
Targeting Zika Virus with New Brain- and Placenta-Crossing Peptide–Porphyrin Conjugates

 ,
,  , , , , , , and
, , , , , , and 
Abstract
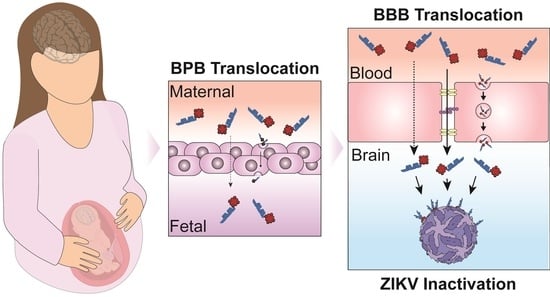
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cells and Cell Culture Reagents
2.3. Peptide Synthesis
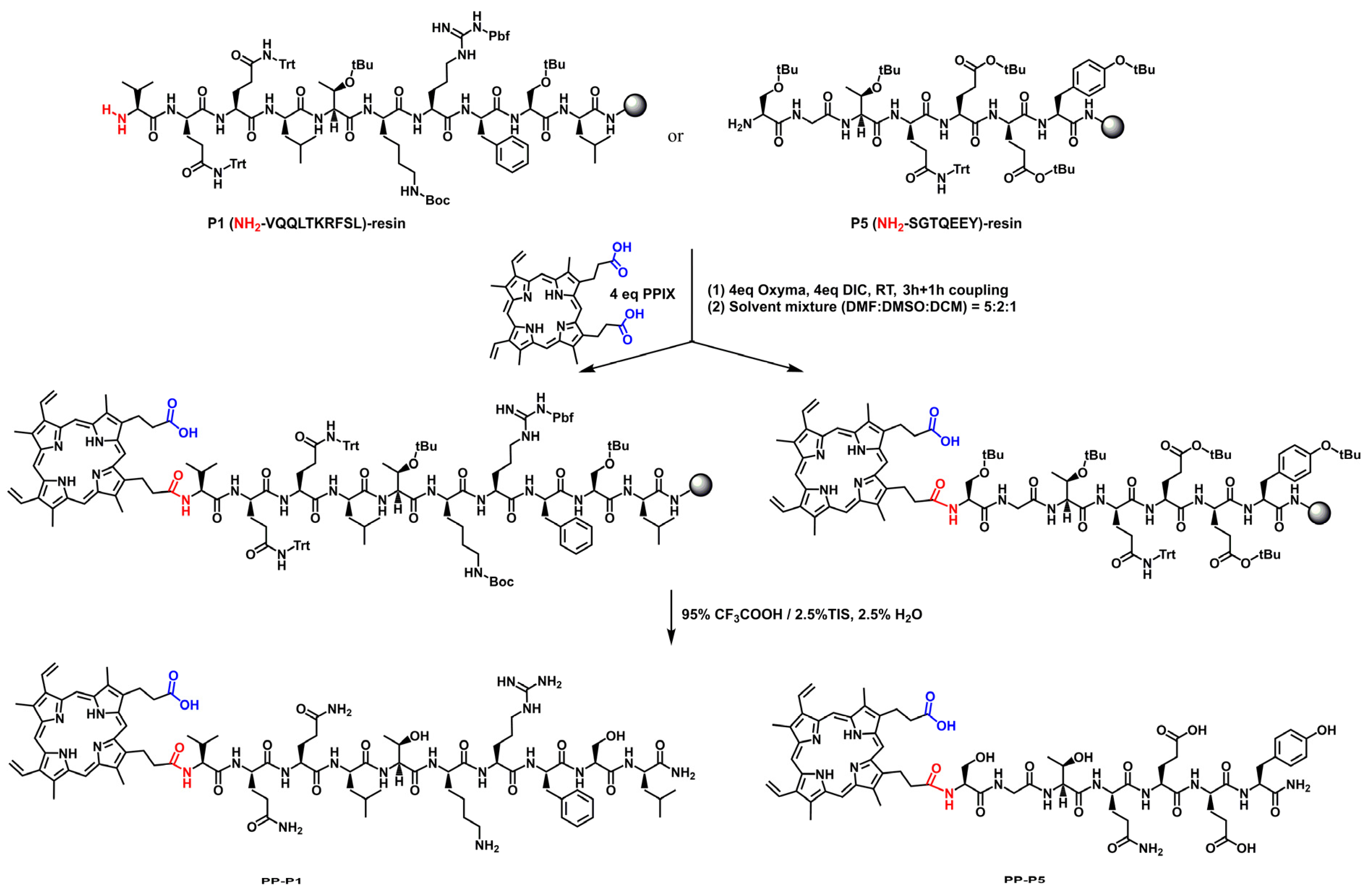
2.4. Conjugation Chemistry
2.5. PPC Purification
2.6. LC-MS Analysis
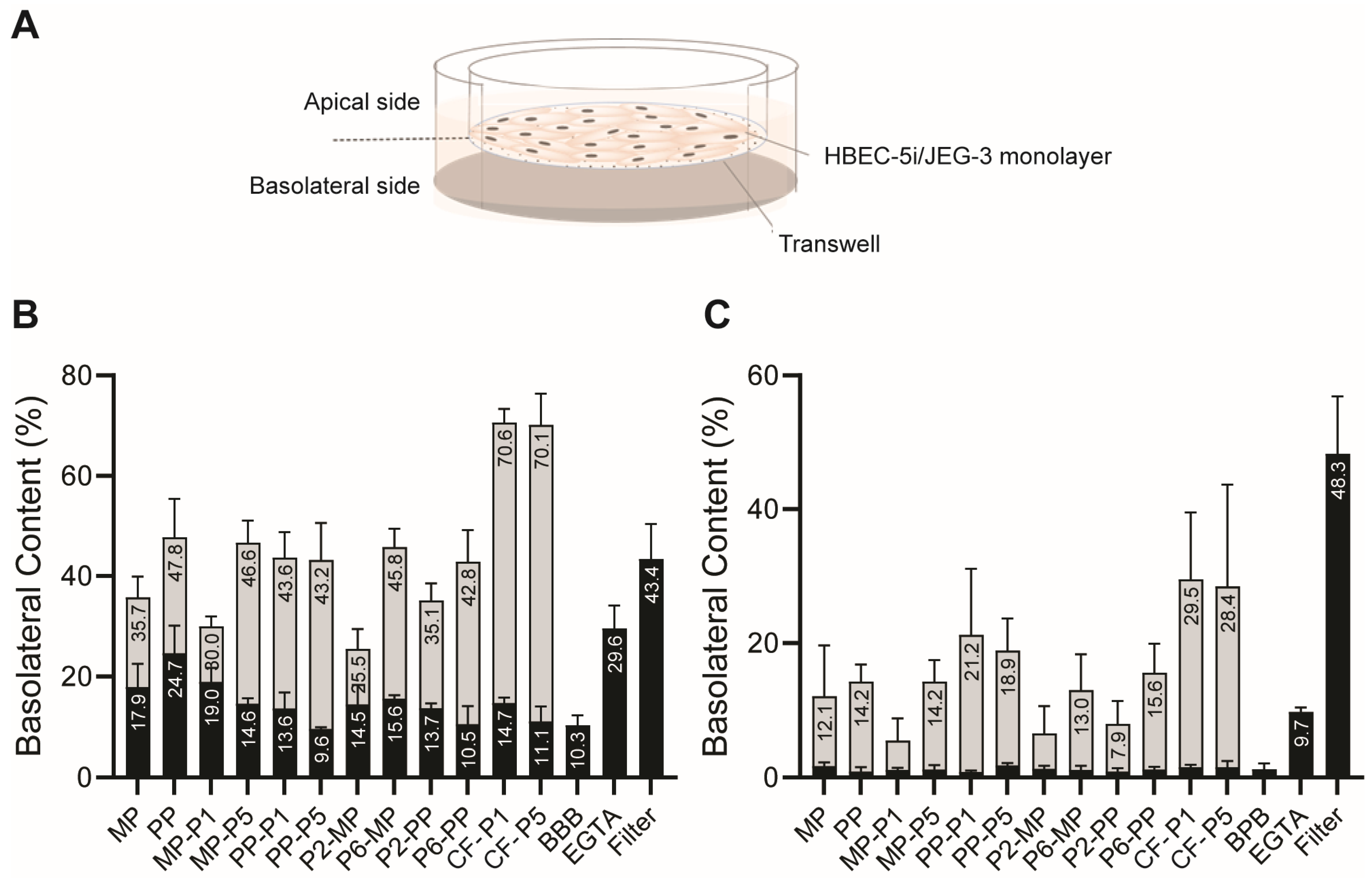
2.7. In Vitro BBB Translocation Assay
2.8. In Vitro BBB Integrity Assay
2.9. In Vitro BPB Translocation and Integrity Assay
2.10. Virus Samples
2.11. ZIKV Inactivation Assay
2.12. Cell Viability Assay
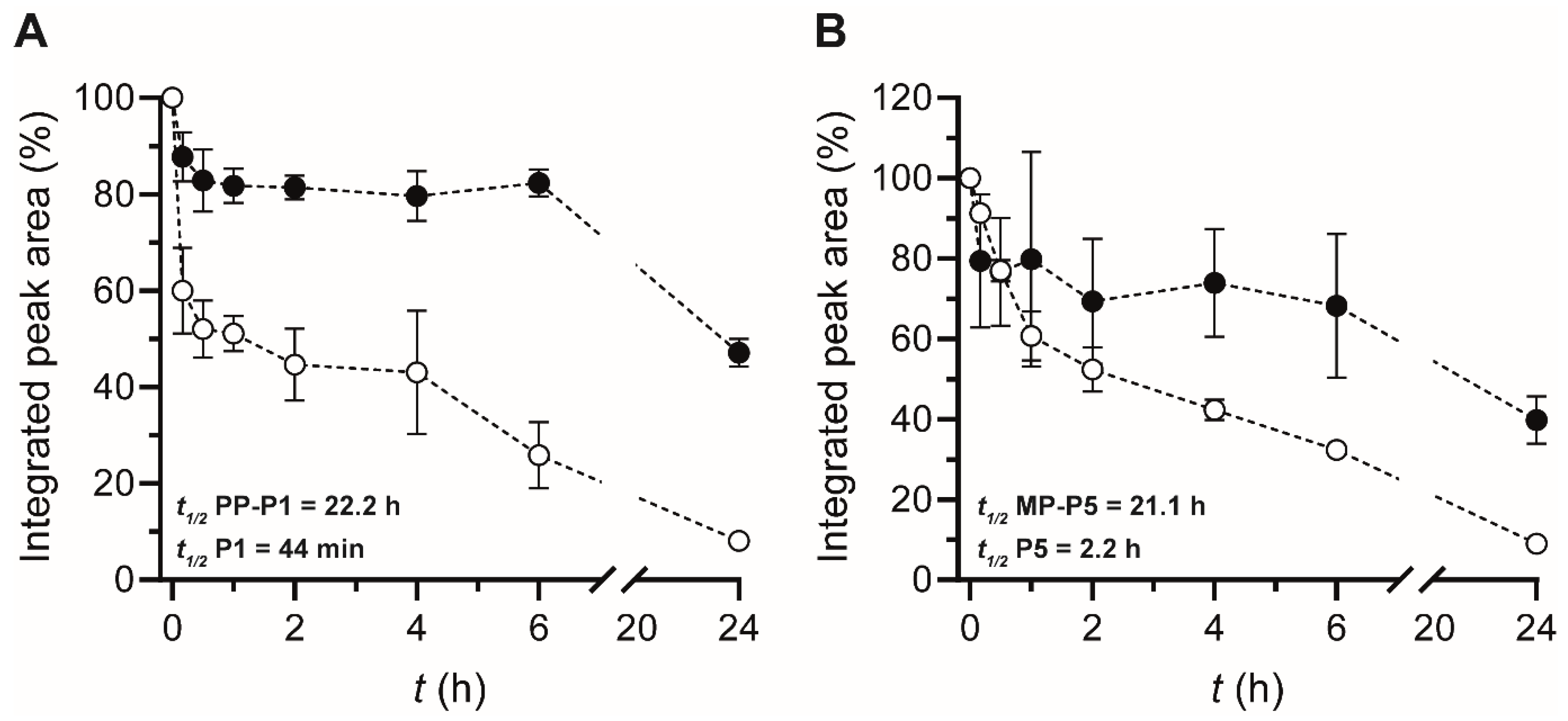
2.13. Serum Stability
3. Results and Discussion
3.1. BBBpS—Porphyrin Conjugate Design
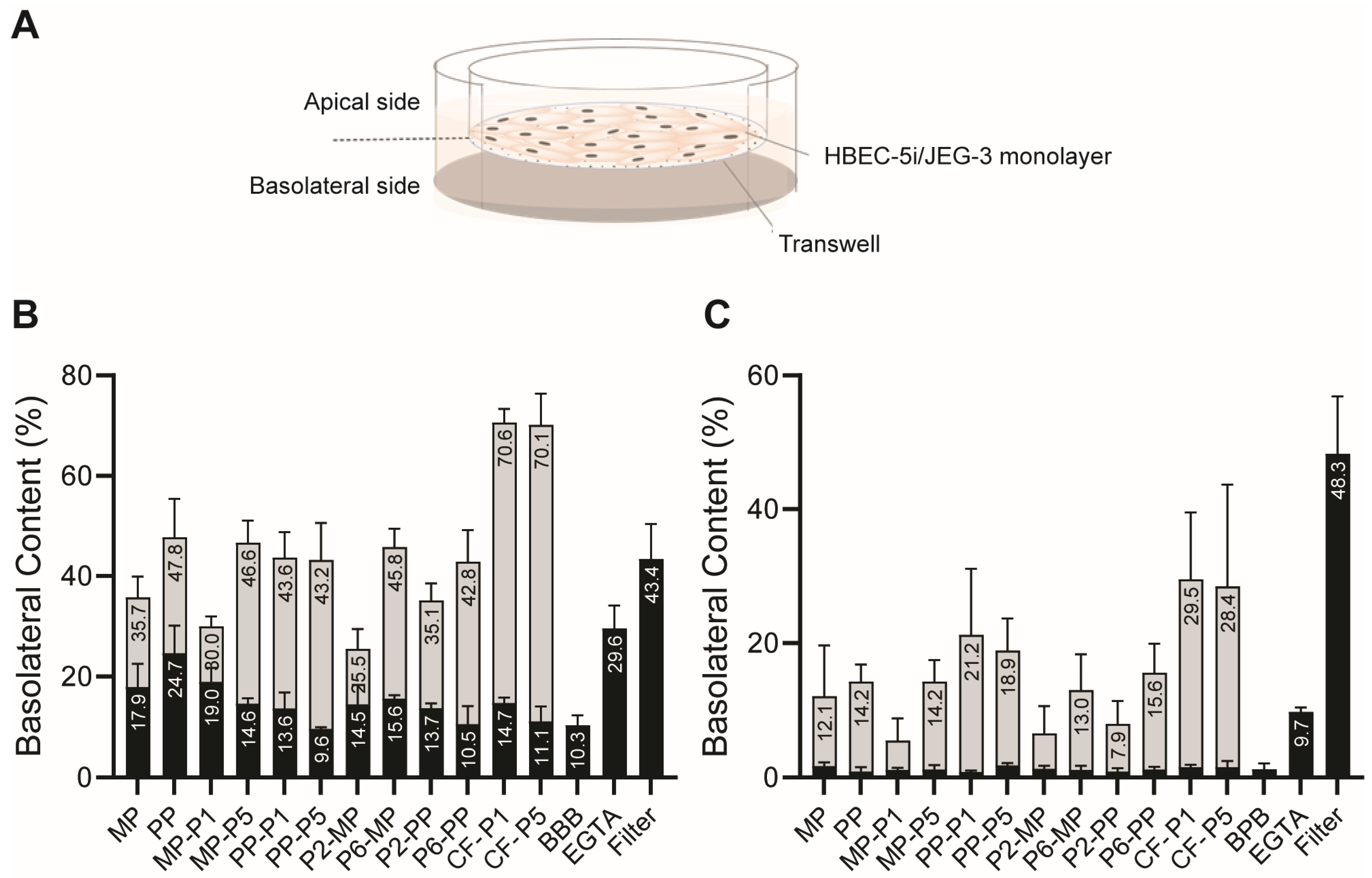
3.2. In Vitro BBB and BPB Translocation Assays
3.3. ZIKV Inactivation
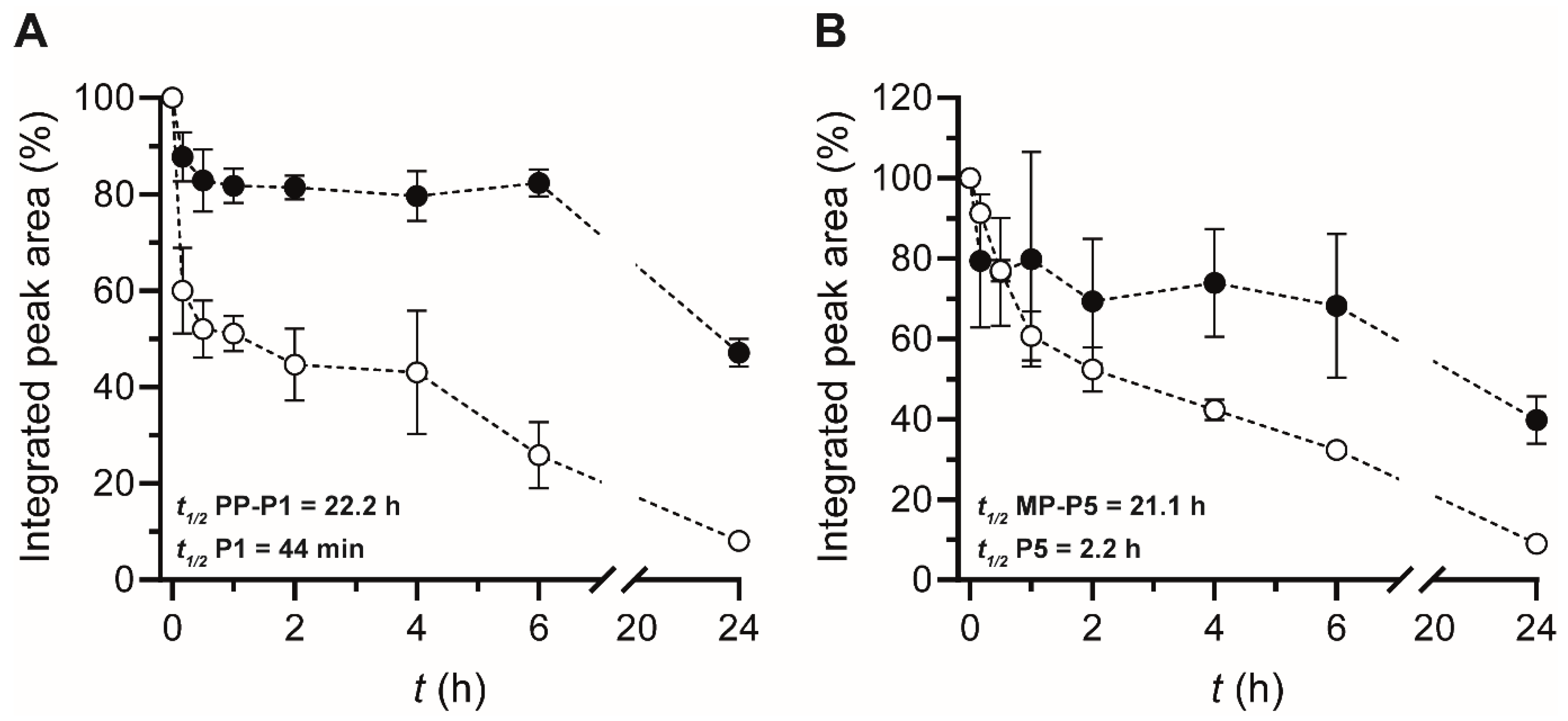
3.4. Serum Stability
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Malone, R.W.; Homan, J.; Callahan, M.V.; Glasspool-Malone, J.; Damodaran, L.; Schneider, A.D.B.; Zimler, R.; Talton, J.; Cobb, R.R.; Ruzic, I.; et al. Zika Virus: Medical Countermeasure Development Challenges. PLoS Negl. Trop. Dis. 2016, 10, e0004530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chimelli, L.; Melo, A.S.O.; Avvad-Portari, E.; Wiley, C.A.; Camacho, A.H.S.; Lopes, V.S.; Machado, H.N.; Andrade, C.V.; Dock, D.C.A.; Moreira, M.E.; et al. The spectrum of neuropathological changes associated with congenital Zika virus infection. Acta Neuropathol. 2017, 133, 983–999. [Google Scholar] [CrossRef] [PubMed]
- Mlakar, J.; Korva, M.; Tul, N.; Popović, M.; Poljšak-Prijatelj, M.; Mraz, J.; Kolenc, M.; Resman Rus, K.; Vesnaver Vipotnik, T.; Fabjan Vodušek, V.; et al. Zika Virus Associated with Microcephaly. N. Engl. J. Med. 2016, 374, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Parra, B.; Lizarazo, J.; Jiménez-Arango, J.A.; Zea-Vera, A.F.; González-Manrique, G.; Vargas, J.; Angarita, J.A.; Zuñiga, G.; Lopez-Gonzalez, R.; Beltran, C.L.; et al. Guillain–Barré Syndrome Associated with Zika Virus Infection in Colombia. N. Engl. J. Med. 2016, 375, 1513–1523. [Google Scholar] [CrossRef] [PubMed]
- Peiter, P.C.; Pereira, R.D.S.; Nunes Moreira, M.C.; Nascimento, M.; Tavares, M.d.F.L.; Franco, V.d.C.; Carvajal Cortês, J.J.; Campos, D.d.S.; Barcellos, C. Zika epidemic and microcephaly in Brazil: Challenges for access to health care and promotion in three epidemic areas. PLoS ONE 2020, 15, e0235010. [Google Scholar] [CrossRef]
- Niemeyer, B.; Niemeyer, R.; Borges, R.; Marchiori, E. Acute Disseminated Encephalomyelitis Following Zika Virus Infection. Eur. Neurol. 2017, 77, 45–46. [Google Scholar] [CrossRef]
- WHO and Experts Prioritize Vaccines, Diagnostics and Innovative Vector Control Tools for Zika R&D. Available online: https://www.who.int/news/item/09-03-2016-who-and-experts-prioritize-vaccines-diagnostics-and-innovative-vector-control-tools-for-zika-r-d (accessed on 19 December 2021).
- Pattnaik, A.; Sahoo, B.R.; Pattnaik, A.K. Current Status of Zika Virus Vaccines: Successes and Challenges. Vaccines 2020, 8, 266. [Google Scholar] [CrossRef] [PubMed]
- Waltz, E. First genetically modified mosquitoes released in the United States. Nature 2021, 593, 175–176. [Google Scholar] [CrossRef] [PubMed]
- Albulescu, I.C.; Kovacikova, K.; Tas, A.; Snijder, E.J.; van Hemert, M.J. Suramin inhibits Zika virus replication by interfering with virus attachment and release of infectious particles. Antivir. Res. 2017, 143, 230–236. [Google Scholar] [CrossRef] [Green Version]
- Sacramento, C.Q.; De Melo, G.R.; De Freitas, C.S.; Rocha, N.; Hoelz, L.V.B.; Miranda, M.; Fintelman-Rodrigues, N.; Marttorelli, A.; Ferreira, A.C.; Barbosa-Lima, G.; et al. The clinically approved antiviral drug sofosbuvir inhibits Zika virus replication. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Demeule, M.; Regina, A.; Ché, C.; Poirier, J.; Nguyen, T.; Gabathuler, R.; Castaigne, J.P.; Béliveau, R. Identification and design of peptides as a new drug delivery system for the brain. J. Pharmacol. Exp. Ther. 2008, 324, 1064–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malakoutikhah, M.; Teixidó, M.; Giralt, E. Toward an optimal blood-brain barrier shuttle by synthesis and evaluation of peptide libraries. J. Med. Chem. 2008, 51, 4881–4889. [Google Scholar] [CrossRef] [PubMed]
- Malakoutikhah, M.; Guixer, B.; Arranz-Gibert, P.; Teixidó, M.; Giralt, E. ‘À la Carte’ Peptide Shuttles: Tools to Increase Their Passage across the Blood-Brain Barrier. ChemMedChem 2014, 9, 1594–1601. [Google Scholar] [CrossRef] [PubMed]
- Oller-Salvia, B.; Sánchez-Navarro, M.; Giralt, E.; Teixidó, M. Blood-brain barrier shuttle peptides: An emerging paradigm for brain delivery. Chem. Soc. Rev. 2016, 45, 4690–4707. [Google Scholar] [CrossRef] [Green Version]
- Pardridge, W.M. Receptor-mediated peptide transport through the blood-brain barrier. Endocr. Rev. 1986, 7, 314–330. [Google Scholar] [CrossRef]
- Cavaco, M.; Frutos, S.; Oliete, P.; Valle, J.; Andreu, D.; Castanho, M.A.R.B.; Vila-Perelló, M.; Neves, V. Conjugation of a Blood Brain Barrier Peptide Shuttle to an Fc Domain for Brain Delivery of Therapeutic Biomolecules. ACS Med. Chem. Lett. 2021, 12, 1663–1668. [Google Scholar] [CrossRef]
- Zhang, B.; Sun, X.; Mei, H.; Wang, Y.; Liao, Z.; Chen, J.; Zhang, Q.; Hu, Y.; Pang, Z.; Jiang, X. LDLR-mediated peptide-22-conjugated nanoparticles for dual-targeting therapy of brain glioma. Biomaterials 2013, 34, 9171–9182. [Google Scholar] [CrossRef]
- Banks, W.A.; Kastin, A.J. Peptides and the blood-brain barrier: Lipophilicity as a predictor of permeability. Brain Res. Bull. 1985, 15, 287–292. [Google Scholar] [CrossRef]
- Kumar, P.; Wu, H.; McBride, J.L.; Jung, K.E.; Hee Kim, M.; Davidson, B.L.; Kyung Lee, S.; Shankar, P.; Manjunath, N. Transvascular delivery of small interfering RNA to the central nervous system. Nature 2007, 448, 39–43. [Google Scholar] [CrossRef]
- Neves, V.; Aires-Da-Silva, F.; Morais, M.; Gano, L.; Ribeiro, E.; Pinto, A.; Aguiar, S.; Gaspar, D.; Fernandes, C.; Correia, J.D.G.; et al. Novel Peptides Derived from Dengue Virus Capsid Protein Translocate Reversibly the Blood-Brain Barrier through a Receptor-Free Mechanism. ACS Chem. Biol. 2017, 12, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Neves-Coelho, S.; Eleutério, R.P.; Enguita, F.J.; Neves, V.; Castanho, M.A.R.B. A new noncanonical anionic peptide that translocates a cellular blood-brain barrier model. Molecules 2017, 22, 1753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendonça, D.A.; Bakker, M.; Cruz-Oliveira, C.; Neves, V.; Jiménez, M.A.; Defaus, S.; Cavaco, M.; Veiga, A.S.; Cadima-Couto, I.; Castanho, M.A.R.B.; et al. Penetrating the Blood-Brain Barrier with New Peptide–Porphyrin Conjugates Having anti-HIV Activity. Bioconjug. Chem. 2021, 32, 1067–1077. [Google Scholar] [CrossRef]
- Sakuma, Y.; Baba, R.; Arita, K.; Morimoto, H.; Fujita, M. Food allergens are transferred intact across the rat blood-placental barrier in vivo. Med. Mol. Morphol. 2014, 47, 14–20. [Google Scholar] [CrossRef]
- Neris, R.L.S.; Figueiredo, C.M.; Higa, L.M.; Araujo, D.F.; Carvalho, C.A.M.; Verçoza, B.R.F.; Silva, M.O.L.; Carneiro, F.A.; Tanuri, A.; Gomes, A.M.O.; et al. Co-protoporphyrin IX and Sn-protoporphyrin IX inactivate Zika, Chikungunya and other arboviruses by targeting the viral envelope. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Delcroix, M.; Riley, L.W. Cell-penetrating peptides for antiviral drug development. Pharmaceuticals 2010, 3, 448–470. [Google Scholar] [CrossRef] [Green Version]
- Li, S.Y.; Cheng, H.; Qiu, W.X.; Liu, L.H.; Chen, S.; Hu, Y.; Xie, B.R.; Li, B.; Zhang, X.Z. Protease-Activable Cell-Penetrating Peptide-Protoporphyrin Conjugate for Targeted Photodynamic Therapy in Vivo. ACS Appl. Mater. Interfaces 2015, 7, 28319–28329. [Google Scholar] [CrossRef]
- Dondi, R.; Yaghini, E.; Tewari, K.M.; Wang, L.; Giuntini, F.; Loizidou, M.; MacRobert, A.J.; Eggleston, I.M. Flexible synthesis of cationic peptide-porphyrin derivatives for light-triggered drug delivery and photodynamic therapy. Org. Biomol. Chem. 2016, 14, 11488–11501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebedeva, N.S.; Gubarev, Y.A.; Koifman, M.O.; Koifman, O.I. The Application of Porphyrins and Their Analogues for Inactivation of Viruses. Molecules 2020, 25, 4368. [Google Scholar] [CrossRef] [PubMed]
- Biscaglia, F.; Gobbo, M. Porphyrin–peptide conjugates in biomedical applications. Pept. Sci. 2018, 110, e24038. [Google Scholar] [CrossRef]
- Assunção-Miranda, I.; Cruz-Oliveira, C.; Neris, R.L.S.; Figueiredo, C.M.; Pereira, L.P.S.; Rodrigues, D.; Araujo, D.F.F.; Da Poian, A.T.; Bozza, M.T. Inactivation of Dengue and Yellow Fever viruses by heme, cobalt-protoporphyrin IX and tin-protoporphyrin IX. J. Appl. Microbiol. 2016, 120, 790–804. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Oliveira, C.; Almeida, A.F.; Freire, J.M.; Caruso, M.B.; Morando, M.A.; Ferreira, V.N.S.; Assunção-Miranda, I.; Gomes, A.M.O.; Castanho, M.A.R.B.; Da Poian, A.T. Mechanisms of Vesicular Stomatitis Virus Inactivation by Protoporphyrin IX, Zinc-Protoporphyrin IX, and Mesoporphyrin IX. Antimicrob. Agents Chemother. 2017, 61, e00053-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eigenmann, D.E.; Jähne, E.A.; Smieško, M.; Hamburger, M.; Oufir, M. Validation of an immortalized human (hBMEC) in vitro blood-brain barrier model. Anal. Bioanal. Chem. 2016, 408, 2095–2107. [Google Scholar] [CrossRef]
- Jayant, R.; Atluri, V.; Agudelo, M.; Sagar, V.; Kaushik, A.; Nair, M. Sustained-release nanoART formulation for the treatment of neuroAIDS. Int. J. Nanomed. 2015, 10, 1077. [Google Scholar] [CrossRef] [Green Version]
- Yin, T.; Yang, L.; Liu, Y.; Zhou, X.; Sun, J.; Liu, J. Sialic acid (SA)-modified selenium nanoparticles coated with a high blood-brain barrier permeability peptide-B6 peptide for potential use in Alzheimer’s disease. Acta Biomater. 2015, 25, 172–183. [Google Scholar] [CrossRef]
- Niewoehner, J.; Bohrmann, B.; Collin, L.; Urich, E.; Sade, H.; Maier, P.; Rueger, P.; Stracke, J.O.; Lau, W.; Tissot, A.C.; et al. Increased Brain Penetration and Potency of a Therapeutic Antibody Using a Monovalent Molecular Shuttle. Neuron 2014, 81, 49–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothbauer, M.; Patel, N.; Gondola, H.; Siwetz, M.; Huppertz, B.; Ertl, P. A comparative study of five physiological key parameters between four different human trophoblast-derived cell lines. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Syme, M.R.; Paxton, J.W.; Keelan, J.A. Drug transfer and metabolism by the human placenta. Clin. Pharmacokinet. 2004, 43, 487–514. [Google Scholar] [CrossRef] [PubMed]
- Schneider, H.; Panigel, M.; Dancis, J. Transfer across the perfused human placenta of antipyrine, sodium, and leucine. Am. J. Obstet. Gynecol. 1972, 114, 822–828. [Google Scholar] [CrossRef]
- Cavaco, M.; Valle, J.; da Silva, R.; Correia, J.D.G.; Castanho, M.A.R.B.; Andreu, D.; Neves, V. D PepH3, an Improved Peptide Shuttle for Receptor-independent Transport Across the Blood-Brain Barrier. Curr. Pharm. Des. 2020, 26, 1495–1506. [Google Scholar] [CrossRef]
- Gallo, M.; Moreno, E.; Defaus, S.; Ortega-Alvaro, A.; Gonzalez, A.; Robledo, P.; Cavaco, M.; Neves, V.; Castanho, M.A.R.B.; Casadó, V.; et al. Orally Active Peptide Vector Allows Using Cannabis to Fight Pain while Avoiding Side Effects. J. Med. Chem. 2021, 64, 6937–6948. [Google Scholar] [CrossRef] [PubMed]
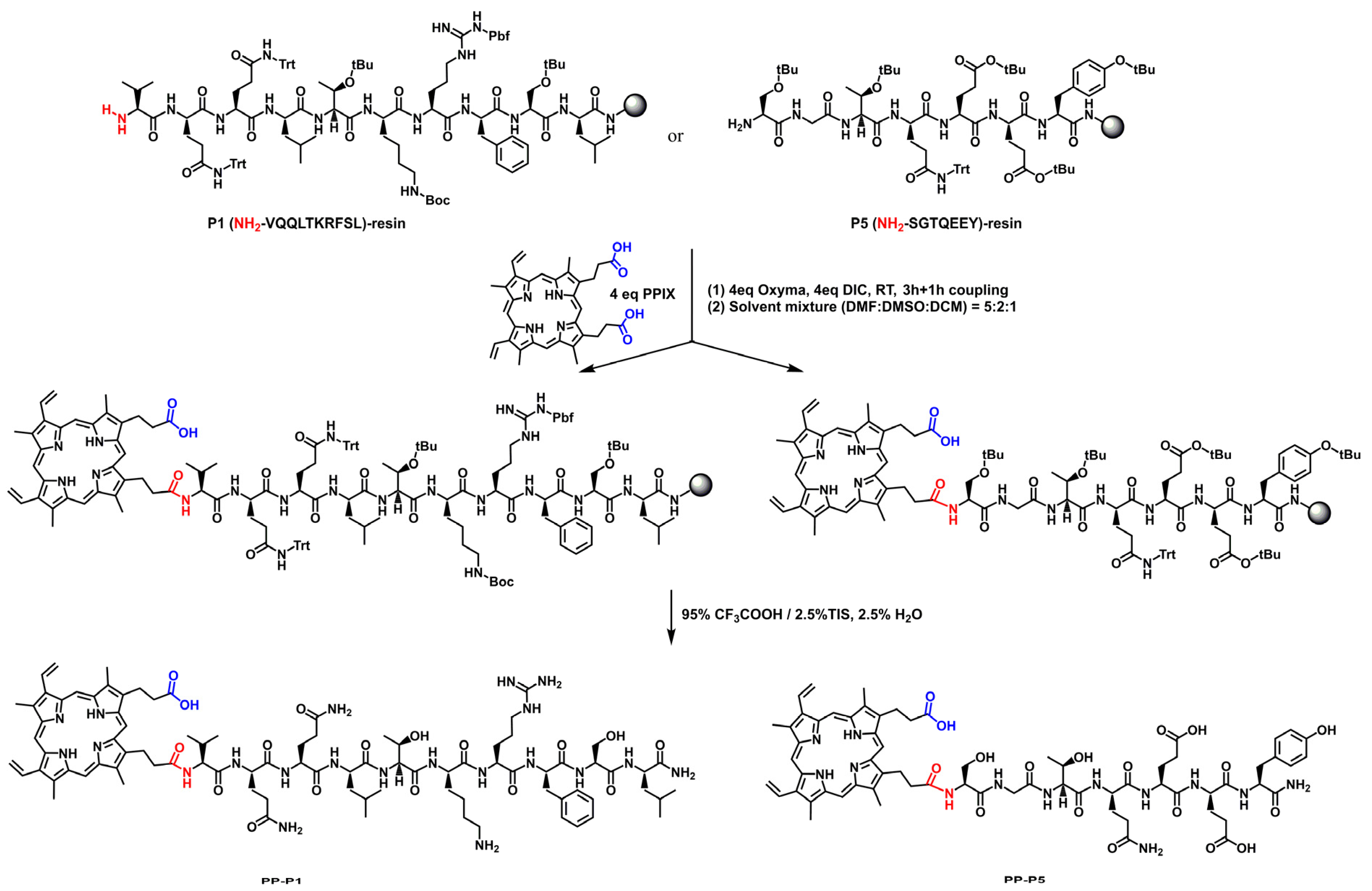

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abbreviation | Structure | [M + H+]+ |
|---|---|---|
| MP-P1 | 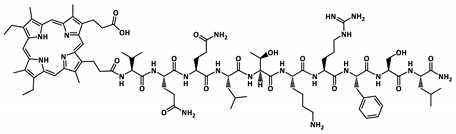 | 1767.0 |
| PP-P1 | 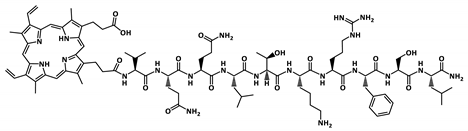 | 1762.9 |
| P2-MP | 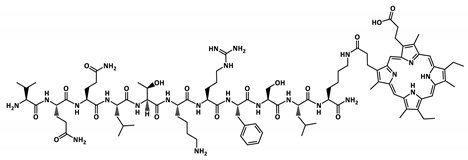 | 1895.1 |
| P2-PP | 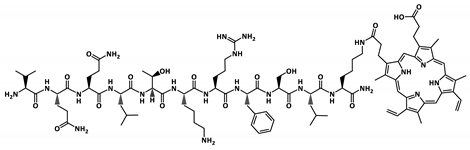 | 1891.1 |
| MP-P5 |  | 1360.6 |
| PP-P5 | 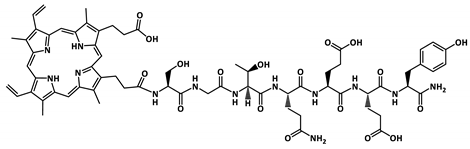 | 1356.6 |
| P6-MP |  | 1488.7 |
| P6-PP |  | 1484.7 |
| IC50 (µM) | ||||
|---|---|---|---|---|
| Virus | MP | PP | MP-P5 | PP-P1 |
| ZIKV | >50 | >50 | 25.07 ± 0.05 | 1.08 ± 0.14 |
| HIV a | >50 | >50 | 33.1 ± 1.38 | >50 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Todorovski, T.; Mendonça, D.A.; Fernandes-Siqueira, L.O.; Cruz-Oliveira, C.; Guida, G.; Valle, J.; Cavaco, M.; Limas, F.I.V.; Neves, V.; Cadima-Couto, Í.; et al. Targeting Zika Virus with New Brain- and Placenta-Crossing Peptide–Porphyrin Conjugates. Pharmaceutics 2022, 14, 738. https://doi.org/10.3390/pharmaceutics14040738
Todorovski T, Mendonça DA, Fernandes-Siqueira LO, Cruz-Oliveira C, Guida G, Valle J, Cavaco M, Limas FIV, Neves V, Cadima-Couto Í, et al. Targeting Zika Virus with New Brain- and Placenta-Crossing Peptide–Porphyrin Conjugates. Pharmaceutics. 2022; 14(4):738. https://doi.org/10.3390/pharmaceutics14040738
Chicago/Turabian StyleTodorovski, Toni, Diogo A. Mendonça, Lorena O. Fernandes-Siqueira, Christine Cruz-Oliveira, Giuseppina Guida, Javier Valle, Marco Cavaco, Fernanda I. V. Limas, Vera Neves, Íris Cadima-Couto, and et al. 2022. "Targeting Zika Virus with New Brain- and Placenta-Crossing Peptide–Porphyrin Conjugates" Pharmaceutics 14, no. 4: 738. https://doi.org/10.3390/pharmaceutics14040738
APA StyleTodorovski, T., Mendonça, D. A., Fernandes-Siqueira, L. O., Cruz-Oliveira, C., Guida, G., Valle, J., Cavaco, M., Limas, F. I. V., Neves, V., Cadima-Couto, Í., Defaus, S., Veiga, A. S., Da Poian, A. T., Castanho, M. A. R. B., & Andreu, D. (2022). Targeting Zika Virus with New Brain- and Placenta-Crossing Peptide–Porphyrin Conjugates. Pharmaceutics, 14(4), 738. https://doi.org/10.3390/pharmaceutics14040738