
Therapeutic Vaccines Targeting Neoantigens to Induce T-Cell Immunity against Cancers



Abstract
:1. Introduction
2. Types of Cancer Antigens
3. Neoantigen-Induced Antitumor Immunity
4. Neoantigen Identification
4.1. Discovery of the Mutanome by Next-Generation Sequencing
4.2. HLA-Epitope Calling by Computational Algorithms
4.3. Identification of HLA Epitopes by Mass Spectrometry (MS)
4.4. Prediction of HLA Epitopes by Machine Learning Algorithms
5. Neoantigen-Derived Cancer Vaccines
5.1. Tumor Lysates and Allogeneic Tumor-Cell-Based Vaccine
5.2. DNA-Based Vaccines
5.3. mRNA-Based Vaccines
5.4. Protein and Peptide Vaccines
5.5. DC-Based Vaccines
6. Opinions and Future Perspectives
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [CrossRef] [PubMed] [Green Version]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Wood, L.D.; Parsons, D.W.; Jones, S.; Lin, J.; Sjoblom, T.; Leary, R.J.; Shen, D.; Boca, S.M.; Barber, T.; Ptak, J.; et al. The genomic landscapes of human breast and colorectal cancers. Science 2007, 318, 1108–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciriello, G.; Miller, M.L.; Aksoy, B.A.; Senbabaoglu, Y.; Schultz, N.; Sander, C. Emerging landscape of oncogenic signatures across human cancers. Nat. Genet. 2013, 45, 1127–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, E.; Robbins, P.F.; Rosenberg, S.A. ‘Final common pathway’ of human cancer immunotherapy: Targeting random somatic mutations. Nat. Immunol. 2017, 18, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, R.E.; Jansen, K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines 2019, 4, 7. [Google Scholar] [CrossRef]
- Coulie, P.G.; Van den Eynde, B.J.; van der Bruggen, P.; Boon, T. Tumour antigens recognized by T lymphocytes: At the core of cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 135–146. [Google Scholar] [CrossRef]
- Van den Eynde, B.J.; van der Bruggen, P. T cell defined tumor antigens. Curr. Opin. Immunol. 1997, 9, 684–693. [Google Scholar] [CrossRef]
- Oesterling, J.E. Prostate specific antigen: A critical assessment of the most useful tumor marker for adenocarcinoma of the prostate. J. Urol. 1991, 145, 907–923. [Google Scholar] [CrossRef]
- Hollingsworth, M.A.; Swanson, B.J. Mucins in cancer: Protection and control of the cell surface. Nat. Rev. Cancer 2004, 4, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Vonderheide, R.H.; Hahn, W.C.; Schultze, J.L.; Nadler, L.M. The telomerase catalytic subunit is a widely expressed tumor-associated antigen recognized by cytotoxic T lymphocytes. Immunity 1999, 10, 673–679. [Google Scholar] [CrossRef] [Green Version]
- Chang, K.; Pastan, I. Molecular cloning of mesothelin, a differentiation antigen present on mesothelium, mesotheliomas, and ovarian cancers. Proc. Natl. Acad. Sci. USA 1996, 93, 136–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finn, O.J.; Gantt, K.R.; Lepisto, A.J.; Pejawar-Gaddy, S.; Xue, J.; Beatty, P.L. Importance of MUC1 and spontaneous mouse tumor models for understanding the immunobiology of human adenocarcinomas. Immunol. Res. 2011, 50, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Correale, P.; Walmsley, K.; Nieroda, C.; Zaremba, S.; Zhu, M.; Schlom, J.; Tsang, K.Y. In vitro generation of human cytotoxic T lymphocytes specific for peptides derived from prostate-specific antigen. J. Natl. Cancer Inst. 1997, 89, 293–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muniyan, S.; Chaturvedi, N.K.; Dwyer, J.G.; LaGrange, C.A.; Chaney, W.G.; Lin, M.-F. Human Prostatic Acid Phosphatase: Structure, Function and Regulation. Int. J. Mol. Sci. 2013, 14, 10438–10464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karbach, J.; Neumann, A.; Atmaca, A.; Wahle, C.; Brand, K.; von Boehmer, L.; Knuth, A.; Bender, A.; Ritter, G.; Old, L.J.; et al. Efficient in vivo priming by vaccination with recombinant NY-ESO-1 protein and CpG in antigen naive prostate cancer patients. Clin. Cancer Res. 2011, 17, 861–870. [Google Scholar] [CrossRef] [Green Version]
- Simpson, A.J.; Caballero, O.L.; Jungbluth, A.; Chen, Y.T.; Old, L.J. Cancer/testis antigens, gametogenesis and cancer. Nat. Rev. Cancer 2005, 5, 615–625. [Google Scholar] [CrossRef]
- Hofmann, O.; Caballero, O.L.; Stevenson, B.J.; Chen, Y.T.; Cohen, T.; Chua, R.; Maher, C.A.; Panji, S.; Schaefer, U.; Kruger, A.; et al. Genome-wide analysis of cancer/testis gene expression. Proc. Natl. Acad. Sci. USA 2008, 105, 20422–20427. [Google Scholar] [CrossRef] [Green Version]
- De Smet, C.; Lurquin, C.; van der Bruggen, P.; De Plaen, E.; Brasseur, F.; Boon, T. Sequence and expression pattern of the human MAGE2 gene. Immunogenetics 1994, 39, 121–129. [Google Scholar] [CrossRef]
- Gnjatic, S.; Cao, Y.; Reichelt, U.; Yekebas, E.F.; Nölker, C.; Marx, A.H.; Erbersdobler, A.; Nishikawa, H.; Hildebrandt, Y.; Bartels, K.; et al. NY-CO-58/KIF2C is overexpressed in a variety of solid tumors and induces frequent T cell responses in patients with colorectal cancer. Int. J. Cancer 2010, 127, 381–393. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.; Selitsky, S.R.; Chai, S.; Armistead, P.M.; Vincent, B.G.; Serody, J.S. Alternative tumour-specific antigens. Nat. Rev. Cancer 2019, 19, 465–478. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, E.F.; Rajasagi, M.; Ott, P.A.; Brusic, V.; Hacohen, N.; Wu, C.J. HLA-binding properties of tumor neoepitopes in humans. Cancer Immunol. Res. 2014, 2, 522–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, F.; Duitama, J.; Al Seesi, S.; Ayres, C.M.; Corcelli, S.A.; Pawashe, A.P.; Blanchard, T.; McMahon, D.; Sidney, J.; Sette, A.; et al. Genomic and bioinformatic profiling of mutational neoepitopes reveals new rules to predict anticancer immunogenicity. J. Exp. Med. 2014, 211, 2231–2248. [Google Scholar] [CrossRef] [PubMed]
- Milicic, A.; Price, D.A.; Zimbwa, P.; Booth, B.L.; Brown, H.L.; Easterbrook, P.J.; Olsen, K.; Robinson, N.; Gileadi, U.; Sewell, A.K.; et al. CD8+ T cell epitope-flanking mutations disrupt proteasomal processing of HIV-1 Nef. J. Immunol. 2005, 175, 4618–4626. [Google Scholar] [CrossRef] [Green Version]
- Wolfers, J.; Lozier, A.; Raposo, G.; Regnault, A.; Thery, C.; Masurier, C.; Flament, C.; Pouzieux, S.; Faure, F.; Tursz, T.; et al. Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat. Med. 2001, 7, 297–303. [Google Scholar] [CrossRef]
- Zitvogel, L.; Casares, N.; Pequignot, M.O.; Chaput, N.; Albert, M.L.; Kroemer, G. Immune response against dying tumor cells. Adv. Immunol. 2004, 84, 131–179. [Google Scholar] [CrossRef]
- Green, D.R.; Ferguson, T.; Zitvogel, L.; Kroemer, G. Immunogenic and tolerogenic cell death. Nat. Rev. Immunol. 2009, 9, 353–363. [Google Scholar] [CrossRef]
- Boon, T.; Cerottini, J.C.; Van den Eynde, B.; van der Bruggen, P.; Van Pel, A. Tumor antigens recognized by T lymphocytes. Annu. Rev. Immunol. 1994, 12, 337–365. [Google Scholar] [CrossRef]
- Bacon, K.; Baggiolini, M.; Broxmeyer, H.; Horuk, R.; Lindley, I.; Mantovani, A.; Maysushima, K.; Murphy, P.; Nomiyama, H.; Oppenheim, J.; et al. Chemokine/chemokine receptor nomenclature. J. Interferon Cytokine Res. 2002, 22, 1067–1068. [Google Scholar] [CrossRef]
- Dubinett, S.M.; Lee, J.M.; Sharma, S.; Mule, J.J. Chemokines: Can effector cells be redirected to the site of the tumor? Cancer J. 2010, 16, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Tureci, O.; Vormehr, M.; Diken, M.; Kreiter, S.; Huber, C.; Sahin, U. Targeting the Heterogeneity of Cancer with Individualized Neoepitope Vaccines. Clin. Cancer Res. 2016, 22, 1885–1896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahles, A.; Ong, C.S.; Zhong, Y.; Ratsch, G. SplAdder: Identification, quantification and testing of alternative splicing events from RNA-Seq data. Bioinformatics 2016, 32, 1840–1847. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.F.; Thomas, J.; Reddy, A.S.; Ben-Hur, A. SpliceGrapher: Detecting patterns of alternative splicing from RNA-Seq data in the context of gene models and EST data. Genome Biol. 2012, 13, R4. [Google Scholar] [CrossRef] [Green Version]
- Denti, L.; Rizzi, R.; Beretta, S.; Vedova, G.D.; Previtali, M.; Bonizzoni, P. ASGAL: Aligning RNA-Seq data to a splicing graph to detect novel alternative splicing events. BMC Bioinform. 2018, 19, 444. [Google Scholar] [CrossRef]
- Ruggles, K.V.; Tang, Z.; Wang, X.; Grover, H.; Askenazi, M.; Teubl, J.; Cao, S.; McLellan, M.D.; Clauser, K.R.; Tabb, D.L.; et al. An Analysis of the Sensitivity of Proteogenomic Mapping of Somatic Mutations and Novel Splicing Events in Cancer. Mol. Cell. Proteom. 2016, 15, 1060–1071. [Google Scholar] [CrossRef] [Green Version]
- Jurtz, V.; Paul, S. NetMHCpan-4.0: Improved Peptide-MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J. Immunol. 2017, 199, 3360–3368. [Google Scholar] [CrossRef]
- Zhang, G.L.; Khan, A.M.; Srinivasan, K.N.; August, J.T.; Brusic, V. MULTIPRED: A computational system for prediction of promiscuous HLA binding peptides. Nucleic Acids Res. 2005, 33, W172–W179. [Google Scholar] [CrossRef] [Green Version]
- Vita, R.; Overton, J.A.; Greenbaum, J.A.; Ponomarenko, J.; Clark, J.D.; Cantrell, J.R.; Wheeler, D.K.; Gabbard, J.L.; Hix, D.; Sette, A.; et al. The immune epitope database (IEDB) 3.0. Nucleic Acids Res. 2015, 43, D405–D412. [Google Scholar] [CrossRef]
- Schubert, B.; Brachvogel, H.P.; Jurges, C.; Kohlbacher, O. EpiToolKit–A web-based workbench for vaccine design. Bioinformatics 2015, 31, 2211–2213. [Google Scholar] [CrossRef] [Green Version]
- Fridman, W.H.; Pagès, F.; Sautès-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreiter, S.; Vormehr, M.; van de Roemer, N.; Diken, M.; Lower, M.; Diekmann, J.; Boegel, S.; Schrors, B.; Vascotto, F.; Castle, J.C.; et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 2015, 520, 692–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, E.; Turcotte, S.; Gros, A.; Robbins, P.F.; Lu, Y.C.; Dudley, M.E.; Wunderlich, J.R.; Somerville, R.P.; Hogan, K.; Hinrichs, C.S.; et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014, 344, 641–645. [Google Scholar] [CrossRef]
- Nielsen, M.; Lund, O. NN-align. An artificial neural network-based alignment algorithm for MHC class II peptide binding prediction. BMC Bioinform. 2009, 10, 296. [Google Scholar] [CrossRef] [Green Version]
- Andreatta, M.; Karosiene, E.; Rasmussen, M.; Stryhn, A.; Buus, S.; Nielsen, M. Accurate pan-specific prediction of peptide-MHC class II binding affinity with improved binding core identification. Immunogenetics 2015, 67, 641–650. [Google Scholar] [CrossRef] [Green Version]
- Hunt, D.F.; Henderson, R.A.; Shabanowitz, J.; Sakaguchi, K.; Michel, H.; Sevilir, N.; Cox, A.L.; Appella, E.; Engelhard, V.H. Characterization of peptides bound to the class I MHC molecule HLA-A2.1 by mass spectrometry. Science 1992, 255, 1261–1263. [Google Scholar] [CrossRef] [Green Version]
- Cravatt, B.F.; Simon, G.M.; Yates Iii, J.R. The biological impact of mass-spectrometry-based proteomics. Nature 2007, 450, 991–1000. [Google Scholar] [CrossRef]
- Kasuga, K. Comprehensive analysis of MHC ligands in clinical material by immunoaffinity-mass spectrometry. Methods Mol. Biol. 2013, 1023, 203–218. [Google Scholar] [CrossRef]
- Mommen, G.P.M.; Frese, C.K.; Meiring, H.D.; van Gaans-van den Brink, J.; de Jong, A.P.J.M.; van Els, C.A.C.M.; Heck, A.J.R. Expanding the detectable HLA peptide repertoire using electron-transfer/higher-energy collision dissociation (EThcD). Proc. Natl. Acad. Sci. USA 2014, 111, 4507–4512. [Google Scholar] [CrossRef] [Green Version]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Rosenberger, G.; Koh, C.C.; Guo, T.; Röst, H.L.; Kouvonen, P.; Collins, B.C.; Heusel, M.; Liu, Y.; Caron, E.; Vichalkovski, A.; et al. A repository of assays to quantify 10,000 human proteins by SWATH-MS. Sci. Data 2014, 1, 140031. [Google Scholar] [CrossRef] [PubMed]
- Veit, J.; Sachsenberg, T.; Chernev, A.; Aicheler, F.; Urlaub, H.; Kohlbacher, O. LFQProfiler and RNP(xl): Open-Source Tools for Label-Free Quantification and Protein-RNA Cross-Linking Integrated into Proteome Discoverer. J. Proteome Res. 2016, 15, 3441–3448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassani-Sternberg, M.; Bräunlein, E.; Klar, R.; Engleitner, T.; Sinitcyn, P.; Audehm, S.; Straub, M.; Weber, J.; Slotta-Huspenina, J.; Specht, K.; et al. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat. Commun. 2016, 7, 13404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regnier, M.; Gourbeyre, P.; Pinton, P.; Napper, S.; Laffite, J.; Cossalter, A.M.; Bailly, J.D.; Lippi, Y.; Bertrand-Michel, J.; Bracarense, A.; et al. Identification of Signaling Pathways Targeted by the Food Contaminant FB1: Transcriptome and Kinome Analysis of Samples from Pig Liver and Intestine. Mol. Nutr. Food Res. 2017, 61, 1700433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abelin, J.G.; Keskin, D.B.; Sarkizova, S.; Hartigan, C.R.; Zhang, W.; Sidney, J.; Stevens, J.; Lane, W.; Zhang, G.L.; Eisenhaure, T.M.; et al. Mass Spectrometry Profiling of HLA-Associated Peptidomes in Mono-allelic Cells Enables More Accurate Epitope Prediction. Immunity 2017, 46, 315–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, M.; Connelley, T.; Ternette, N. Improved Prediction of Bovine Leucocyte Antigens (BoLA) Presented Ligands by Use of Mass-Spectrometry-Determined Ligand and in Vitro Binding Data. J. Proteome Res. 2018, 17, 559–567. [Google Scholar] [CrossRef] [Green Version]
- Andreatta, M.; Lund, O.; Nielsen, M. Simultaneous alignment and clustering of peptide data using a Gibbs sampling approach. Bioinformatics 2013, 29, 8–14. [Google Scholar] [CrossRef] [Green Version]
- Bulik-Sullivan, B.; Busby, J.; Palmer, C.D.; Davis, M.J.; Murphy, T.; Clark, A.; Busby, M.; Duke, F.; Yang, A.; Young, L.; et al. Deep learning using tumor HLA peptide mass spectrometry datasets improves neoantigen identification. Nat. Biotechnol. 2019, 37, 55–63. [Google Scholar] [CrossRef]
- Caron, E.; Aebersold, R.; Banaei-Esfahani, A.; Chong, C.; Bassani-Sternberg, M. A Case for a Human Immuno-Peptidome Project Consortium. Immunity 2017, 47, 203–208. [Google Scholar] [CrossRef]
- Karosiene, E.; Rasmussen, M.; Blicher, T.; Lund, O.; Buus, S.; Nielsen, M. NetMHCIIpan-3.0, a common pan-specific MHC class II prediction method including all three human MHC class II isotypes, HLA-DR, HLA-DP and HLA-DQ. Immunogenetics 2013, 65, 711–724. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, T.J.; Rubinsteyn, A.; Bonsack, M.; Riemer, A.B.; Laserson, U.; Hammerbacher, J. MHCflurry: Open-Source Class I MHC Binding Affinity Prediction. Cell Syst. 2018, 7, 129–132.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.; Kim, D. Deep convolutional neural networks for pan-specific peptide-MHC class I binding prediction. BMC Bioinform. 2017, 18, 585. [Google Scholar] [CrossRef] [PubMed]
- Alvaro-Benito, M.; Morrison, E.; Abualrous, E.T.; Kuropka, B.; Freund, C. Quantification of HLA-DM-Dependent Major Histocompatibility Complex of Class II Immunopeptidomes by the Peptide Landscape Antigenic Epitope Alignment Utility. Front. Immunol. 2018, 9, 872. [Google Scholar] [CrossRef] [Green Version]
- Bjerregaard, A.M.; Nielsen, M.; Hadrup, S.R.; Szallasi, Z.; Eklund, A.C. MuPeXI: Prediction of neo-epitopes from tumor sequencing data. Cancer Immunol. Immunother. 2017, 66, 1123–1130. [Google Scholar] [CrossRef]
- Schenck, R.O.; Lakatos, E.; Gatenbee, C.; Graham, T.A.; Anderson, A.R.A. NeoPredPipe: High-throughput neoantigen prediction and recognition potential pipeline. BMC Bioinform. 2019, 20, 264. [Google Scholar] [CrossRef] [Green Version]
- Chowell, D.; Krishna, S.; Becker, P.D. TCR contact residue hydrophobicity is a hallmark of immunogenic CD8+ T cell epitopes. Proc. Natl. Acad. Sci. 2015, 112, E1754–E1762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Kim, H.S.; Kim, E.; Lee, M.G.; Shin, E.C.; Paik, S.; Kim, S. Neopepsee: Accurate genome-level prediction of neoantigens by harnessing sequence and amino acid immunogenicity information. Ann. Oncol. 2018, 29, 1030–1036. [Google Scholar] [CrossRef]
- Chan, A.D.; Morton, D.L. Active immunotherapy with allogeneic tumor cell vaccines: Present status. Semin. Oncol. 1998, 25, 611–622. [Google Scholar]
- Simons, J.W.; Mikhak, B. Ex-vivo gene therapy using cytokine-transduced tumor vaccines: Molecular and clinical pharmacology. Semin. Oncol. 1998, 25, 661–676. [Google Scholar]
- Phan, V.; Errington, F.; Cheong, S.C.; Kottke, T.; Gough, M.; Altmann, S.; Brandenburger, A.; Emery, S.; Strome, S.; Bateman, A.; et al. A new genetic method to generate and isolate small, short-lived but highly potent dendritic cell-tumor cell hybrid vaccines. Nat. Med. 2003, 9, 1215–1219. [Google Scholar] [CrossRef] [PubMed]
- Vermorken, J.B.; Claessen, A.M.; van Tinteren, H.; Gall, H.E.; Ezinga, R.; Meijer, S.; Scheper, R.J.; Meijer, C.J.; Bloemena, E.; Ransom, J.H.; et al. Active specific immunotherapy for stage II and stage III human colon cancer: A randomised trial. Lancet 1999, 353, 345–350. [Google Scholar] [CrossRef]
- Arlen, P.M.; Mohebtash, M.; Madan, R.A.; Gulley, J.L. Promising novel immunotherapies and combinations for prostate cancer. Future Oncol. 2009, 5, 187–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, A.A.; Bever, K.M.; Ho, W.J.; Fertig, E.J.; Niu, N.; Zheng, L.; Parkinson, R.M.; Durham, J.N.; Onners, B.; Ferguson, A.K.; et al. A Phase II Study of Allogeneic GM-CSF–Transfected Pancreatic Tumor Vaccine (GVAX) with Ipilimumab as Maintenance Treatment for Metastatic Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 5129–5139. [Google Scholar] [CrossRef] [PubMed]
- Sondak, V.K.; Sosman, J.A. Results of clinical trials with an allogenic melanoma tumor cell lysate vaccine: Melacine. Semin. Cancer Biol. 2003, 13, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Hsueh, E.C.; Morton, D.L. Antigen-based immunotherapy of melanoma: Canvaxin therapeutic polyvalent cancer vaccine. Semin. Cancer Biol. 2003, 13, 401–407. [Google Scholar] [CrossRef]
- Gleisner, M.A.; Pereda, C.; Tittarelli, A. A heat-shocked melanoma cell lysate vaccine enhances tumor infiltration by prototypic effector T cells inhibiting tumor growth. J. Immunother. Cancer 2020, 8, e000999. [Google Scholar] [CrossRef]
- Nayerossadat, N.; Maedeh, T.; Ali, P. Viral and nonviral delivery systems for gene delivery. Adv. Biomed. Res. 2012, 1, 27. [Google Scholar] [CrossRef]
- Xiang, S.D.; Selomulya, C.; Ho, J.; Apostolopoulos, V.; Plebanski, M. Delivery of DNA vaccines: An overview on the use of biodegradable polymeric and magnetic nanoparticles. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2010, 2, 205–218. [Google Scholar] [CrossRef]
- Yarchoan, M.; Gane, E.; Marron, T.; Rochestie, S.; Cooch, N.; Peters, J.; Csiki, I.; Perales-Puchalt, A.; Sardesai, N. 453 Personalized DNA neoantigen vaccine (GNOS-PV02) in combination with plasmid IL-12 and pembrolizumab for the treatment of patients with advanced hepatocellular carcinoma. J. Immunother. Cancer 2021, 9, A481. [Google Scholar] [CrossRef]
- He, T.C.; Zhou, S.; da Costa, L.T.; Yu, J.; Kinzler, K.W.; Vogelstein, B. A simplified system for generating recombinant adenoviruses. Proc. Natl. Acad. Sci. USA 1998, 95, 2509–2514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falsey, A.R.; Sobieszczyk, M.E.; Hirsch, I.; Sproule, S.; Robb, M.L.; Corey, L.; Neuzil, K.M.; Hahn, W.; Hunt, J.; Mulligan, M.J.; et al. Phase 3 Safety and Efficacy of AZD1222 (ChAdOx1 nCoV-19) COVID-19 Vaccine. N. Engl. J. Med. 2021, 385, 2348–2360. [Google Scholar] [CrossRef] [PubMed]
- Tapia, M.D.; Sow, S.O.; Mbaye, K.D.; Thiongane, A.; Ndiaye, B.P.; Ndour, C.T.; Mboup, S.; Keshinro, B.; Kinge, T.N.; Vernet, G.; et al. Safety, reactogenicity, and immunogenicity of a chimpanzee adenovirus vectored Ebola vaccine in children in Africa: A randomised, observer-blind, placebo-controlled, phase 2 trial. Lancet Infect. Dis. 2020, 20, 719–730. [Google Scholar] [CrossRef]
- Shiratsuchi, T.; Rai, U.; Kaneko, I.; Zhang, M.; Iwanaga, S.; Yuda, M.; Tsuji, M. A potent malaria vaccine based on adenovirus with dual modifications at Hexon and pVII. Vaccine 2017, 35, 6990–7000. [Google Scholar] [CrossRef]
- Overman, M.; Fakih, M.; Le, D.; Shields, A.; Pedersen, K.; Shah, M.; Mukherjee, S.; Faivre, T.; Leoni, G.; D’Alise, A.M.; et al. 410 Phase I interim study results of Nous-209, an off-the-shelf immunotherapy, with pembrolizumab, for the treatment of tumors with a deficiency in mismatch repair/microsatellite instability (dMMR/MSI). J. Immunother. Cancer 2021, 9, A441. [Google Scholar] [CrossRef]
- Floudas, C.; Strauss, J.; Allen, C.; Donahue, R.; Jochems, C.; Steinberg, S.; Cordes, L.; Brough, D.; Lankford, A.; McMahon, S.; et al. 483 Initial safety results and immune responses induced by a novel human papillomavirus (HPV)-specific gorilla adenovirus immunotherapy vaccine, PRGN-2009, in patients with advanced HPV-associated cancers. J. Immunother. Cancer 2021, 9, A513. [Google Scholar] [CrossRef]
- Barouch, D.H.; Pau, M.G.; Custers, J.H.; Koudstaal, W.; Kostense, S.; Havenga, M.J.; Truitt, D.M.; Sumida, S.M.; Kishko, M.G.; Arthur, J.C.; et al. Immunogenicity of recombinant adenovirus serotype 35 vaccine in the presence of pre-existing anti-Ad5 immunity. J. Immunol. 2004, 172, 6290–6297. [Google Scholar] [CrossRef]
- Guo, J.; Mondal, M.; Zhou, D. Development of novel vaccine vectors: Chimpanzee adenoviral vectors. Hum. Vaccines Immunother. 2018, 14, 1679–1685. [Google Scholar] [CrossRef] [Green Version]
- Haigentz, M.; Ramalingam, S.S.; Gerstner, G.J.; Halmos, B.; Morganstein, N.; Vangala, S.; Parsi, M.; Kabala, V.; Simkhada, D.; Metran, C.; et al. A phase 1 study of an off-the shelf, multi-neoantigen vector (ADXS-503) in subjects with metastatic non-small cell lung cancer (NSCLC) progressing on pembrolizumab as last therapy. J. Clin. Oncol. 2021, 39, 2616. [Google Scholar] [CrossRef]
- Hecht, J.R.; Goldman, J.W.; Hayes, S.; Balli, D.; Princiotta, M.F.; Dennie, J.G.; Heyburn, J.; Sands, T.; Sheeri, S.; Petit, R.; et al. Abstract CT007: Safety and immunogenicity of a personalized neoantigen—Listeria vaccine in cancer patients. Cancer Res. 2019, 79, CT007. [Google Scholar] [CrossRef]
- Pilishvili, T.; Gierke, R.; Fleming-Dutra, K.E.; Farrar, J.L.; Mohr, N.M.; Talan, D.A.; Krishnadasan, A.; Harland, K.K.; Smithline, H.A.; Hou, P.C.; et al. Effectiveness of mRNA COVID-19 Vaccine among U.S. Health Care Personnel. N. Engl. J. Med. 2021, 385, e90. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.-P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A.; Patel, M.R.; Cho, D.C.; Clarke, J.M.; Gutierrez, M.; Zaks, T.Z.; Frederick, J.; Hopson, K.; Mody, K.; Binanti-Berube, A.; et al. A phase I multicenter study to assess the safety, tolerability, and immunogenicity of mRNA-4157 alone in patients with resected solid tumors and in combination with pembrolizumab in patients with unresectable solid tumors. J. Clin. Oncol. 2019, 37, 2523. [Google Scholar] [CrossRef]
- Bauman, J.; Burris, H.; Clarke, J.; Patel, M.; Cho, D.; Gutierrez, M.; Julian, R.; Scott, A.; Cohen, P.; Frederick, J.; et al. 798 Safety, tolerability, and immunogenicity of mRNA-4157 in combination with pembrolizumab in subjects with unresectable solid tumors (KEYNOTE-603): An update. J. Immunother. Cancer 2020, 8, A477. [Google Scholar] [CrossRef]
- Cafri, G.; Gartner, J.J.; Zaks, T.; Hopson, K.; Levin, N.; Paria, B.C.; Parkhurst, M.R.; Yossef, R.; Lowery, F.J.; Jafferji, M.S.; et al. mRNA vaccine-induced neoantigen-specific T cell immunity in patients with gastrointestinal cancer. J. Clin. Inverstig. 2020, 130, 5976–5988. [Google Scholar] [CrossRef]
- Sahin, U.; Karikó, K.; Türeci, Ö. mRNA-based therapeutics—Developing a new class of drugs. Nat. Rev. Drug Discov. 2014, 13, 759–780. [Google Scholar] [CrossRef]
- Baiersdörfer, M.; Boros, G.; Muramatsu, H.; Mahiny, A.; Vlatkovic, I.; Sahin, U.; Karikó, K. A Facile Method for the Removal of dsRNA Contaminant from In Vitro-Transcribed mRNA. Mol. Ther. Nucleic Acids 2019, 15, 26–35. [Google Scholar] [CrossRef] [Green Version]
- Rauch, S.; Lutz, J.; Kowalczyk, A.; Schlake, T.; Heidenreich, R. RNActive® Technology: Generation and Testing of Stable and Immunogenic mRNA Vaccines. Methods Mol. Biol. 2017, 1499, 89–107. [Google Scholar] [CrossRef]
- Luo, M.; Wang, H.; Wang, Z.; Cai, H.; Lu, Z.; Li, Y.; Du, M.; Huang, G.; Wang, C.; Chen, X.; et al. A STING-activating nanovaccine for cancer immunotherapy. Nat. Nanotechnol. 2017, 12, 648–654. [Google Scholar] [CrossRef]
- Kranz, L.M.; Diken, M.; Haas, H.; Kreiter, S.; Loquai, C.; Reuter, K.C.; Meng, M.; Fritz, D.; Vascotto, F.; Hefesha, H.; et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016, 534, 396–401. [Google Scholar] [CrossRef]
- BioNTech Receives FDA Fast Track Designation for Its FixVac Candidate BNT111 in Advanced Melanoma. Available online: https://investors.biontech.de/news-releases/news-release-details/biontech-receives-fda-fast-track-designation-its-fixvac (accessed on 19 November 2021).
- Shemesh, C.S.; Hsu, J.C.; Hosseini, I.; Shen, B.Q.; Rotte, A.; Twomey, P.; Girish, S.; Wu, B. Personalized Cancer Vaccines: Clinical Landscape, Challenges, and Opportunities. Mol. Ther. 2021, 29, 555–570. [Google Scholar] [CrossRef] [PubMed]
- Keskin, D.B.; Anandappa, A.J.; Sun, J.; Tirosh, I.; Mathewson, N.D.; Li, S.; Oliveira, G.; Giobbie-Hurder, A.; Felt, K.; Gjini, E.; et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2019, 565, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Hilf, N.; Kuttruff-Coqui, S.; Frenzel, K.; Bukur, V.; Stevanović, S.; Gouttefangeas, C.; Platten, M.; Tabatabai, G.; Dutoit, V.; van der Burg, S.H.; et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 2019, 565, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Platten, M.; Bunse, L.; Wick, A.; Bunse, T.; Le Cornet, L.; Harting, I.; Sahm, F.; Sanghvi, K.; Tan, C.L.; Poschke, I.; et al. A vaccine targeting mutant IDH1 in newly diagnosed glioma. Nature 2021, 592, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Hu-Lieskovan, S.; Chmielowski, B.; Govindan, R.; Naing, A.; Bhardwaj, N.; Margolin, K.; Awad, M.M.; Hellmann, M.D.; Lin, J.J.; et al. A Phase Ib Trial of Personalized Neoantigen Therapy Plus Anti-PD-1 in Patients with Advanced Melanoma, Non-small Cell Lung Cancer, or Bladder Cancer. Cell 2020, 183, 347–362.e24. [Google Scholar] [CrossRef] [PubMed]
- Gillison, M.L.; Awad, M.M.; Twardowski, P.; Sukari, A.; Johnson, M.L.; Stein, M.N.; Hernandez, R.; Price, J.; Mancini, K.J.; Shainheit, M.; et al. Long term results from a phase 1 trial of GEN-009, a personalized neoantigen vaccine, combined with PD-1 inhibition in advanced solid tumors. J. Clin. Oncol. 2021, 39, 2613. [Google Scholar] [CrossRef]
- Nabhan, C. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 1966–1967. [Google Scholar] [CrossRef]
- Carreno, B.M.; Magrini, V.; Becker-Hapak, M.; Kaabinejadian, S.; Hundal, J.; Petti, A.A.; Ly, A.; Lie, W.-R.; Hildebrand, W.H.; Mardis, E.R.; et al. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 2015, 348, 803–808. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Zou, Z.; Du, J.; Su, S.; Shao, J.; Meng, F.; Yang, J.; Xu, Q.; Ding, N.; Yang, Y.; et al. Neoantigen identification strategies enable personalized immunotherapy in refractory solid tumors. J. Clin. Inverstig. 2019, 129, 2056–2070. [Google Scholar] [CrossRef]
- Ding, Z.; Li, Q.; Zhang, R.; Xie, L.; Shu, Y.; Gao, S.; Wang, P.; Su, X.; Qin, Y.; Wang, Y.; et al. Personalized neoantigen pulsed dendritic cell vaccine for advanced lung cancer. Signal Transduct. Target. Ther. 2021, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.; Blanc, C.; Granier, C.; Saldmann, A.; Tanchot, C.; Tartour, E. Therapeutic cancer vaccine: Building the future from lessons of the past. Semin. Immunopathol. 2019, 41, 69–85. [Google Scholar] [CrossRef] [PubMed]
- van der Burg, S.H. Correlates of immune and clinical activity of novel cancer vaccines. Semin. Immunol. 2018, 39, 119–136. [Google Scholar] [CrossRef] [PubMed]
- Masopust, D.; Ha, S.J.; Vezys, V.; Ahmed, R. Stimulation history dictates memory CD8 T cell phenotype: Implications for prime-boost vaccination. J. Immunol. 2006, 177, 831–839. [Google Scholar] [CrossRef] [Green Version]
- Wirth, T.C.; Xue, H.H.; Rai, D.; Sabel, J.T.; Bair, T.; Harty, J.T.; Badovinac, V.P. Repetitive antigen stimulation induces stepwise transcriptome diversification but preserves a core signature of memory CD8(+) T cell differentiation. Immunity 2010, 33, 128–140. [Google Scholar] [CrossRef] [Green Version]
- Fraser, K.A.; Schenkel, J.M.; Jameson, S.C.; Vezys, V.; Masopust, D. Preexisting high frequencies of memory CD8+ T cells favor rapid memory differentiation and preservation of proliferative potential upon boosting. Immunity 2013, 39, 171–183. [Google Scholar] [CrossRef] [Green Version]
- Alspach, E.; Lussier, D.M.; Miceli, A.P.; Kizhvatov, I.; DuPage, M.; Luoma, A.M.; Meng, W.; Lichti, C.F.; Esaulova, E.; Vomund, A.N.; et al. MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature 2019, 574, 696–701. [Google Scholar] [CrossRef]
- Nembrini, C.; Stano, A.; Dane, K.Y.; Ballester, M.; van der Vlies, A.J.; Marsland, B.J.; Swartz, M.A.; Hubbell, J.A. Nanoparticle conjugation of antigen enhances cytotoxic T-cell responses in pulmonary vaccination. Proc. Natl. Acad. Sci. USA 2011, 108, E989–E997. [Google Scholar] [CrossRef] [Green Version]
- Li, A.V.; Moon, J.J.; Abraham, W.; Suh, H.; Elkhader, J.; Seidman, M.A.; Yen, M.; Im, E.J.; Foley, M.H.; Barouch, D.H.; et al. Generation of effector memory T cell-based mucosal and systemic immunity with pulmonary nanoparticle vaccination. Sci. Transl. Med. 2013, 5, 204ra130. [Google Scholar] [CrossRef] [Green Version]
- Kuai, R.; Ochyl, L.J.; Bahjat, K.S.; Schwendeman, A.; Moon, J.J. Designer vaccine nanodiscs for personalized cancer immunotherapy. Nat. Mater. 2017, 16, 489–496. [Google Scholar] [CrossRef]
- Spranger, S.; Gajewski, T.F. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat. Rev. Cancer 2018, 18, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.T.; Moffett, H.F.; Stephan, S.B.; Opel, C.F.; Dumigan, A.G.; Jiang, X.; Pillarisetty, V.G.; Pillai, S.P.S.; Wittrup, K.D.; Stephan, M.T. Biopolymers codelivering engineered T cells and STING agonists can eliminate heterogeneous tumors. J. Clin. Invest. 2017, 127, 2176–2191. [Google Scholar] [CrossRef] [PubMed]
- Stephan, S.B.; Taber, A.M.; Jileaeva, I.; Pegues, E.P.; Sentman, C.L.; Stephan, M.T. Biopolymer implants enhance the efficacy of adoptive T-cell therapy. Nat. Biotechnol. 2015, 33, 97–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Wang, C.; Zhang, X.; Chen, G.; Hu, Q.; Li, H.; Wang, J.; Wen, D.; Zhang, Y.; Lu, Y.; et al. In situ sprayed bioresponsive immunotherapeutic gel for post-surgical cancer treatment. Nat. Nanotechnol. 2019, 14, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Gosselin, E.A.; Eppler, H.B.; Bromberg, J.S.; Jewell, C.M. Designing natural and synthetic immune tissues. Nat. Mater. 2018, 17, 484–498. [Google Scholar] [CrossRef] [PubMed]
- Pavesi, A.; Tan, A.T.; Koh, S.; Chia, A.; Colombo, M.; Antonecchia, E.; Miccolis, C.; Ceccarello, E.; Adriani, G.; Raimondi, M.T.; et al. A 3D microfluidic model for preclinical evaluation of TCR-engineered T cells against solid tumors. JCI Insight 2017, 2, e89762. [Google Scholar] [CrossRef] [PubMed]
- Ando, Y.; Siegler, E.L.; Ta, H.P.; Cinay, G.E.; Zhou, H.; Gorrell, K.A.; Au, H.; Jarvis, B.M.; Wang, P.; Shen, K. Evaluating CAR-T Cell Therapy in a Hypoxic 3D Tumor Model. Adv. Healthc. Mater. 2019, 8, e1900001. [Google Scholar] [CrossRef]
- Wallstabe, L.; Göttlich, C.; Nelke, L.C.; Kühnemundt, J.; Schwarz, T.; Nerreter, T.; Einsele, H.; Walles, H.; Dandekar, G.; Nietzer, S.L.; et al. ROR1-CAR T cells are effective against lung and breast cancer in advanced microphysiologic 3D tumor models. JCI Insight 2019, 4, e126345. [Google Scholar] [CrossRef] [Green Version]
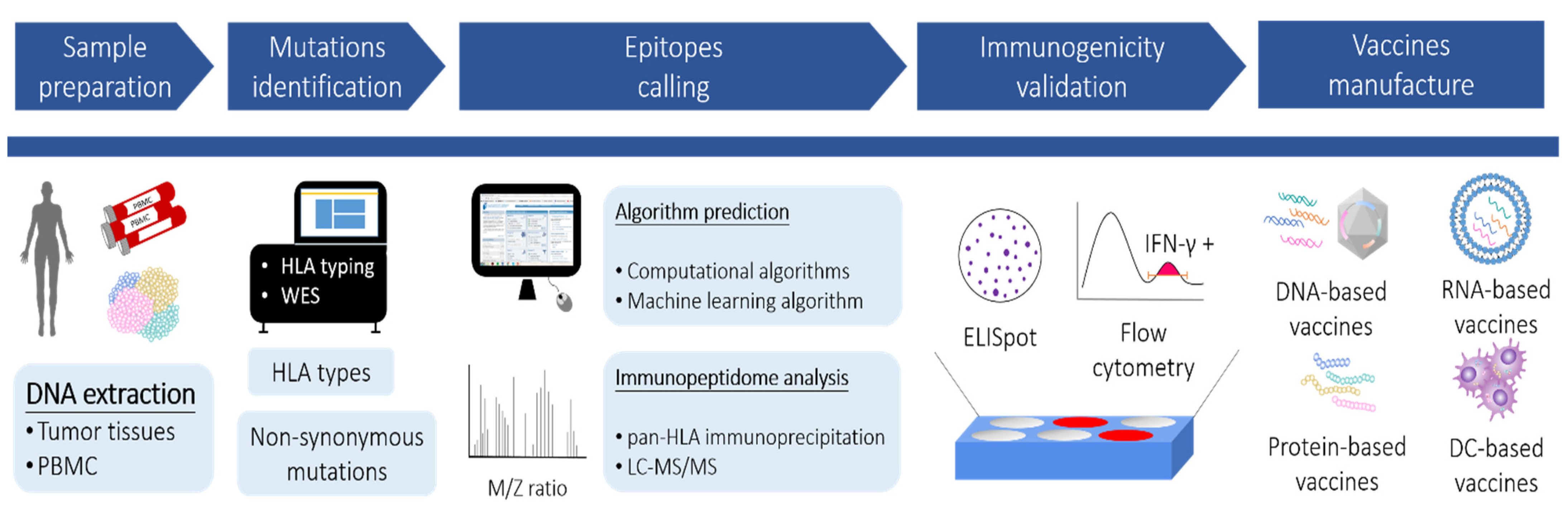

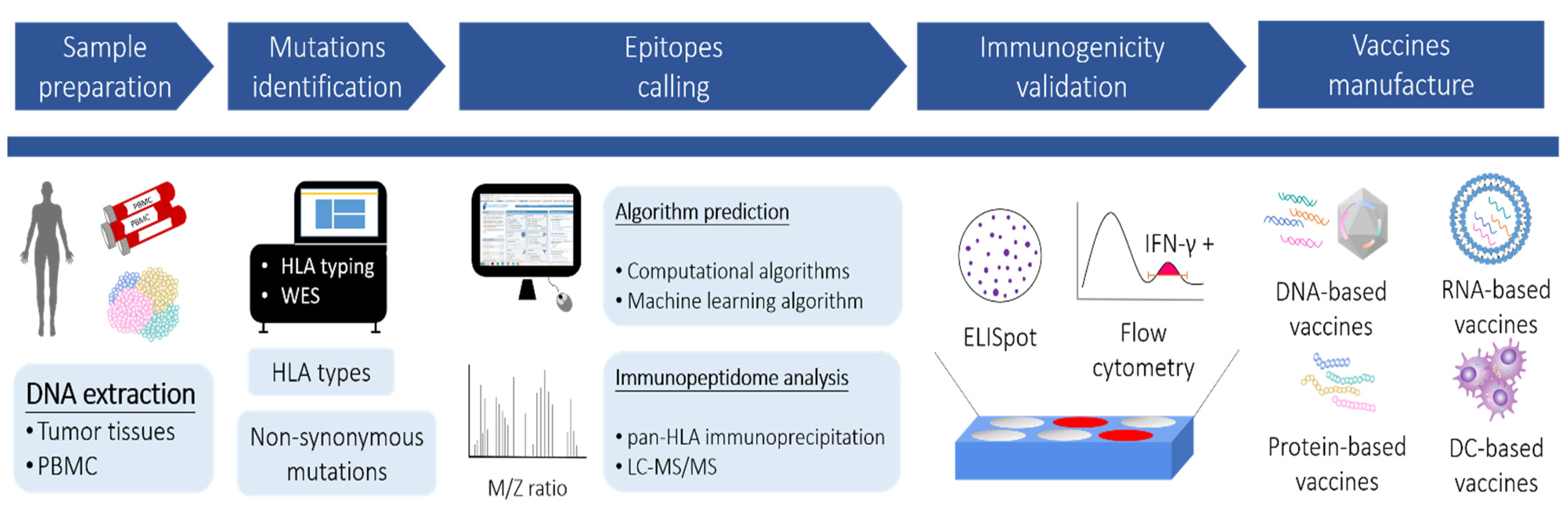{kind=link}
| Method [Ref] | Principle | Year |
|---|---|---|
| NetMHCpan [37] | Comparison of epitope sequences by artificial neural networks that provide peptide–MHC-I-affinity predictions | 2016 |
| NetMHCIIpan [61] | Pan-specific predictor able to predict binding affinities for all HLA-class-II molecules based on neural networks | 2013 |
| MHCflurry [62] | Neutral networks including mass-spectrometry datasets for predicting peptide–MHC-I affinities | 2018 |
| ConvMHC [63] | peptide–MHC interactions encoded into image-like array data and analyzed by deep convolutional neural network | 2017 |
| PLAtEAU [64] | Defines shared consensus epitopes arising from a series of eluted nested peptides and quantified by mass spectrometry | 2018 |
| MuPeXI [65] | Integration of somatic mutation calls, list of HLA types, an optional gene-expression profile, and NetMHCpan 3.0 to provide immunogenicity score based on similarity to non-mutated wild-type peptide | 2017 |
| NeoPrepPipe [66] | Predicts neoantigen burdens and provide insights into the tumor heterogeneity, somatic mutation calls, and patient HLA haplotypes | 2019 |
| EpitopeHunter [67] | Integrates expression of RNA with artificial neutral networks of immunogenicity-prediction algorithm based on the hydrophobicity of the TCR contact residues | 2015 |
| Neopepsee [68] | Integrates sequence and amino-acid-immunogenicity information, including antigen processing and presentation to reduce the false-discovery rate | 2018 |
| Trial No. (Brand Name) | Target | Indication | Format/Route of Administration | Combination Therapy | Status |
|---|---|---|---|---|---|
| NCT03122106 | Personalized NeoAg + Mesothelin | Pancreatic Cancer | Plasmid DNA/Electroporation + IM injection | N/A | Phase 1, Active, Not Recruiting |
| NCT04015700 (GNOS-PV01) | Personalized NeoAg | Unmethylated Glioblastoma | Plasmid DNA/Electroporation + IM injection | Pembrolizumab, Plasmid encoded IL-12 (INO-9012) | Phase 1, Recruiting |
| NCT04251117 (GNOS-PV02) | Personalized NeoAg + Mesothelin | HCC | Plasmid DNA/Electroporation + IM injection | Pembrolizumab, Plasmid encoded IL-12 (INO-9012) | Phase 1/2a, Recruiting |
| NCT04990479 (Nous-PEV) | Personalized NeoAg | Melanoma, NSCLC | Adenovirus vector + Vaccinia virus vector/IM injection | Pembrolizumab | Phase 1, Recruiting |
| NCT04041310 (Nous-209) | Personalized NeoAg | MSI-H CRC, gastric, G-E junction tumors | Adenovirus vector + vaccinia virus vector/IM injection | Pembrolizumab | Phase 1/2, Active, Not Recruiting |
| NCT05018273 (VB10.NEO) | Personalized NeoAg | Solid Tumors | Plasmid DNA/IM injection | Atezolizumab | Phase 1b, Recruiting |
| NCT02348320 | Personalized NeoAg | Triple-Negative Breast Cancer | Plasmid DNA/Electroporation + IM injection | N/A | Phase 1, Completed |
| NCT03953235 (SLATE) | Shared Neoantigen | Shared neoantigen positive tumors | Adenovirus vector + RNA vector/Not specific | Nivolumab, Ipilimumab | Phase 1/2, Recruiting |
| NCT03265080 (ADXS-NEO) | Personalized NeoAg | Colon Cancer, Head & Neck Cancer, NSCLC, Urothelial Carcinoma, Melanoma | Lm-based vector/I.V. infusion | Pembrolizumab (selectively) | Phase 1, Active, Not Recruiting |
| NCT03847519 (ADXS-503) | Personalized NeoAg | NSCLC, Metastatic SCC, Metastatic NSCLC | Lm-based vector/I.V. infusion | Pembrolizumab (selectively) | Phase 1/2, Recruiting |
| Trial No. (Brand Name) | Target | Indication | Format/Route of Administration | Combination Therapy | Status |
|---|---|---|---|---|---|
| RO7198457 | |||||
| NCT03289962 | Personalized NeoAg | Solid tumors | RNA-Lipoplex/I.V. | Atezolizumab | Phase 1a/1b, Recruiting |
| NCT03815058 | Personalized NeoAg | Advanced Melanoma | RNA-Lipoplex/I.V. | Pembrolizumab | Phase 2, Recruiting |
| NCT04486378 | Personalized NeoAg | Colorectal Cancer Stage II, III | RNA-Lipoplex/I.V. | N/A | Phase 2, Recruiting |
| NCT04161755 | Personalized NeoAg | Pancreatic Cancer | RNA-Lipoplex/I.V. | Atezolizumab, mFOLFIRINOX | Phase 1, Recruiting |
| IVAC mutanome | |||||
| NCT02035956 | Personalized NeoAg | Melanoma | Not specific/Intra-nodal | RBL001/RBL002 (TAA RNA Vaccine) | Phase 1, Completed |
| NCT02316457 | Personalized NeoAg | Breast Cancer (TNBC) | Nanoparticulate lipoplex RNA/I.V. | IVAC_W_bre1_uID (TAA RNA vaccine) | Phase 1, Active, Not Recruiting |
| mRNA-4157 | |||||
| NCT03897881 | Personalized NeoAg | Melanoma | lipid encapsulated RNA/I.M. | Pembrolizumab | Phase 2, Active, Not Recruiting |
| NCT03313778 | Personalized NeoAg | Solid tumors | lipid encapsulated RNA/I.M. | Pembrolizumab | Phase 1, Recruiting |
| mRNA-5671 | |||||
| NCT03948763 | KRAS common mutations | Solid Tumors | lipid encapsulated RNA/I.M. | Pembrolizumab (selectively) | Phase 1, Recruiting |
| Trial No. (Brand Name) | Target | Indication | Format/Route of Administration | Combination Therapy | Status |
| NCT04799431 | Personalized NeoAg | MMR-p Colon Cancer Pancreatic Ductal Cancer | Peptide + poly-ICLC/subcutaneous | Retifanlimab | Phase 1, Not Yet Recruiting |
| NCT03956056 | Personalized NeoAg + Mesothelin | Pancreatic Cancer | Peptide + poly-ICLC/ subcutaneous | N/A | Phase 1, Recruiting |
| NCT04248569 | DNAJB1- PRKACA fusion | Fibrolamellar Hepatocellular Carcinoma | Peptide + poly-ICLC | Nivolumab, Ipilimumab | Phase 1, Recruiting |
| NCT04117087 | Common mutant KRAS | Colorectal Cancer Pancreatic Cancer | Peptide + poly-ICLC | Nivolumab, Ipilimumab | Phase 1, Recruiting |
| NCT04749641 | Histone H3.3-K27M mutant | Diffuse Intrinsic Pontine Glioma | Peptide + poly-ICLC/subcutaneous | N/A | Phase 1, Recruiting |
| NCT03715985 (NeoPepVac) | Personalized NeoAg | Melanoma, NSCLC, Bladder, Urothelial Carcinoma, | Peptide + CAF09b/I.P. + I.M. | N/A | Phase 1, Recruiting |
| NCT03359239 (PGV-001) | Personalized NeoAg | Urothelial/Bladder Cancer | Peptide + poly-ICLC | Atezolizumab | Phase 1, Recruiting |
| NCT02149225 (GAPVAC) | Personalized NeoAg | Glioblastoma | Peptide + poly-ICLC/not specific | TAA peptide vaccine, GM-CSF | Phase 1, Completed |
| NeoVax | |||||
| NCT01970358 | Personalized NeoAg | Melanoma | Peptide + poly-ICLC/subcutaneous | N/A | Phase 1, Completed |
| NCT02950766 | Personalized NeoAg | Kidney cancer | Peptide + poly-ICLC/subcutaneous | Nivolumab, Ipilimumab | Phase 1, Recruiting |
| NCT02287428 | Personalized NeoAg | Glioblastoma | Peptide + poly-ICLC | Pembrolizumab Temozolomide (Both selectively) | Phase 1, Recruiting |
| NCT03929029 | Personalized NeoAg | Melanoma | Peptide + poly-ICLC + Montanide | Nivolumab Ipilimumab | Phase 1b, Recruiting |
| NCT0402487 | Personalized NeoAg | Ovarian Cancer | Peptide + poly-ICLC | Nivolumab | Phase 1, Recruiting |
| NCT03219450 | Personalized NeoAg | Lymphocytic Leukemia | Peptide + poly-ICLC | Pembrolizumab Cyclophosphamide (both selectively) | Phase 1, Recruiting |
| Neo-PV-01 | |||||
| NCT03380871 | Personalized NeoAg | Lung cancer | Peptide + poly-ICLC/subcutaneous | Pembrolizumab Carboplatin Pemetrexed | Phase 1, Completed |
| NCT02897765 | Personalized NeoAg | Urinary Bladder Cancer Melanoma Lung Cancer | Peptide + poly-ICLC/subcutaneous | Nivolumab | Phase 1, Completed |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pao, S.-C.; Chu, M.-T.; Hung, S.-I. Therapeutic Vaccines Targeting Neoantigens to Induce T-Cell Immunity against Cancers. Pharmaceutics 2022, 14, 867. https://doi.org/10.3390/pharmaceutics14040867
Pao S-C, Chu M-T, Hung S-I. Therapeutic Vaccines Targeting Neoantigens to Induce T-Cell Immunity against Cancers. Pharmaceutics. 2022; 14(4):867. https://doi.org/10.3390/pharmaceutics14040867
Chicago/Turabian StylePao, Shih-Cheng, Mu-Tzu Chu, and Shuen-Iu Hung. 2022. "Therapeutic Vaccines Targeting Neoantigens to Induce T-Cell Immunity against Cancers" Pharmaceutics 14, no. 4: 867. https://doi.org/10.3390/pharmaceutics14040867
APA StylePao, S.-C., Chu, M.-T., & Hung, S.-I. (2022). Therapeutic Vaccines Targeting Neoantigens to Induce T-Cell Immunity against Cancers. Pharmaceutics, 14(4), 867. https://doi.org/10.3390/pharmaceutics14040867