–4320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cristofoletti, R.; Hens, B.; Patel, N.; Esteban, V.V.; Schmidt, S.; Dressman, J. Integrating Drug- and Formulation-Related Properties with Gastrointestinal Tract Variability Using a Product-Specific Particle Size Approach: Case Example Ibuprofen. J. Pharm. Sci. 2019, 108, 3842–3847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loisios-Konstantinidis, I.; Cristofoletti, R.; Jamei, M.; Turner, D.; Dressman, J. Physiologically Based Pharmacokinetic/Pharmacodynamic Modeling to Predict the Impact of CYP2C9 Genetic Polymorphisms, Co-Medication and Formulation on the Pharmacokinetics and Pharmacodynamics of Flurbiprofen. Pharmaceutics 2020, 12, 1049. [Google Scholar] [CrossRef] [PubMed]
- Pepin, X.; Goetschy, M.; Abrahmsen-Alami, S. Mechanistic Models for USP2 Dissolution Apparatus, Including Fluid Hydrodynamics and Sedimentation. J. Pharm. Sci. 2022, 111, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Siepmann, J.; Peppas, N.A. Mathematical modeling of controlled drug delivery. Adv. Drug Deliv. Rev. 2001, 48, 137–138. [Google Scholar] [CrossRef]
- Siepmann, J.; Peppas, N.A. Modeling of drug release from delivery systems based on hydroxypropyl methylcellulose (HPMC). Adv. Drug Deliv. Rev. 2001, 48, 139–157. [Google Scholar] [CrossRef]
- Lu, A.T.; Frisella, M.E.; Johnson, K.C. Dissolution modeling: Factors affecting the dissolution rates of polydisperse powders. Pharm. Res. 1993, 10, 1308–1314. [Google Scholar] [CrossRef]
- Takano, R.; Sugano, K.; Higashida, A.; Hayashi, Y.; Machida, M.; Aso, Y.; Yamashita, S. Oral absorption of poorly water-soluble drugs: Computer simulation of fraction absorbed in humans from a miniscale dissolution test. Pharm. Res. 2006, 23, 1144–1156. [Google Scholar] [CrossRef]
- Pepin, X.J.H.; Sanderson, N.J.; Blanazs, A.; Grover, S.; Ingallinera, T.G.; Mann, J.C. Bridging in vitro dissolution and in vivo exposure for acalabrutinib. Part, I. Mechanistic modelling of drug product dissolution to derive a P-PSD for PBPK model input. Eur. J. Pharm. Biopharm. 2019, 142, 421–434. [Google Scholar] [CrossRef]
- Pepin, X.J.H.; Andrea, J.M.; Mann, J.C.; Sanderson, N.J.; Barker, R.; Meehan, E.; Plumb, A.P.; Bailey, G.R.; Murphy, D.S.; Krejsa, C.M.; et al. Bridging in vitro dissolution and in vivo exposure for acalabrutinib. Part II. A mechanistic PBPK model for IR formulation comparison, proton pump inhibitor drug interactions, and administration with acidic juices. Eur. J. Pharm. Biopharm. 2019, 142, 435–448. [Google Scholar] [CrossRef]
- Mitra, A.; Petek, B.; Bajc, A.; Velagapudi, R.; Legen, I. Physiologically based absorption modeling to predict bioequivalence of controlled release and immediate release oral products. Eur. J. Pharm. Biopharm. 2019, 134, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wen, H.; Fan, J.; Vince, B.; Li, T.; Gao, W.; Kinjo, M.; Brown, J.; Sun, W.; Jiang, W.; et al. Integrating In Vitro, Modeling, and In Vivo Approaches to Investigate Warfarin Bioequivalence. CPT Pharm. Syst. Pharmacol. 2017, 6, 523–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stillhart, C.; Pepin, X.; Tistaert, C.; Good, D.; Van Den Bergh, A.; Parrott, N.; Kesisoglou, F. PBPK Absorption Modeling: Establishing the In Vitro-In Vivo Link-Industry Perspective. AAPS J. 2019, 21, 19. [Google Scholar] [CrossRef]
- Kesisoglou, F.; Xia, B.; Agrawal, N.G.B. Comparison of Deconvolution-Based and Absorption Modeling IVIVC for Extended Release Formulations of a BCS III Drug Development Candidate. AAPS J. 2015, 17, 1492–1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodacre, B.C.; Murray, P.J. A mathematical model of drug absorption. J. Clin. Hosp. Pharm. 1981, 6, 117–133. [Google Scholar] [CrossRef]
- Dressman, J.B.; Fleisher, D.; Amidon, G.L. Physicochemical model for dose-dependent drug absorption. J. Pharm. Sci. 1984, 73, 1274–1279. [Google Scholar] [CrossRef]
- Dressman, J.B.; Fleisher, D. Mixing-tank model for predicting dissolution rate control or oral absorption. J. Pharm. Sci. 1986, 75, 109–116. [Google Scholar] [CrossRef]
- Vertzoni, M.; Sulaiman, S.; Goumas, K.; Kersten, E.; Anlahr, J.; Muenster, U.; Reppas, C. Characteristics of Contents of Lower intestine in the 65–74 Years of Age Range Could Impact the Performance of Safe and Efficacious Modified Release Products. J. Pharm. Sci. 2021, 110, 251–258. [Google Scholar] [CrossRef]
- Couto, N.; Al-Majdoub, Z.M.; Gibson, S.; Davies, P.J.; Achour, B.; Harwood, M.D.; Carlson, G.; Barber, J.; Rostami-Hodjegan, A.; Warhurst, G. Quantitative Proteomics of Clinically Relevant Drug-Metabolizing Enzymes and Drug Transporters and Their Intercorrelations in the Human Small Intestine. Drug Metab. Dispos. 2020, 48, 245–254. [Google Scholar] [CrossRef] [Green Version]
- Hens, B.; Pathak, S.M.; Mitra, A.; Patel, N.; Liu, B.; Patel, S.; Jamei, M.; Brouwers, J.; Augustijns, P.; Turner, D.B. In Silico Modeling Approach for the Evaluation of Gastrointestinal Dissolution, Supersaturation, and Precipitation of Posaconazole. Mol. Pharm. 2017, 14, 4321–4333. [Google Scholar] [CrossRef]
- Achour, B.; Al-Majdoub, Z.M.; Grybos-Gajniak, A.; Lea, K.; Kilford, P.; Zhang, M.; Knight, D.; Barber, J.; Schageman, J.; Rostami-Hodjegan, A. Liquid Biopsy Enables Quantification of the Abundance and Interindividual Variability of Hepatic Enzymes and Transporters. Clin. Pharmacol. Ther. 2021, 109, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.G.; Aarons, L.; Augustijns, P.; Brouwers, J.; Darwich, A.S.; De Waal, T.; Garbacz, G.; Hansmann, S.; Hoce, D.; Ivanovag, A.; et al. Integration of advanced methods and models to study drug absorption and related processes: An UNGAP perspective. Eur. J. Pharm. Sci. 2022, 172, 106100. [Google Scholar] [CrossRef] [PubMed]
- Rostami-Hodjegan, A. Reverse Translation in PBPK and QSP: Going Backwards in Order to Go Forward with Confidence. Clin. Pharmacol. Ther. 2018, 103, 224–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doki, K.; Darwich, A.S.; Patel, N.; Rostami-Hodjegan, A. Virtual bioequivalence for achlorhydric subjects: The use of PBPK modelling to assess the formulation-dependent effect of achlorhydria. Eur. J. Pharm. Sci. 2017, 109, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Olivares-Morales, A.; Ghosh, A.; Aarons, L.; Rostami-Hodjegan, A. Development of a Novel Simplified PBPK Absorption Model to Explain the Higher Relative Bioavailability of the OROS(R) Formulation of Oxybutynin. AAPS J. 2016, 18, 1532–1549. [Google Scholar] [CrossRef] [Green Version]
- Tsakalozou, E.; Babiskin, A.; Zhao, L. Physiologically-based pharmacokinetic modeling to support bioequivalence and approval of generic products: A case for diclofenac sodium topical gel, 1. CPT Pharmacomet. Syst. Pharm. 2021, 10, 399–411. [Google Scholar] [CrossRef]
- Wang, K.; Parrott, N.; Olivares-Morales, A.; Dudal, S.; Singer, T.; Lave, T.; Ribba, B. Real-World Data and Physiologically-Based Mechanistic Models for Precision Medicine. Clin. Pharmacol. Ther. 2020, 107, 694–696. [Google Scholar] [CrossRef]
- Lesko, L.J.; Fang, L.; Schmidt, S.; Trame, M.N. An Integrated Bioinformatics and Quantitative Modeling Approach to Investigate Potential Claims of Oral Generic Drug Product Bioinequivalence: Introduction. J. Clin. Pharmacol. 2019, 59, 1245–1248. [Google Scholar] [CrossRef]
- Melillo, M.; Aarons, L.; Magni, P.; Darwich, A.S. Variance based global sensitivity analysis of physiologically based pharmacokinetic absorption models for BCS I–IV drugs. J. Pharmacokinet. Pharmacodyn. 2019, 46, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Melillo, N.; Darwich, A.S. A latent variable approach to account for correlated inputs in global sensitivity analysis. J. Pharmacokinet. Pharmacodyn. 2021, 48, 671–686. [Google Scholar] [CrossRef]
- Bennett-Lenane, H.; O’Shea, J.P.; O’Driscoll, C.M.; Griffin, B.T. A Retrospective Biopharmaceutical Analysis of >800 Approved Oral Drug Products: Are Drug Properties of Solid Dispersions and Lipid-Based Formulations Distinctive? J. Pharm. Sci. 2020, 109, 3248–3261. [Google Scholar] [CrossRef] [PubMed]
- Kuentz, M.; Holm, R.; Kronseder, C.; Saal, C.; Griffin, B.T. Rational Selection of Bio-Enabling Oral Drug Formulations—A PEARRL Commentary. J. Pharm. Sci. 2021, 110, 1921–1930. [Google Scholar] [CrossRef] [PubMed]
- Henze, L.J.; Koehl, N.J.; O’Shea, J.P.; Kostewicz, E.S.; Holm, R.; Griffin, B.T. The pig as a preclinical model for predicting oral bioavailability and in vivo performance of pharmaceutical oral dosage forms: A PEARRL review. J. Pharm. Pharmacol. 2019, 71, 581–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musther, H.; Olivares-Morales, A.; Hatley, O.J.D.; Liu, B.; Rostami Hodjegan, A. Animal versus human oral drug bioavailability: Do they correlate? Eur. J. Pharm. Sci. 2014, 57, 280–291. [Google Scholar] [CrossRef]
- Henze, L.J.; Koehl, N.J.; Bennett-Lenane, H.; Holm, R.; Grimm, M.; Schneider, F.; Weitschies, W.; Koziolek, M.; Griffin, B.T. Characterization of gastrointestinal transit and luminal conditions in pigs using a telemetric motility capsule. Eur. J. Pharm. Sci. 2021, 156, 105627. [Google Scholar] [CrossRef]
- Henze, L.J.; Koehl, N.J.; Jansen, R.; Holm, R.; Vertzoni, M.; Whitfield, P.D.; Griffin, B.T. Development and evaluation of a biorelevant medium simulating porcine gastrointestinal fluids. Eur. J. Pharm. Biopharm. 2020, 154, 116–126. [Google Scholar] [CrossRef]
- Henze, L.J.; Koehl, N.J.; O’Shea, J.P.; Holm, R.; Vertzoni, M.; Griffin, B.T. Combining species specific in vitro & in silico models to predict in vivo food effect in a preclinical stage–case study of Venetoclax. Eur. J. Pharm. Sci. 2021, 162, 105840. [Google Scholar]
- Kalantzi, L.; Goumas, K.; Kalioras, V.; Abrahamsson, B.; Dressman, J.B.; Reppas, C. Characterization of the human upper gastrointestinal contents under conditions simulating bioavailability/bioequivalence studies. Pharm. Res. 2006, 23, 165–176. [Google Scholar] [CrossRef]
- Reppas, C.; Karatza, E.; Goumas, C.; Markopoulos, C.; Vertzoni, M. Characterization of contents of distal ileum and cecum to which drugs/drug products are exposed during bioavailability/bioequivalence studies in healthy adults. Pharm. Res. 2015, 32, 3338–3349. [Google Scholar] [CrossRef]
- Diakidou, A.; Vertzoni, M.; Goumas, K.; Soderlind, E.; Abrahamsson, B.; Dressman, J.; Reppas, C. Characterization of the contents of ascending colon to which drugs are exposed after oral administration to healthy adults. Pharm. Res. 2009, 26, 2141–2151. [Google Scholar] [CrossRef]
- Pentafragka, C.S.M.; McAllister, M.; Dressman, J.; Vertzoni, M.; Reppas, C. The impact of food intake on the luminal environment and performance of oral drug products with a view to in vitro and in silico simulations: A PEARRL review. J. Pharm. Pharmacol. 2019, 71, 557–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dressman, J.B.; Amidon, G.L.; Reppas, C.; Shah, V.P. Dissolution testing as a prognostic tool for oral drug absorption: Immediate release dosage forms. Pharm. Res. 1998, 15, 11–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jantratid, E.; Janssen, N.; Reppas, C.; Dressman, J.B. Dissolution Media Simulating Conditions in the Proximal Human Gastrointestinal Tract: An Update. Pharm. Res. 2008, 25, 1663–1676. [Google Scholar] [CrossRef] [PubMed]
- Andreas, C.J.; Tomaszewska, I.; Muenster, U.; van der Mey, D.; Mueck, W. Can dosage form-dependent food effects be predicted using biorelevant dissolution tests? Case example extended release nifedipine. Eur. J. Pharm. Biopharm. 2016, 105, 193–202. [Google Scholar] [CrossRef]
- Litou, C.P.N.; Turner, D.B.; Kostewicz, E.; Kuentz, M.; Box, K.J.; Dressman, J. Combining biorelevant in vitro and in silico tools to simulate and better understand the in vivo performance of a nano-sized formulation of aprepitant in the fasted and fed states. Eur. J. Pharm. Sci. 2019, 138, 105031. [Google Scholar] [CrossRef] [Green Version]
- Paraiso, R.L.M.; Watanabe, A.; Andreas, C.J.; Turner, D.; Zane, P.; Dressman, J. In-vitro–in-silico investigation of the negative food effect of zolpidem when administered as immediate-release tablets. J. Pharm. Pharmacol. 2019, 71, 1663–1676. [Google Scholar] [CrossRef]
- Segregur, D.; Flanagan, T.; Mann, J.; Moir, A.; Karlsson, E.M.; Hoch, M.; Carlile, D. Impact of acid-reducing agents on gastrointestinal physiology and design of biorelevant dissolution tests to reflect these changes. J. Pharm. Sci. 2019, 108, 3461–3477. [Google Scholar] [CrossRef]
- Segregur, D.M.J.; Moir, A.; Karlsson, E.M.; Dressman, J. Prediction of plasma profiles of a weakly basic drug after administration of omeprazole using PBPK modelling. Eur. J. Pharm. Sci. 2020, 158, 105656. [Google Scholar] [CrossRef]
- Segregur, D.; Barker, R.; Mann, J.; Moir, A.; Karlsson, E.M.; Turner, D.B.; Arora, S. Evaluating the impact of acid-reducing agents on drug absorption using biorelevant in vitro tools and PBPK modeling-case example dipyridamole. Eur. J. Pharm. Sci. 2021, 160, 105750. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Strategy and Data Input | Design of Studies (Clinical or Preclinical) | Data Utilisation/Regulatory Impact |
|---|---|---|
| Understand/define which CQAs/CBAs can be explored and/or mitigated using PBBM | Understand/define which CQAs/CBAs ones to test in clinical studies (relBA or BE?) | Understanding CQAs/CBAs is critical to putting together a well-designed biopharmaceutical risk assessment |
A biorelevant method is not necessarily clinically relevant until a link to in vivo performance has been shown.
| In vivo clinical study setup to claim a dissolution safe space
| Use of the terms biorelevant/clinically relevant; is the terminology consistent yet? If and how are biorelevant dissolution methods used in the development space and how can this information be shared at the IMPD or market application stage to enable efficient life cycle management? |
| Ways of including dissolution data into PBBM models: Z factor or API PSD/P-PSD and success rates of both? Will the input depend on the question? Use of PBDT data or QC data in PBBM models—should we approach this depending on BCS/DCS?
| Utilisation of totality of clinical relBA/BE type studies in the model verification step, together with CBA/CQA specifically designed studies Justifying studies in companies that share accountability for product quality across divisions is easier than for companies where development and manufacturing are disconnected—would industry agree? | Interaction with agencies to discuss compound strategy advocated. Experience is time to submit/receive feedback is quite lengthy. Can we look at how to improve this to encourage more interactions? Industry examples demonstrate that CRDS/PBBM modelling approaches are being used with some success. |
| Model should be fit for purpose/build to address the question. Are we clear on the level of model validation expectations?
| In which circumstances can we use the models in place of clinical trials, or will the models only be accepted for specification setting and post-approval changes? | Guidance on model setup vs. model verification/validation essential Differences in global regulatory acceptance of mechanistic absorption models in specification setting, IVIVC acceptance compared with IVIVR acceptance; agencies continue to learn together with industry |
Understand sources of variability
| Mechanistic Modelling is a key area of growth. | Reduce the number of animal experiments when it is clear that the best model for humans is human; learn more about drug–drug interactions at the level of absorption (i.e., PPIs), as this can impact labelling/dosing times. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McAllister, M.; Flanagan, T.; Cole, S.; Abend, A.; Kotzagiorgis, E.; Limberg, J.; Mead, H.; Mangas-Sanjuan, V.; Dickinson, P.A.; Moir, A.; et al. Developing Clinically Relevant Dissolution Specifications (CRDSs) for Oral Drug Products: Virtual Webinar Series. Pharmaceutics 2022, 14, 1010. https://doi.org/10.3390/pharmaceutics14051010
McAllister M, Flanagan T, Cole S, Abend A, Kotzagiorgis E, Limberg J, Mead H, Mangas-Sanjuan V, Dickinson PA, Moir A, et al. Developing Clinically Relevant Dissolution Specifications (CRDSs) for Oral Drug Products: Virtual Webinar Series. Pharmaceutics. 2022; 14(5):1010. https://doi.org/10.3390/pharmaceutics14051010
Chicago/Turabian StyleMcAllister, Mark, Talia Flanagan, Susan Cole, Andreas Abend, Evangelos Kotzagiorgis, Jobst Limberg, Heather Mead, Victor Mangas-Sanjuan, Paul A. Dickinson, Andrea Moir, and et al. 2022. "Developing Clinically Relevant Dissolution Specifications (CRDSs) for Oral Drug Products: Virtual Webinar Series" Pharmaceutics 14, no. 5: 1010. https://doi.org/10.3390/pharmaceutics14051010
APA StyleMcAllister, M., Flanagan, T., Cole, S., Abend, A., Kotzagiorgis, E., Limberg, J., Mead, H., Mangas-Sanjuan, V., Dickinson, P. A., Moir, A., Pepin, X., Zhou, D., Tistaert, C., Dokoumetzidis, A., Anand, O., Le Merdy, M., Turner, D. B., Griffin, B. T., Darwich, A., ... Mackie, C. (2022). Developing Clinically Relevant Dissolution Specifications (CRDSs) for Oral Drug Products: Virtual Webinar Series. Pharmaceutics, 14(5), 1010. https://doi.org/10.3390/pharmaceutics14051010