
Simultaneous Quantification and Pharmacokinetic Characterization of Doxapram and 2-Ketodoxapram in Porcine Plasma and Brain Tissue

 , , , , , , , and
, , , , , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Study and Sample Generation
2.2. Reagents and Solvents
2.3. Preparation of Standard Solutions
2.4. Sample Preparation
2.5. Instrumental Analysis
2.6. Method Validation
2.7. Protein Binding
2.8. Calculations
3. Results and Discussion
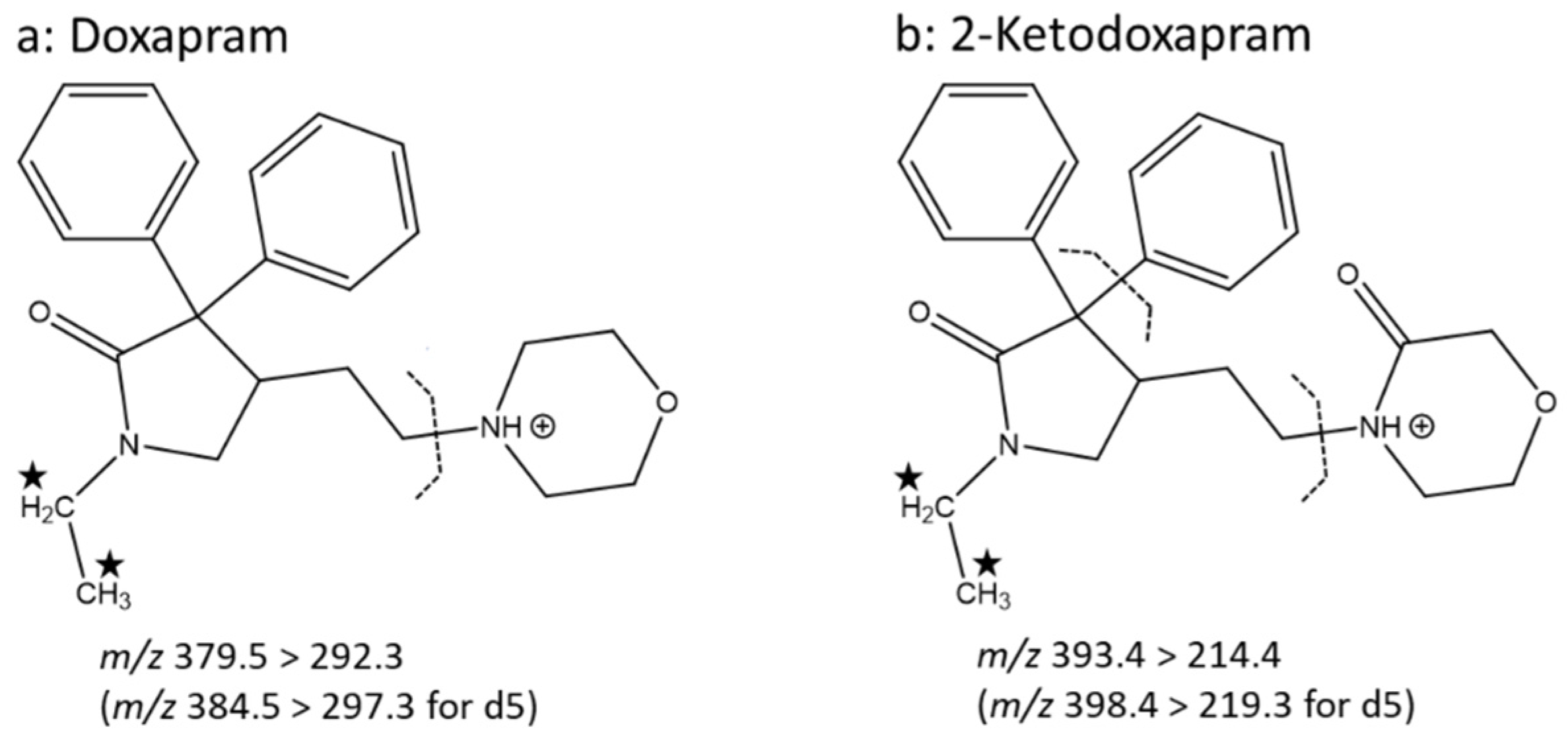
3.1. Mass Spectrometry and Chromatography
3.2. Sample Preparation
3.3. Method Validation
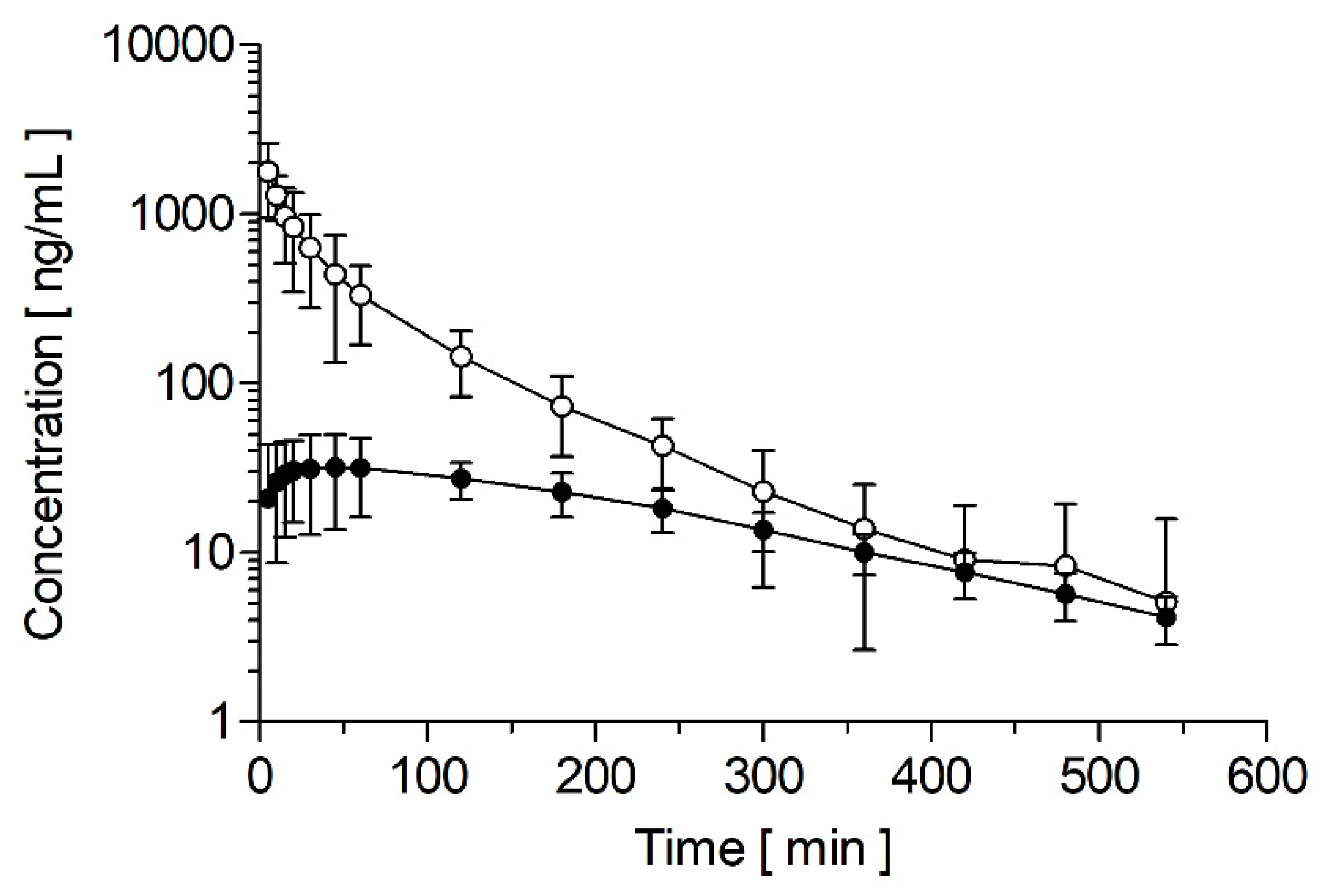
3.4. Pharmacokinetics
3.5. Protein Binding
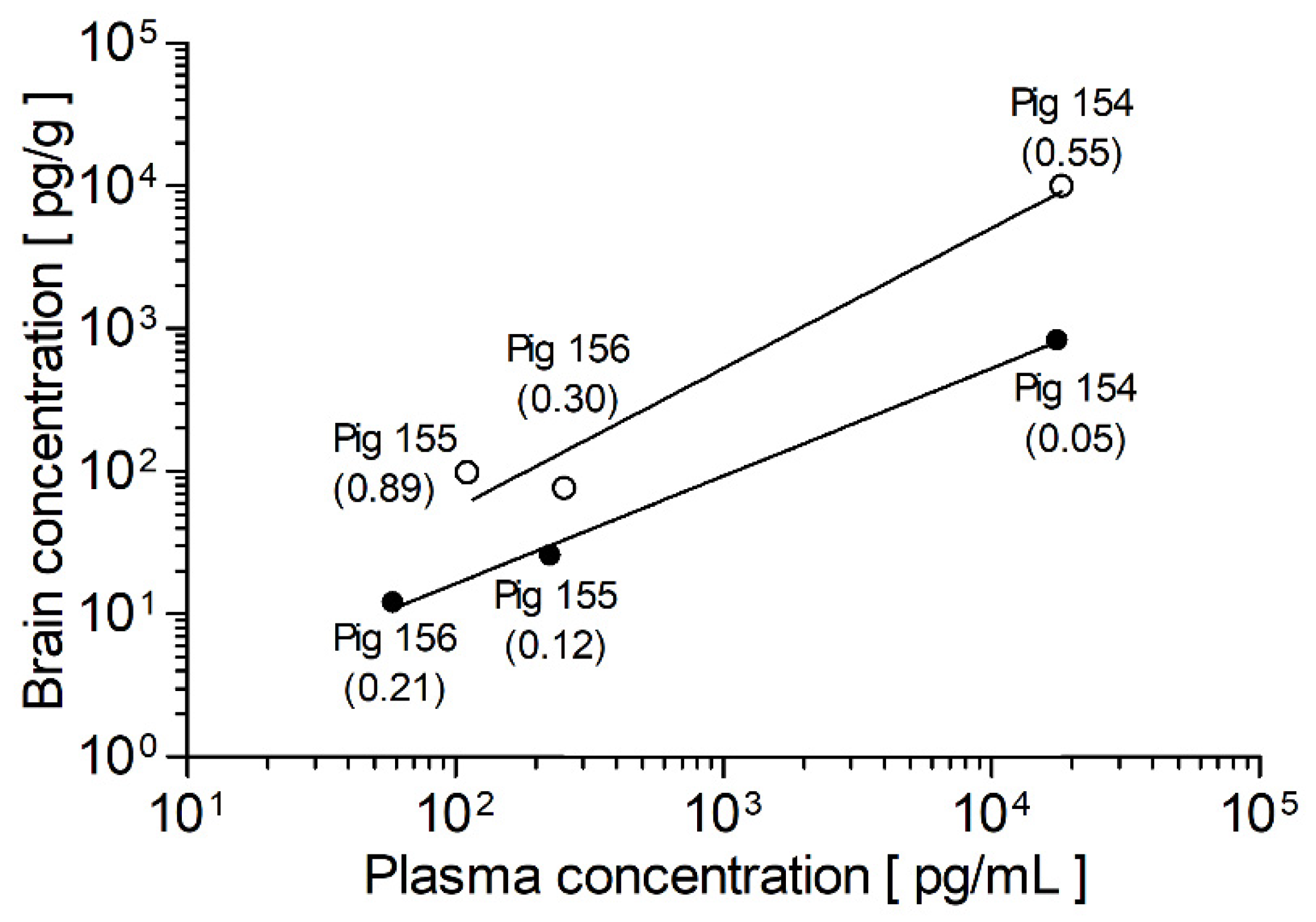
3.6. Brain-to-Plasma Ratio
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chugh, S.S.; Havmoeller, R.; Narayanan, K.; Singh, D.; Rienstra, M.; Benjamin, E.J.; Gillum, R.F.; Kim, Y.; McAnulty, J.H., Jr.; Zheng, Z.; et al. Worldwide epidemiology of atrial fibrillation: A Global Burden of Disease 2010 Study. Circulation 2014, 129, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Hindricks, G.; Potpara, T.; Dagres, N.; Arbelo, E.; Bax, J.J.; Blomström-Lundqvist, C.; Boriani, G.; Castella, M.; Dan, G.; Dilaveris, P.E.; et al. 2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association for Cardio-Thoracic Surgery (EACTS) The Task Force for the diagnosis and management of atrial fibrillation of the European Society of Cardiology (ESC) Developed with the special contribution of the European Heart Rhythm Association (EHRA) of the ESC. Eur. Heart J. 2021, 42, 373–498. [Google Scholar] [CrossRef] [PubMed]
- Ravens, U. Atrial-selective K+ channel blockers: Potential antiarrhythmic drugs in atrial fibrillation? Can. J. Physiol. Pharmacol. 2017, 95, 1313–1318. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C.; Wiedmann, F.; Voigt, N.; Zhou, X.B.; Heijman, J.; Lang, S.; Albert, V.; Kallenberger, S.; Ruhparwar, A.; Szabó, G.; et al. Upregulation of K2P3. 1 K+ current causes action potential shortening in patients with chronic atrial fibrillation. Circulation 2015, 132, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Wiedmann, F.; Beyersdorf, C.; Zhou, X.B.; Kraft, M.; Paasche, A.; Jávorszky, N.; Rinné, S.; Sutanto, H.; Büscher, A.; Foerster, K.I.; et al. Treatment of atrial fibrillation with doxapram: TASK-1 potassium channel inhibition as a novel pharmacological strategy. Cardiovasc. Res. 2021, cvab177:1–cvab177:14. [Google Scholar] [CrossRef]
- Wiedmann, F.; Beyersdorf, C.; Zhou, X.B.; Kraft, M.; Foerster, K.I.; El-Battrawy, I.; Lang, S.; Borggrefe, M.; Haefeli, W.E.; Frey, N.; et al. The Experimental TASK-1 Potassium Channel Inhibitor A293 Can Be Employed for Rhythm Control of Persistent Atrial Fibrillation in a Translational Large Animal Model. Front. Physiol. 2021, 11, 629421:1–629421:13. [Google Scholar] [CrossRef]
- Wiedmann, F.; Beyersdorf, C.; Zhou, X.B.; Büscher, A.; Kraft, M.; Nietfeld, J.; Walz, T.P.; Unger, L.A.; Loewe, A.; Schmack, B.; et al. Pharmacologic TWIK-Related Acid-Sensitive K+ Channel (TASK-1) Potassium Channel Inhibitor A293 Facilitates Acute Cardioversion of Paroxysmal Atrial Fibrillation in a Porcine Large Animal Model. J. Am. Heart Assoc. 2020, 9, e015751:1–e015751:16. [Google Scholar] [CrossRef]
- Schmidt, C.; Wiedmann, F.; Langer, C.; Tristram, F.; Anand, P.; Wenzel, W.; Lugenbiel, P.; Schweizer, P.; Katus, H.A.; Thomas, D. Cloning, functional characterization, and remodeling of K2P3.1 (TASK-1) potassium channels in a porcine model of atrial fibrillation and heart failure. Heart Rhythm 2014, 11, 1798–1805. [Google Scholar] [CrossRef]
- Yost, C.S. A New Look at the Respiratory Stimulant Doxapram. CNS Drug Rev. 2006, 12, 236–249. [Google Scholar] [CrossRef]
- Baxter, A.D. Side effects of doxapram infusion. Eur. J. Intensive Care Med. 1976, 2, 87–88. [Google Scholar] [CrossRef]
- US Department of Health and Human Services FDA. Bioanalytical Method Validation Guidance for Industry. May 2018. Available online: https://www.fda.gov/files/drugs/published/Bioanalytical-Method-Validation-Guidance-for-Industry.pdf (accessed on 24 October 2021).
- European Medicines Agency. Guideline on Bioanalytical Method Validation. EMEA/CHMP/EWP/192217/2009 Rev. 1 Corr. 2. July 2011. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-bioanalytical-method-validation_en.pdf (accessed on 24 October 2021).
- Matuszewski, B.K.; Constanzer, M.L.; Chavez-Eng, C.M. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Anal. Chem. 2003, 75, 3019–3030. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Kamada, N.; Ohno, H.; Abe, Y.; Endo, T.; Kobayashi, M. Development and validation of an LC–MS/MS method for simultaneously determining doxapram and keto-doxapram in human serum. Bioanalysis 2017, 9, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Flint, R.B.; Bahmany, S.; van der Nagel, B.C.; Koch, B.C. Simultaneous quantification of fentanyl, sufentanil, cefazolin, doxapram and keto-doxapram in plasma using liquid chromatography–tandem mass spectrometry. Biomed. Chromatogr. 2018, 32, e4290:1–e4290:8. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, R.; Sengupta, P.; Cherynak, G.; Wadhwa, A.; Sessler, D.I.; Liu, J.; Hurst, H.E.; Lenhardt, R. Doxapram only slightly reduces the shivering threshold in healthy volunteers. Anesth. Analg. 2005, 101, 1368–1373. [Google Scholar] [CrossRef]
- Aranda, J.V.; Beharry, K.; Rex, J.; Linder, N.; Blanchard, P. High pressure liquid chromatographic microassay for simultaneous measurement of doxapram and its metabolites in premature newborn infants. J. Liq. Chromatogr. 1988, 11, 2983–2991. [Google Scholar] [CrossRef]
- Barbé, F.; Hansen, C.; Badonnel, Y.; Legagneur, H.; Vert, P.; Boutroy, M.J. Severe side effects and drug plasma concentrations in preterm infants treated with doxapram. Ther. Drug Monit. 1999, 21, 547–552. [Google Scholar] [CrossRef]
- Coutts, R.T.; Jamali, F.; Malek, F.; Peliowski, A.; Finer, N.N. Urinary metabolites of doxapram in premature neonates. Xenobiotica 1991, 21, 1407–1418. [Google Scholar] [CrossRef]
- LeGatt, D.F.; Beaudry, M.A.; Bradley, J.M. Simultaneous determination of doxapram and 2-ketodoxapram in plasma of neonates by gas chromatography. J. Chromatogr. 1986, 378, 478–481. [Google Scholar] [CrossRef]
- Lin, G.; Ma, J.; Hu, L.; Wang, X.; Zhu, J.; Wang, X. Determination of doxapram hydrochloride in rabbit plasma by LC-MS-MS and its application. Chromatographia 2011, 73, 183–187. [Google Scholar] [CrossRef]
- Nichol, H.; Vine, J.; Thomas, J.; Moore, R.G. Quantitation of doxapram in blood, plasma and urine. J. Chromatogr. 1980, 182, 191–200. [Google Scholar] [CrossRef]
- Ogawa, Y.; Irikura, M.; Kobaru, Y.; Tomiyasu, M.; Kochiyama, Y.; Uriu, M.; Ishitsuka, Y.; Kondo, Y.; Yukawa, E.; Kamada, N.; et al. Population pharmacokinetics of doxapram in low-birth-weight Japanese infants with apnea. Eur. J. Pediatr. 2015, 174, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Robson, R.H.; Prescott, L.F. Rapid gas-liquid chromatographic estimation of doxapram in plasma. J. Chromatogr. 1977, 143, 527–529. [Google Scholar] [CrossRef]
- Roozekrans, M.; Olofsen, E.; van der Schrier, R.; Boom, M.; Mooren, R.; Dahan, A. Doxapram-mediated Increase in Cardiac Output Reduces Opioid Plasma Concentrations: A Pharmacokinetic/Pharmacodynamic–Pharmacokinetic/Pharmacodynamic Modeling Study in Healthy Volunteers. Clin. Pharmacol. Ther. 2017, 102, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Sams, R.A.; Detra, R.L.; Muir, W.W., III. Pharmacokinetics and metabolism of intravenous doxapram in horses. Equine Vet. J. 1992, 24, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Torok-Both, G.A.; Coutts, R.T.; Jamali, F.; Pasutto, F.M.; Barrington, K.J. Sensitive nitrogen-phosphorus capillary gas chromatographic assay for doxapram in premature infants. J. Chromatogr. 1985, 344, 372–377. [Google Scholar] [CrossRef]
- Robson, R.H.; Prescott, L.F. A pharmacokinetic study of doxapram in patients and volunteers. Br. J. Clin. Pharmacol. 1979, 7, 81–87. [Google Scholar] [CrossRef][Green Version]
- Clements, J.A.; Robson, R.H.; Prescott, L.F. The disposition of intravenous doxapram in man. Eur. J. Clin. Pharmacol. 1979, 16, 411–416. [Google Scholar] [CrossRef]
- Bairam, A.; Blanchard, P.W.; Mullahoo, K.; Beharry, K.; Laudignon, N.; Aranda, J.V. Pharmacodynamic effects and pharmacokinetic profiles of keto-doxapram and doxapram in newborn lambs. Pediatr. Res. 1990, 28, 142–146. [Google Scholar] [CrossRef]
- Schmidt, W.; Jähnchen, E. Species-dependent stereospecific serum protein binding of the oral anticoagulant drug phenprocoumon. Experientia 1978, 34, 1323–1325. [Google Scholar] [CrossRef]
- Kosa, T.; Maruyama, T.; Otagiri, M. Species Differences of Serum Albumins: I. Drug Binding Sites. Pharm. Res. 1997, 14, 1607–1612. [Google Scholar] [CrossRef]
- Kumita, H.; Mizuno, S.; Shinohara, M.; Ichikawa, T.; Yamazaki, T. Low-dose doxapram therapy in premature infants and its CSF and serum concentrations. Acta. Paediatr. Scand. 1991, 80, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Bruce, R.B.; Pitts, J.E.; Pinchbeck, F.; Newman, J. Excretion, distribution, and metabolism of doxapram hydrochloride. J. Med. Chem. 1965, 8, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A. Characteristics of compounds that cross the blood-brain barrier. BMC Neurol. 2009, 9, S3:1–S3:5. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Study | Instruments | Range [ng/mL] | Analytes | Sample Matrix | Sample Volume | Run Time | Internal Standard | Sample Preparation |
|---|---|---|---|---|---|---|---|---|
| Aranda et al. (1988) [17] | HPLC-UV | 1000–15,000 | Doxapram, ketodoxapram, AHR 5904, AHR 0914 | Human serum | 50 µL | 15 min | Beta-hydroxy-phenyl-theophylline | LLE |
| Barbé et al. (1999) [18] | HPLC-UV/VIS | 100–20,000 | Doxapram, ketodoxapram, AHR 5904, AHR 0914 | Human plasma | 60 µL | 10 min | Butobarbital | LLE |
| Coutts et al. (1991) [19] | GC-N/P-D | - 1 | Doxapram and many metabolites | Human urine | 0.5–1 mL | - 2 | - | LLE |
| Flint et al. (2018) [15] | UPLC-MS/MS | 50–4500 | Doxapram | Human plasma | 50 µL | 5 min | Fentanyl-d5 | PP |
| 50–5000 | Ketodoxapram | |||||||
| Komatsu et al. (2005) [16] | GC-MS | 250–5000 | Doxapram | Human plasma | - 2 | - 2 | Diazepam | SPE |
| LeGatt et al. (1986) [20] | GC-N/P-D | 100–10,000 | Doxapram and ketodoxapram | Human plasma | 200 µL | 6 min 3 | AHR-755 (doxapram analog) | LLE |
| Lin et al. (2011) [21] | LC-MS/MS | 2–1000 | Doxapram | Rabbit plasma | 100 µL | 10 min | Urapidil | PP |
| Nichol et al. (1980) [22] | GC-MS | -2 | Doxapram | Human blood, plasma | 50–100 µL | - 2 | Dextromoramide | LLE |
| GC-FID | Human urine | 0.5–2 mL | ||||||
| Ogawa et al. (2015) [23] | HPLC-UV/VIS | 30–? 4 | Doxapram | Human serum | 50 µL | 25 min 3 | Butobarbital | LLE |
| 10–? 4 | Ketodoxapram | |||||||
| Robson and Prescott (1977) [24] | GC-N-D | 10–5000 | Doxapram | Human plasma | 2 mL | - 2 | Naftidrofuryl oxalte | LLE |
| 250–5000 | Ketodoxapram | |||||||
| Roozekrans et al. (2017) [25] | LC-MS/MS | 2–5000 | Doxapram | Human plasma | - 2 | - 2 | - 2 | PP |
| Sams et al. (1992) [26] | GC-N/P-D | 25–5000 | Doxapram | Horse plasma | 1 mL | - 2 | Diazepam | LLE |
| Suzuki et al. (2017) [14] | LC-MS/MS | 20–5000 | Doxapram and ketodoxapram | Human serum | 25 µL | 17 min | Propranolol | PP |
| Torok-Both et al. (1985) [27] | GC-N/P-D | - 2 | Doxapram | Human plasma | 20–100 µL | 8 min 3 | Diazepam | PP and LLE |
| Human urine | 2–5 µL |
| Animal | Cmax [ng/mL] | AUC [ng/mL*h] | VSS [L] | Cl [mL/min/kg] | t1/2 [h] | tmax [h] |
|---|---|---|---|---|---|---|
| Doxapram | ||||||
| Pig 154 | 2149 | 1169 | 34.4 | 14.2 | 1.18 | - |
| Pig 155 | 1490 | 986 | 51.5 | 16.9 | 1.26 | - |
| Pig 156 | 1701 | 1403 | 37.1 | 11.9 | 1.69 | - |
| Mean | 1780 ± 275 | 1186 ± 170 | 41.0 ± 7.5 | 14.3 ± 2.0 | 1.38 ± 0.22 | - |
| 2-Ketodoxapram | ||||||
| Pig 154 | 31.2 | 157 | - | - | 2.47 | 0.33 |
| Pig 155 | 26.2 | 139 | - | - | 2.36 | 1.00 |
| Pig 156 | 39.5 | 185 | - | - | 2.42 | 0.75 |
| Mean | 32.3 ± 5.5 | 160 ± 19 | - | - | 2.42 ± 0.04 | 0.69 ± 0.28 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kraft, M.; Foerster, K.I.; Wiedmann, F.; Sauter, M.; Paasche, A.; Blochberger, P.L.; Yesilgöz, B.; L’hoste, Y.; Frey, N.; Haefeli, W.E.; et al. Simultaneous Quantification and Pharmacokinetic Characterization of Doxapram and 2-Ketodoxapram in Porcine Plasma and Brain Tissue. Pharmaceutics 2022, 14, 762. https://doi.org/10.3390/pharmaceutics14040762
Kraft M, Foerster KI, Wiedmann F, Sauter M, Paasche A, Blochberger PL, Yesilgöz B, L’hoste Y, Frey N, Haefeli WE, et al. Simultaneous Quantification and Pharmacokinetic Characterization of Doxapram and 2-Ketodoxapram in Porcine Plasma and Brain Tissue. Pharmaceutics. 2022; 14(4):762. https://doi.org/10.3390/pharmaceutics14040762
Chicago/Turabian StyleKraft, Manuel, Kathrin I. Foerster, Felix Wiedmann, Max Sauter, Amelie Paasche, Pablo L. Blochberger, Baran Yesilgöz, Yannick L’hoste, Norbert Frey, Walter E. Haefeli, and et al. 2022. "Simultaneous Quantification and Pharmacokinetic Characterization of Doxapram and 2-Ketodoxapram in Porcine Plasma and Brain Tissue" Pharmaceutics 14, no. 4: 762. https://doi.org/10.3390/pharmaceutics14040762
APA StyleKraft, M., Foerster, K. I., Wiedmann, F., Sauter, M., Paasche, A., Blochberger, P. L., Yesilgöz, B., L’hoste, Y., Frey, N., Haefeli, W. E., Burhenne, J., & Schmidt, C. (2022). Simultaneous Quantification and Pharmacokinetic Characterization of Doxapram and 2-Ketodoxapram in Porcine Plasma and Brain Tissue. Pharmaceutics, 14(4), 762. https://doi.org/10.3390/pharmaceutics14040762