
Biological Evaluation of New Thienopyridinium and Thienopyrimidinium Derivatives as Human Choline Kinase Inhibitors

 , , , ,
, , , ,  , ,
, ,  , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
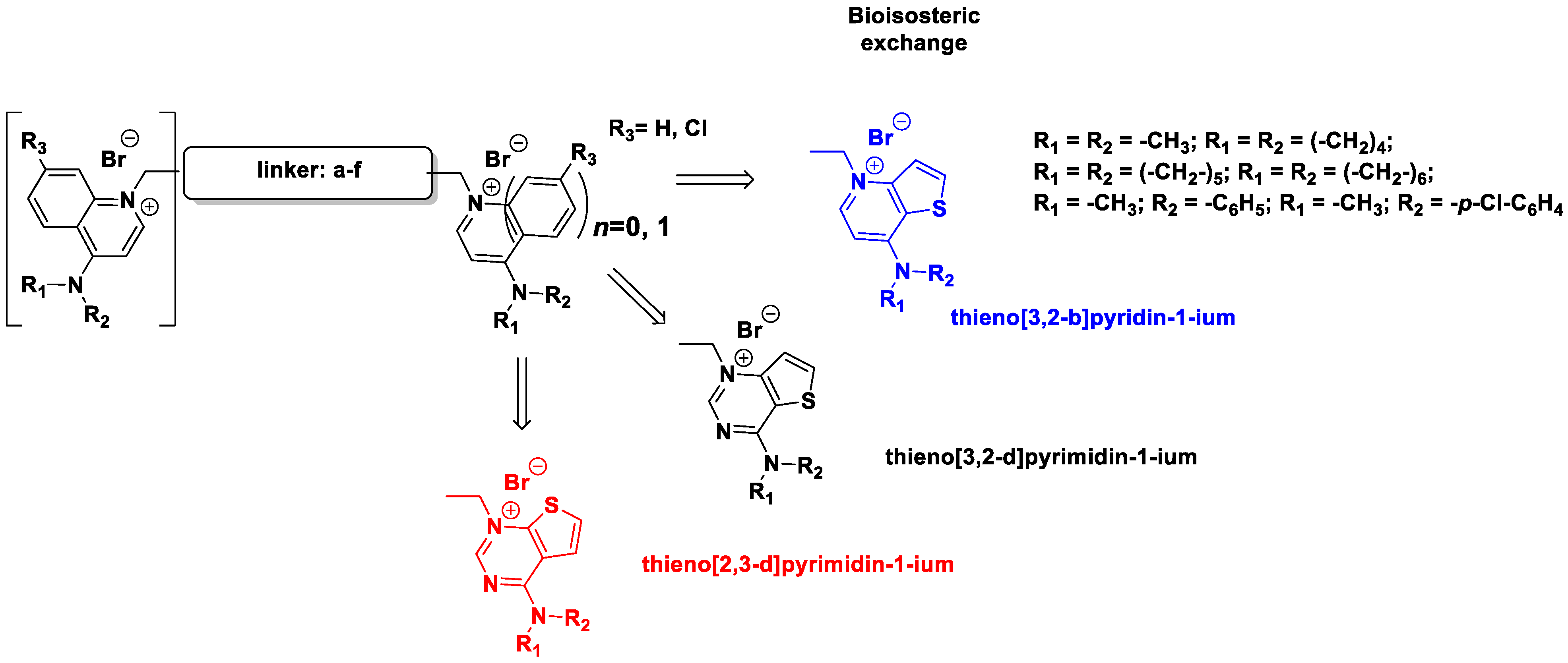
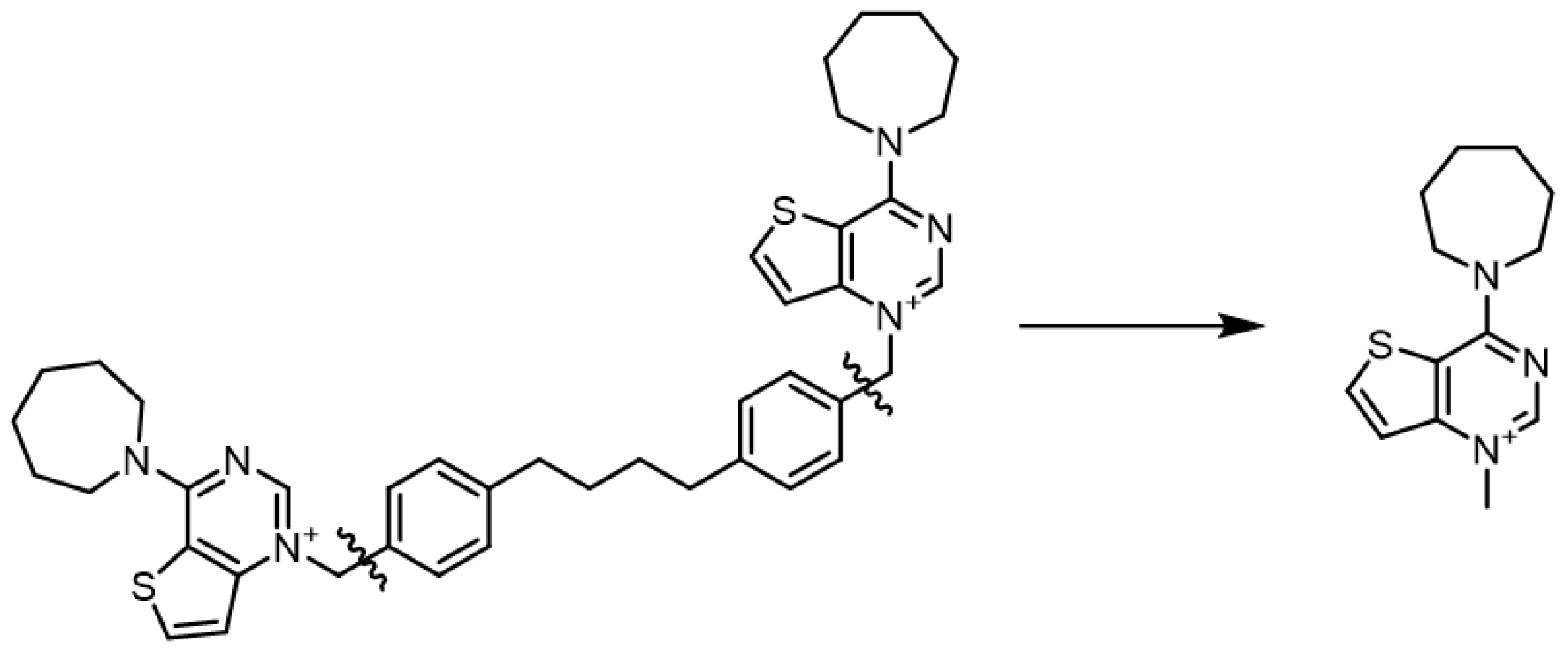
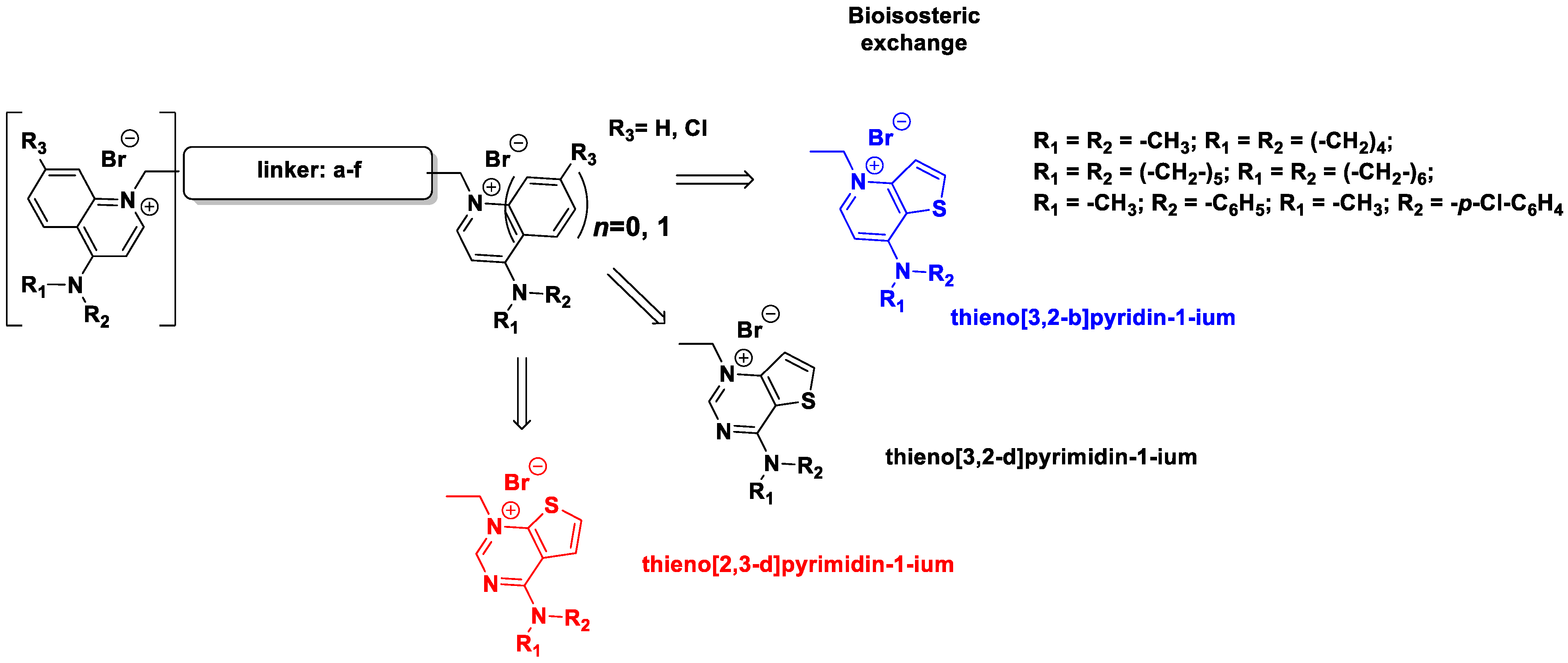
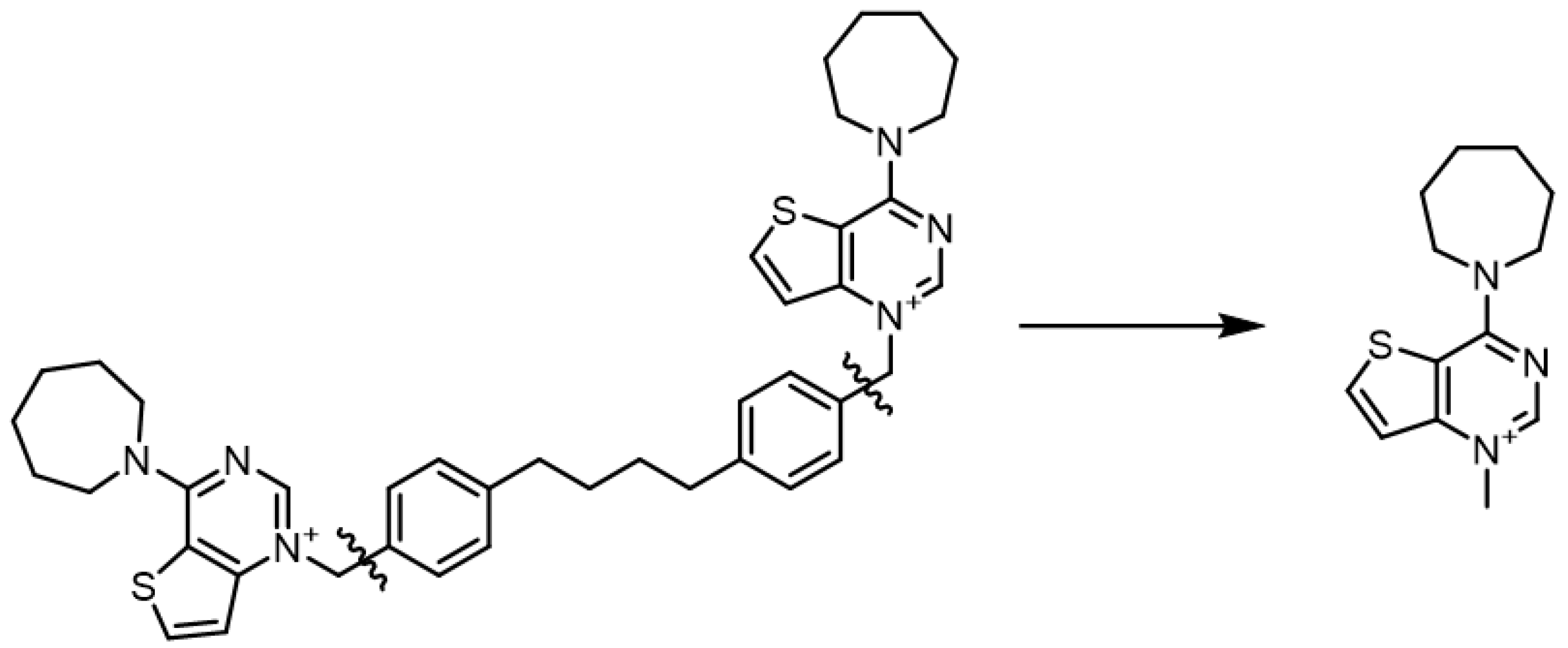
2.1. Chemistry
2.2. Cloning, Protein Expression, and Purification of CK
2.3. Choline Kinase Assay
2.4. Docking Calculations
2.5. DFT Calculations
2.6. Antiproliferative Activity
2.7. Choline Uptake Assay
2.8. Cell Cycle Analysis
2.9. Measurement of Apoptosis by Flow Cytometry
2.10. Predicted Parameters Related to the ADME (Absorption, Distribution, Metabolism, and Excretion) and PAINS (Pan Assay Interferences Structures)
3. Results and Discussion
3.1. Biological Assays
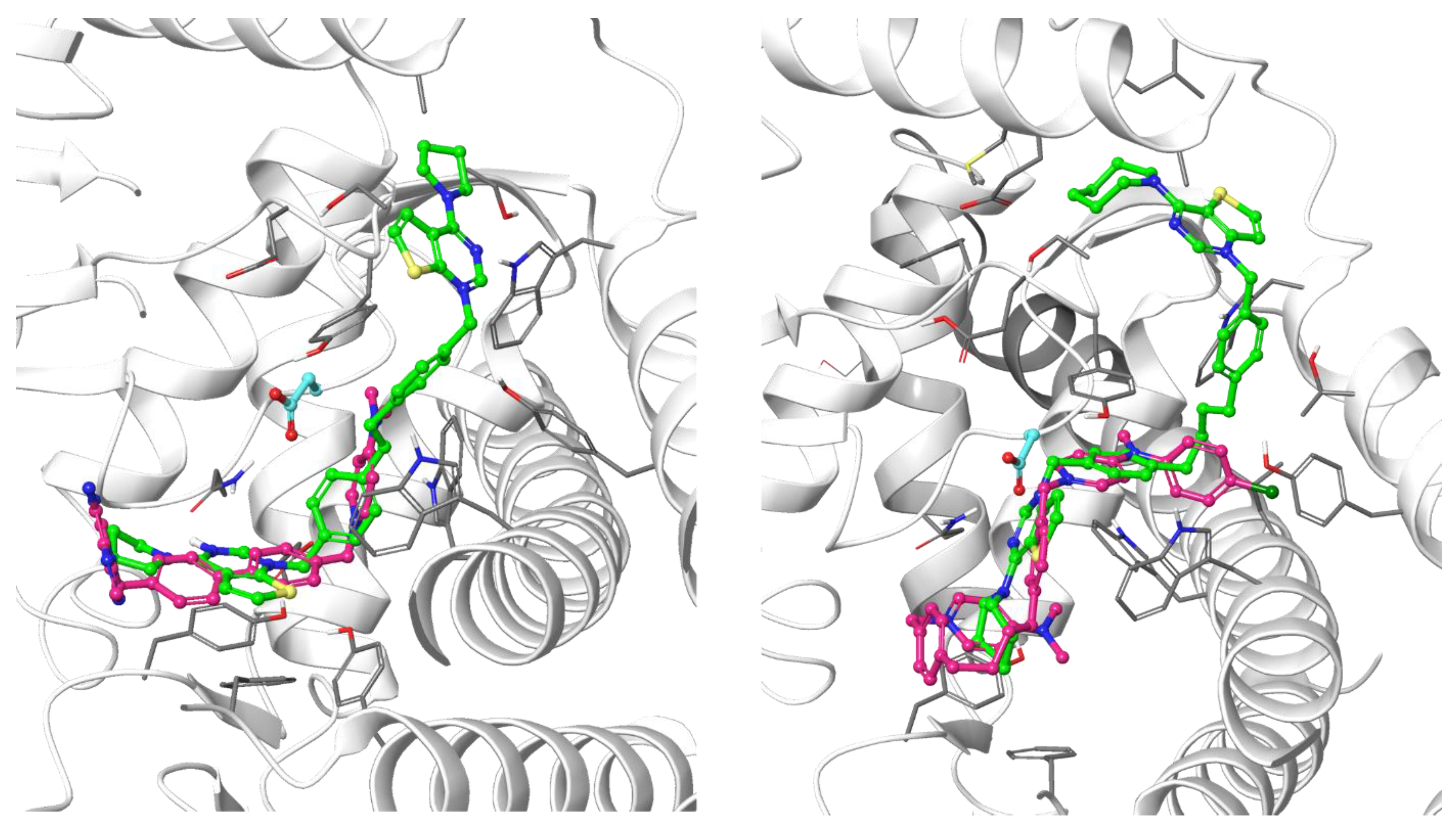
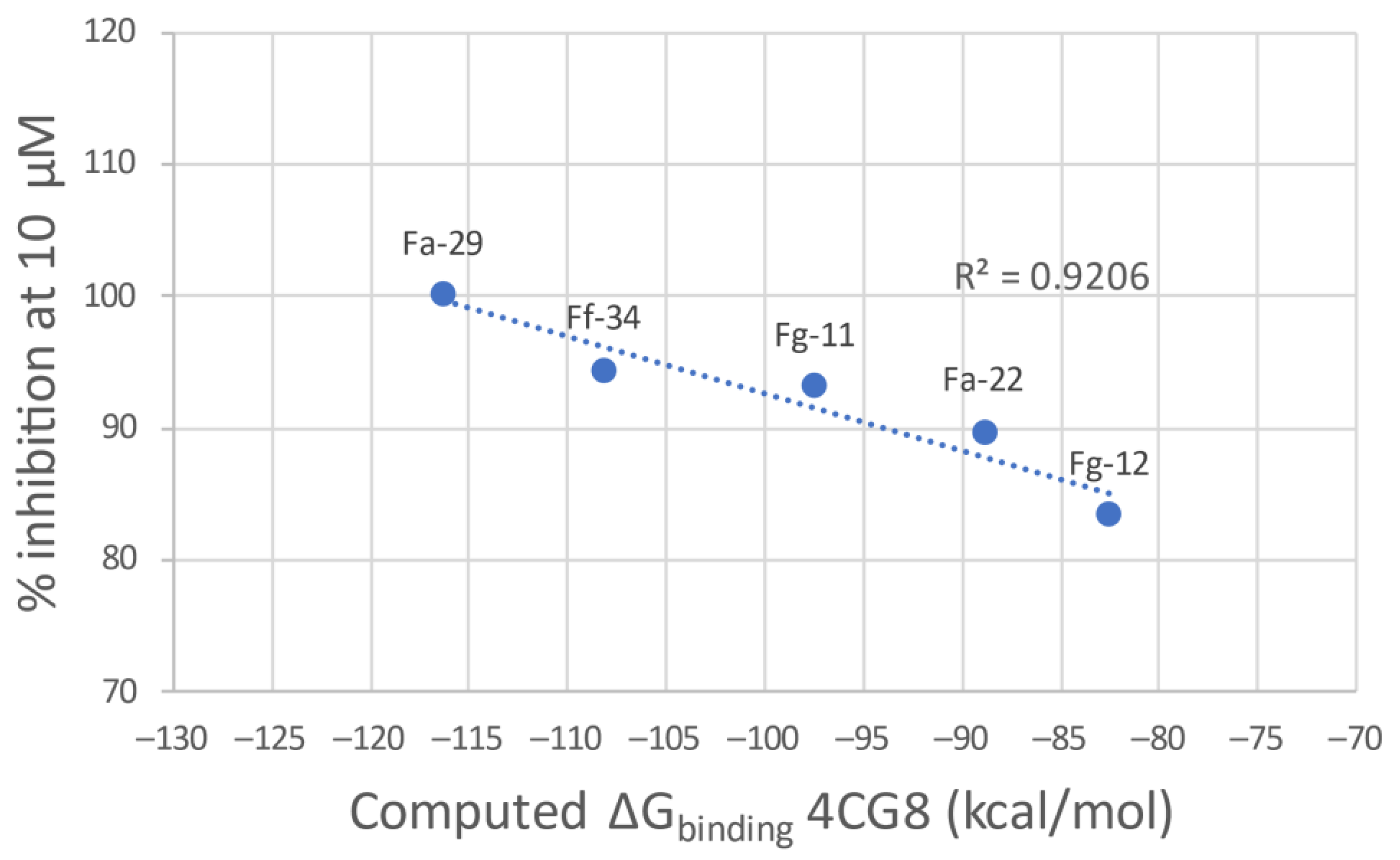
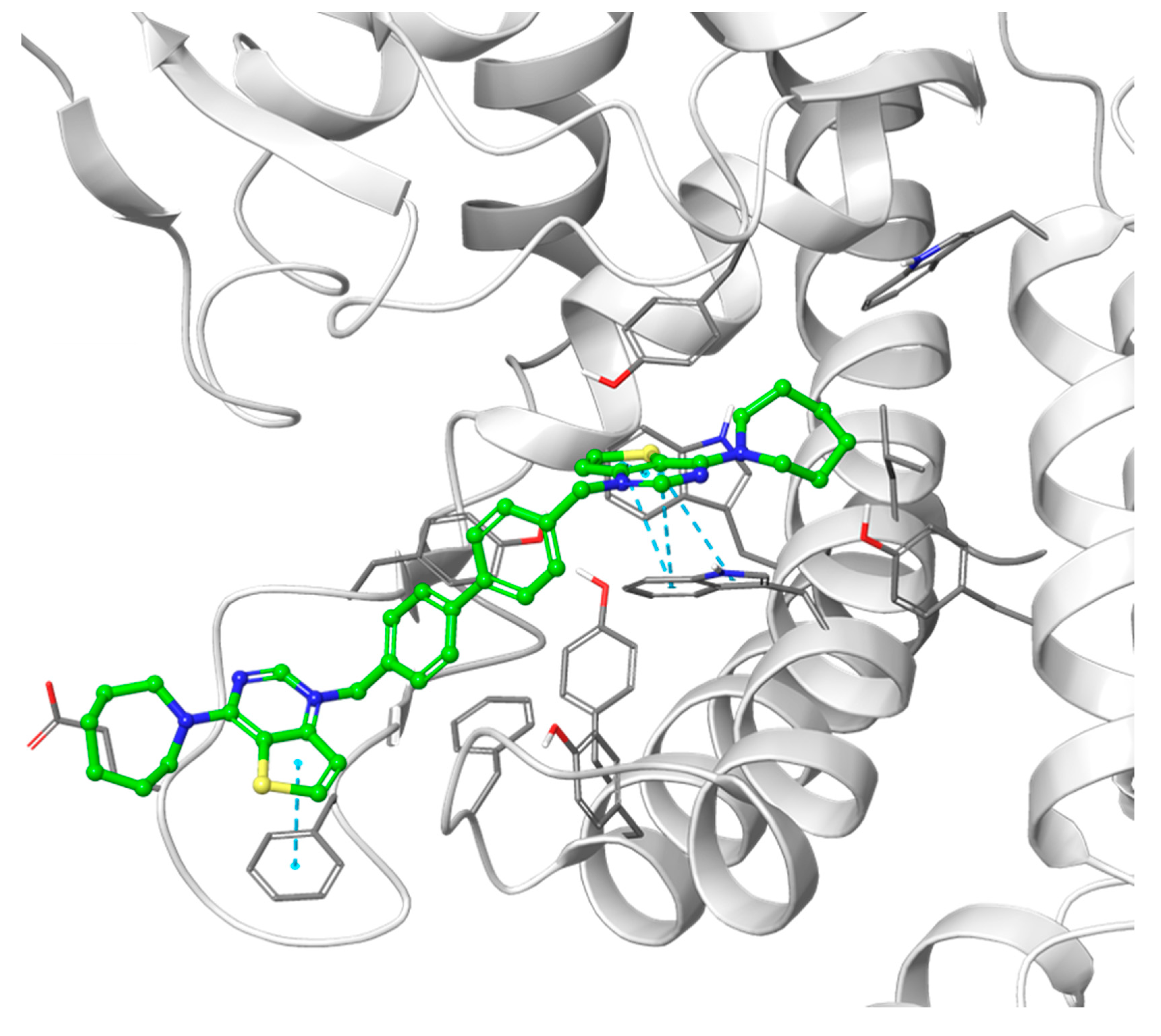
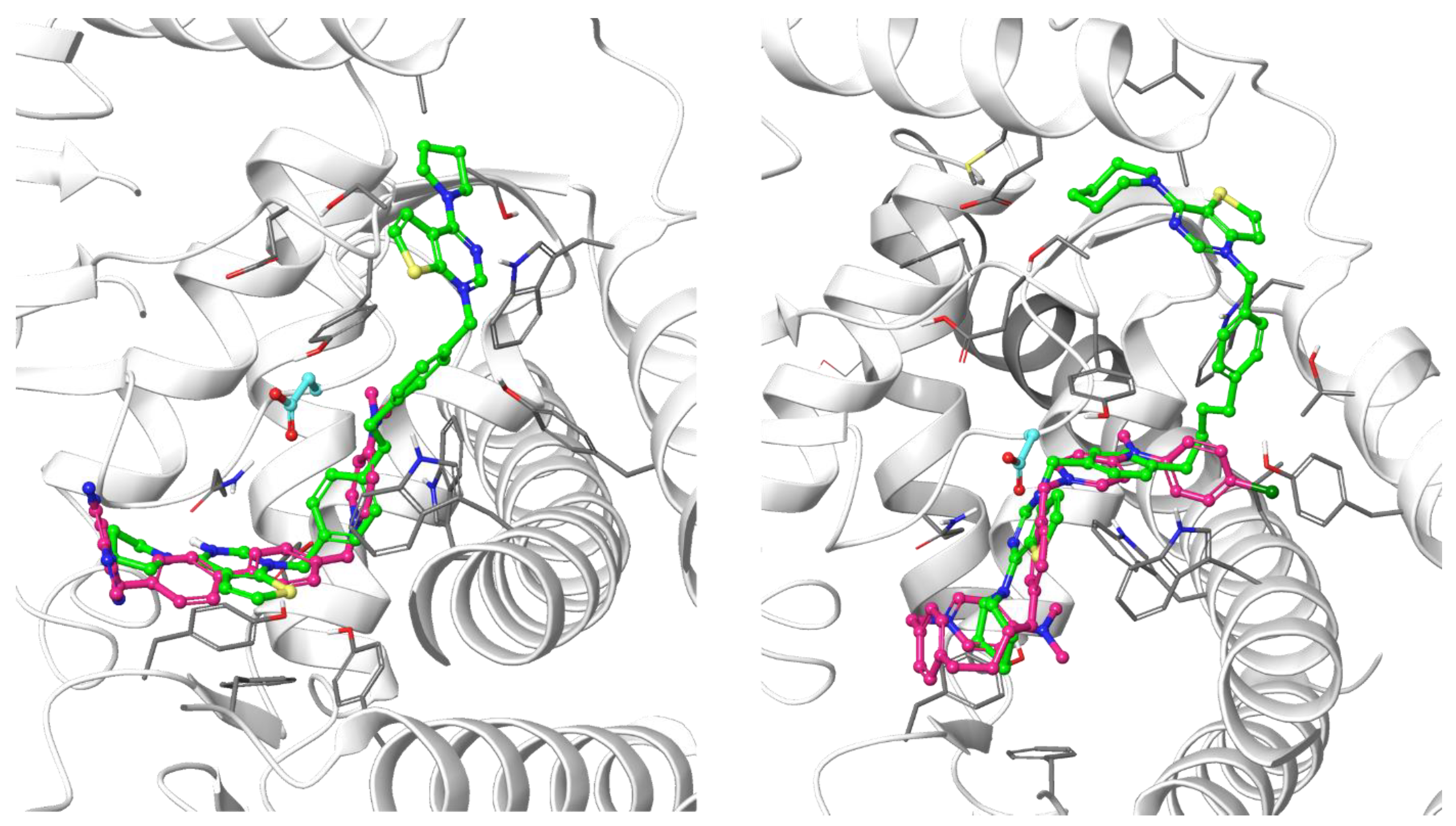
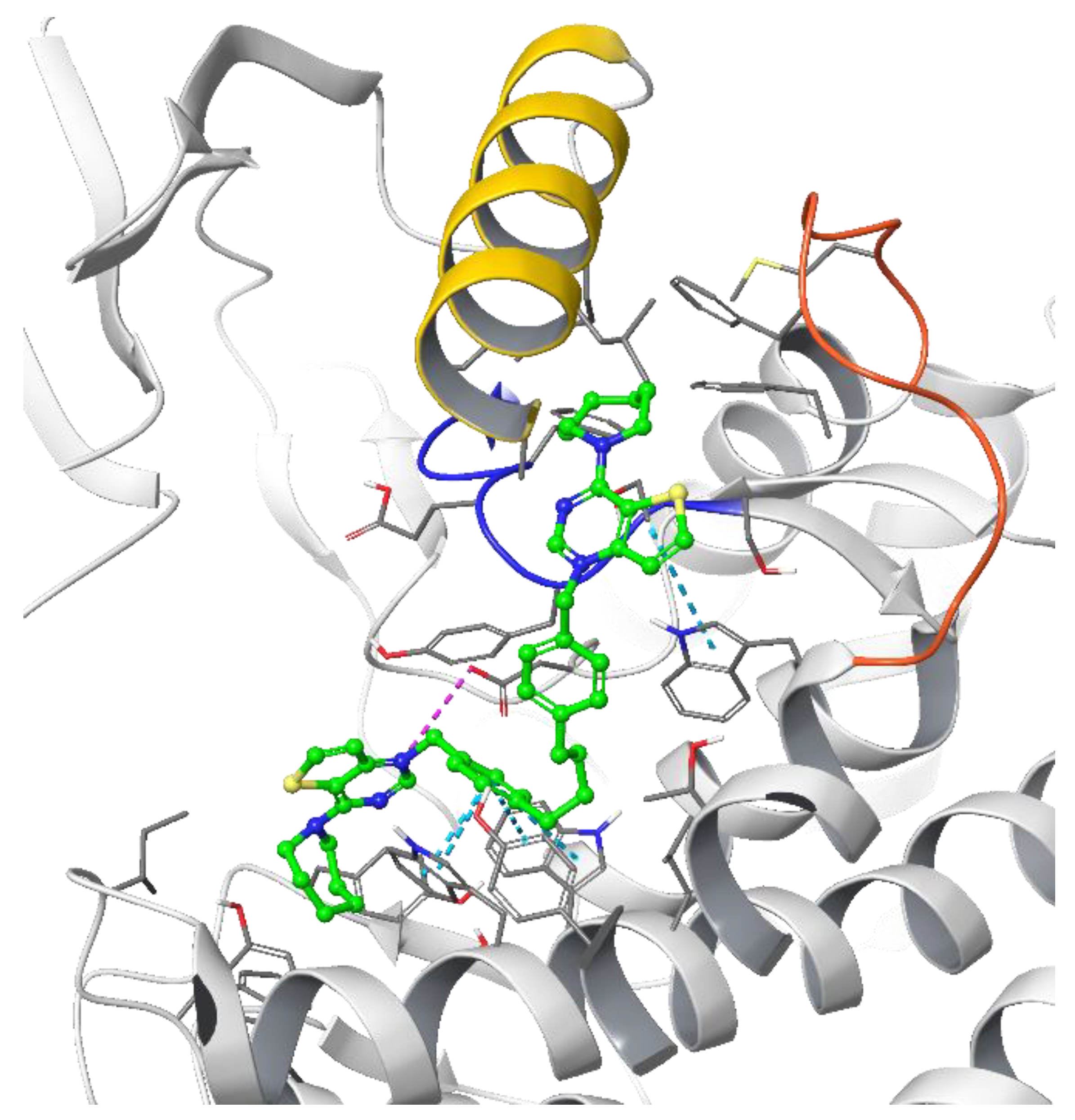
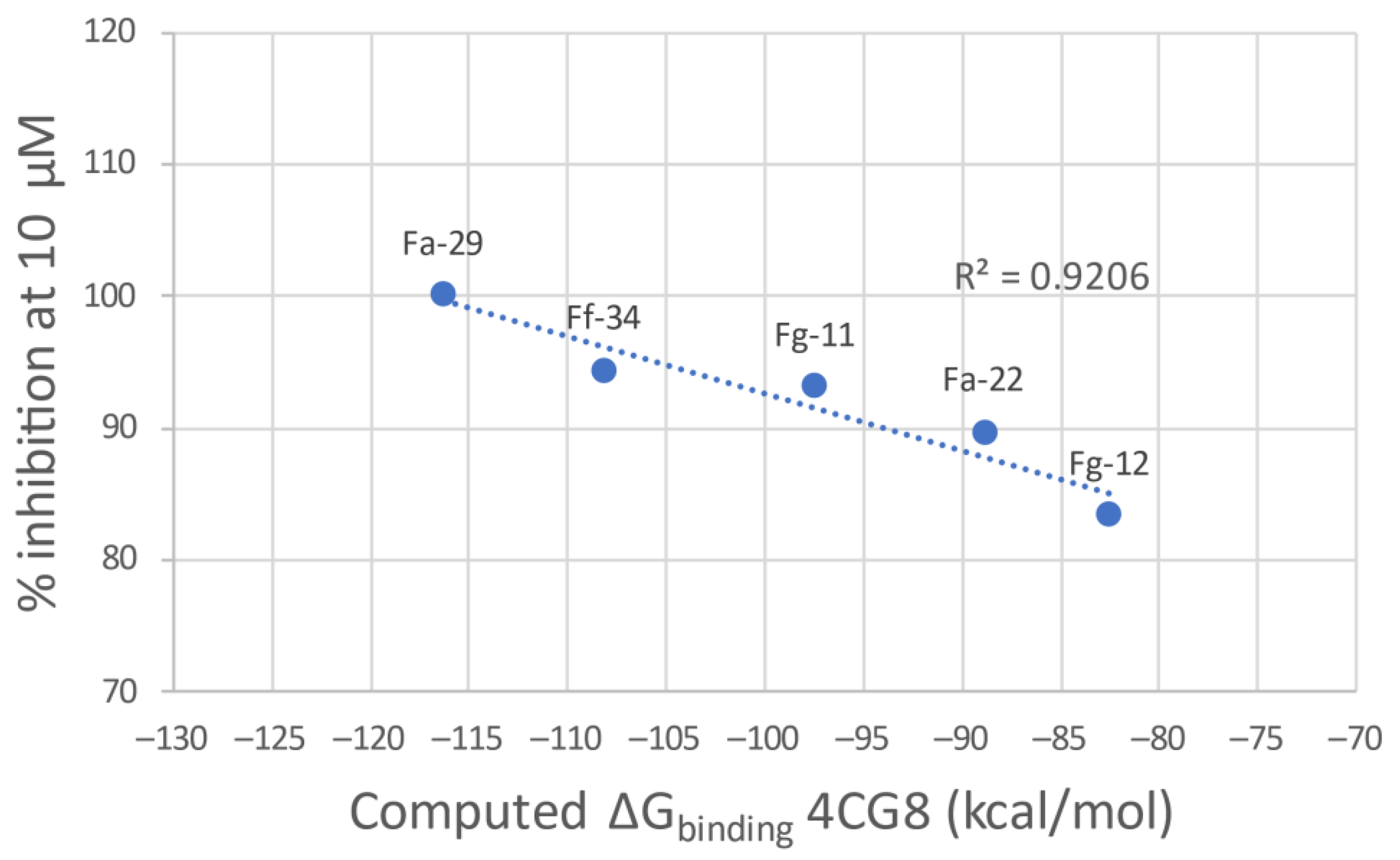
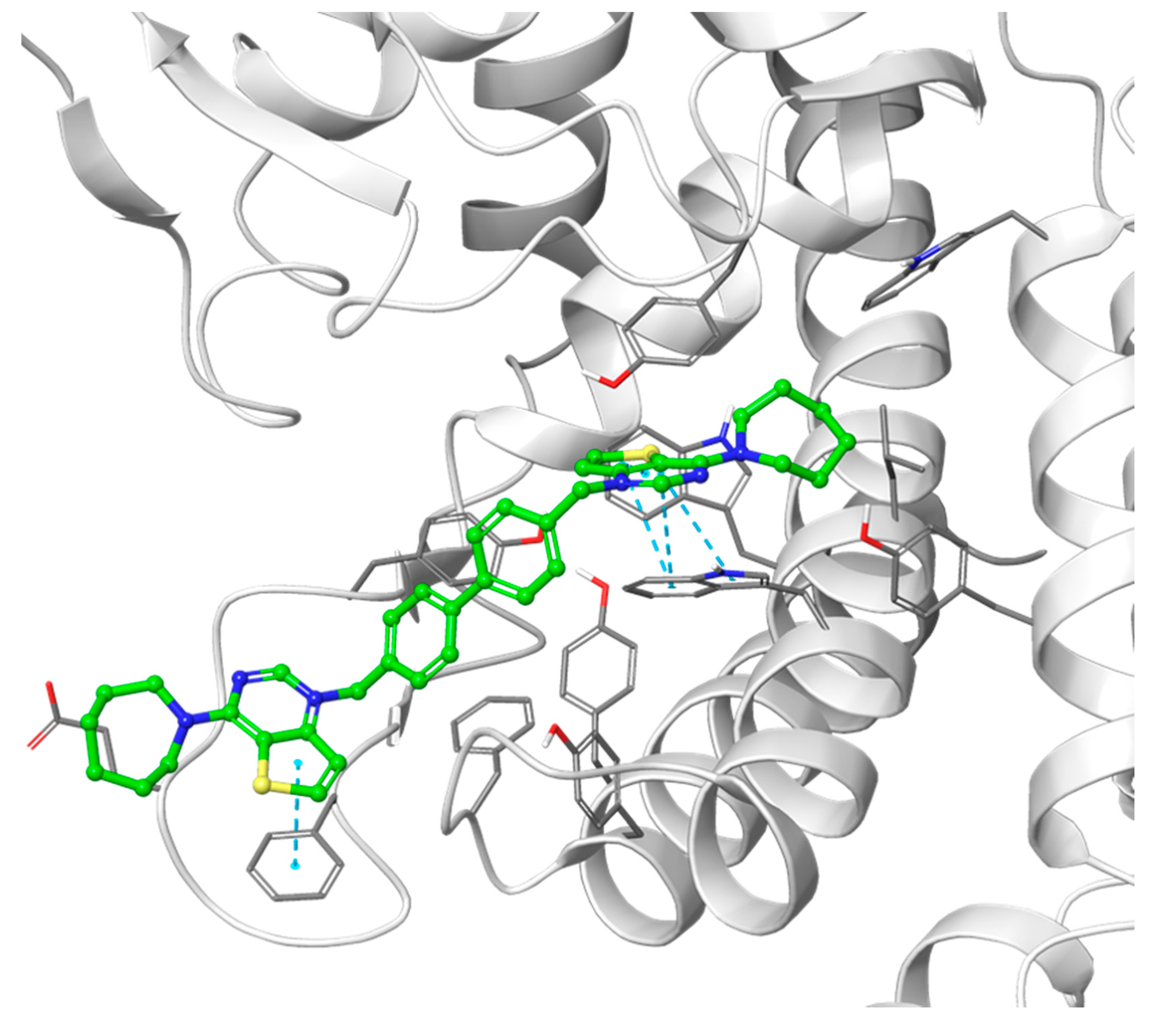
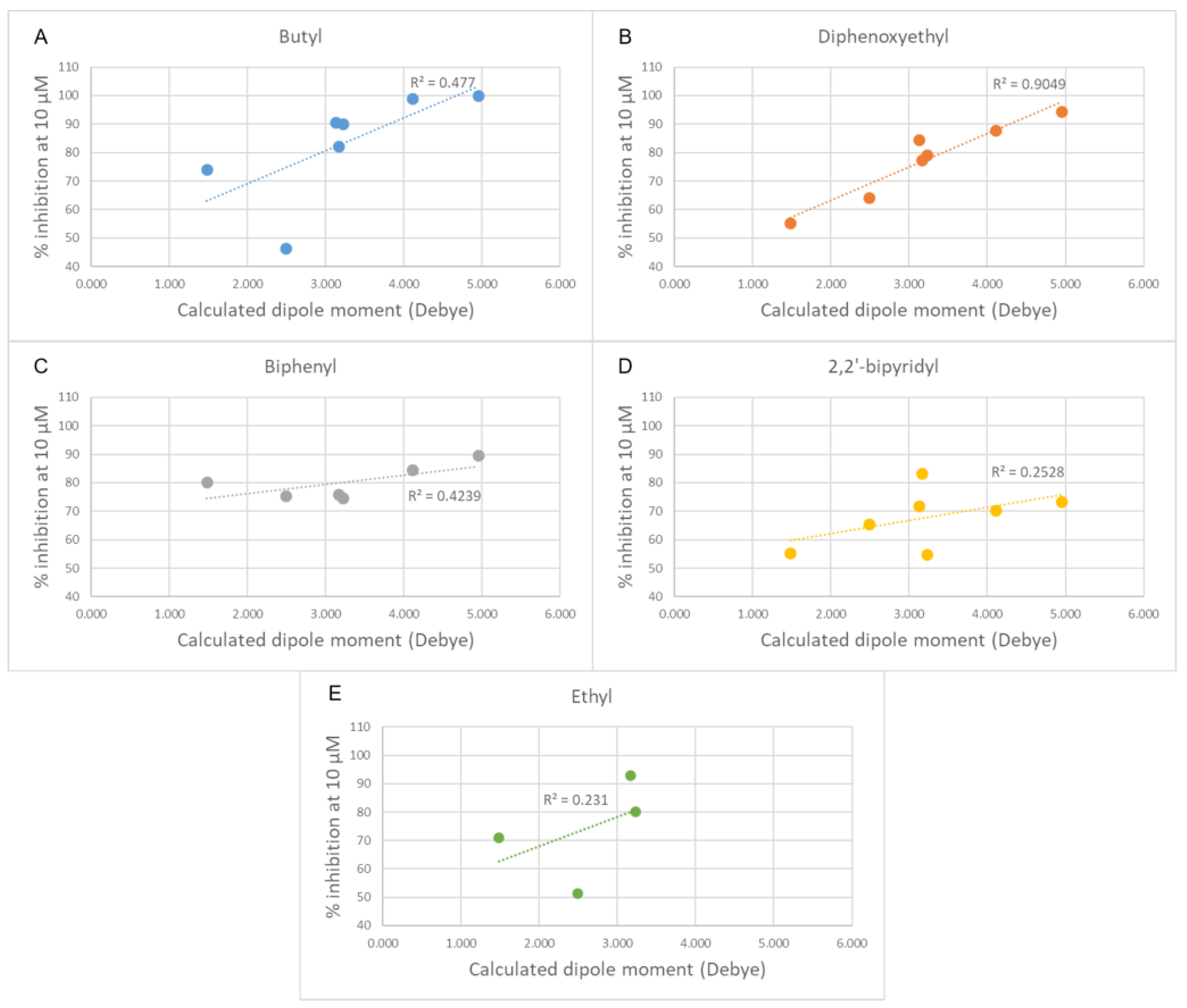
3.1.1. Docking Results
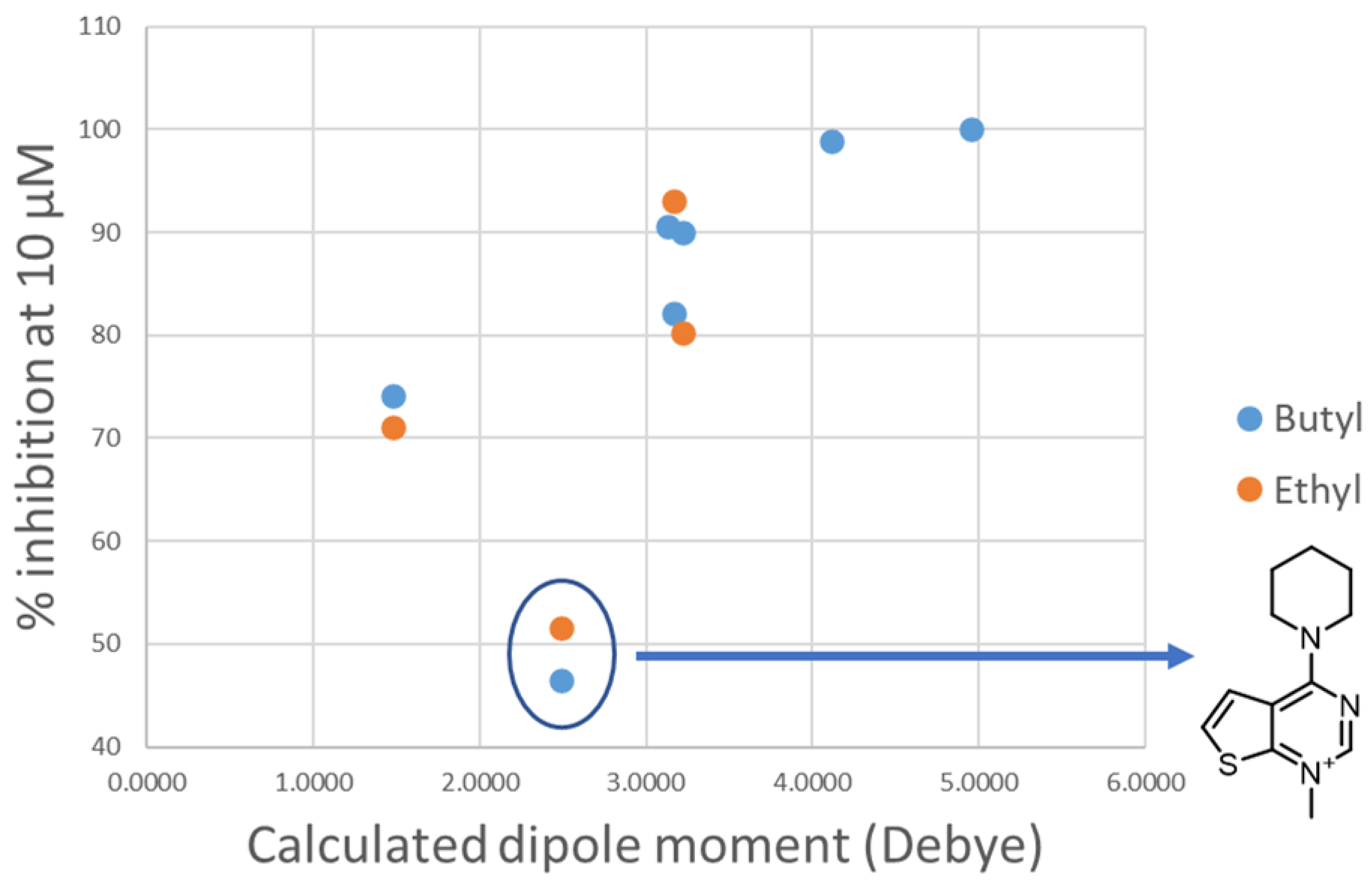
- Decreasing the cycloalkyl ring size (from azepane to piperidine to pyrrolidine) could reduce the occupancy of the hydrophobic pockets found in the choline binding site and near the choline binding motif.
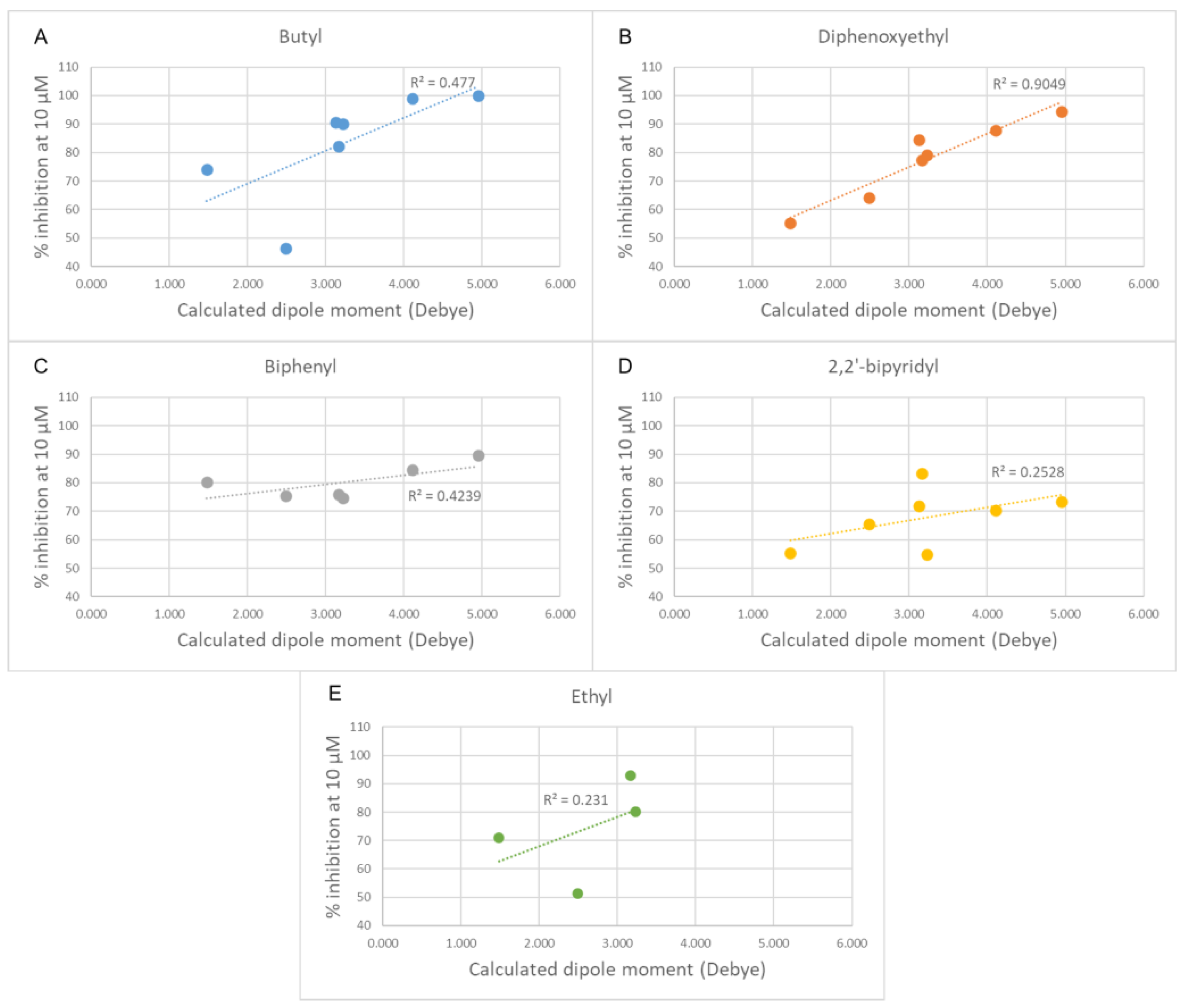
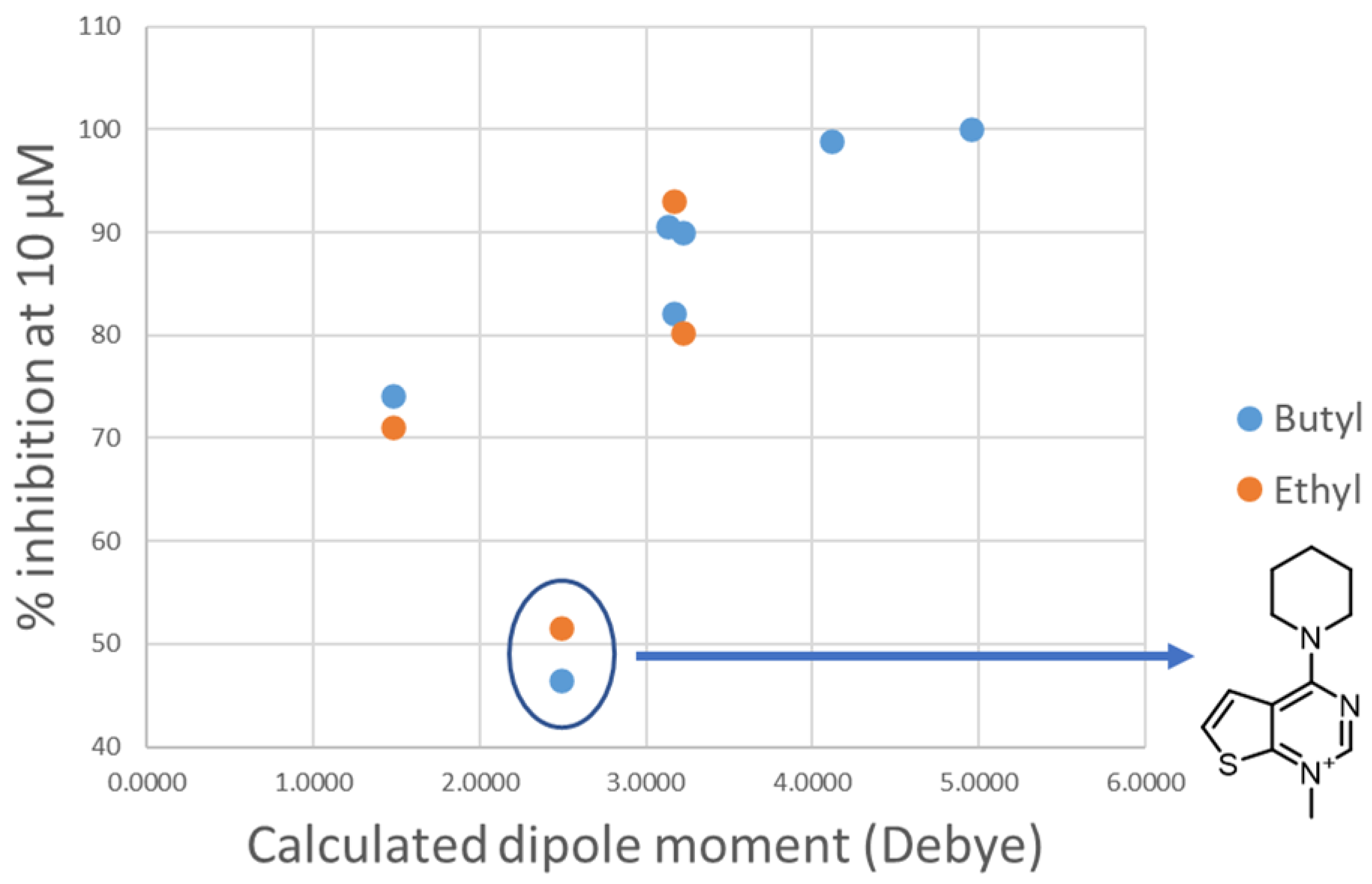
- Changing the butyl linker to a diphenoxyethyl one could have three distinct effects on the binding pose: i. The linker could become less flexible due to the Hydrogen Bond Acceptor (HBA) nature of the added oxygen atoms, restricting the linker in a less elongated conformation; ii. The delocalization of the oxygen lone pair into the aromatic ring could increase the energy barrier for the rotation around the Caromatic-O bond; iii. The increased electron density in the phenyl rings could weaken the π-π interactions with the electron-rich side chains of the Trp and Tyr residues due to higher electrostatic repulsion.
- Shortening the linker from a length of 4 carbon atoms to 2 (butyl to ethyl linker) can lead to a not optimal position for the cationic head, which would not reach the secondary pocket.
3.1.2. Antiproliferative Activity and Inhibition of Choline Uptake
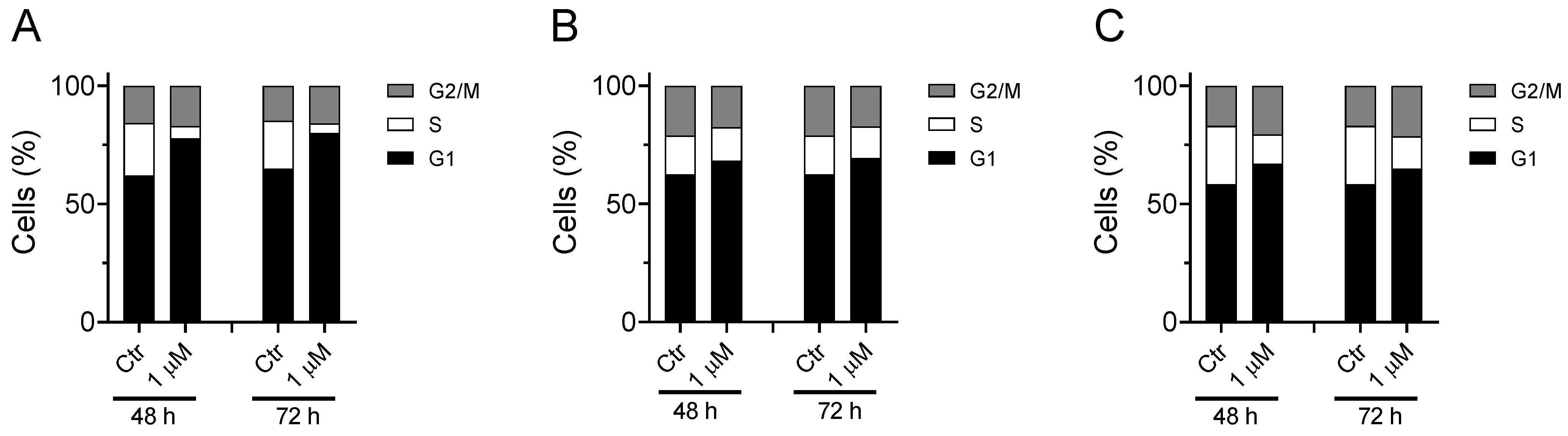
3.1.3. Cell Cycle Analysis
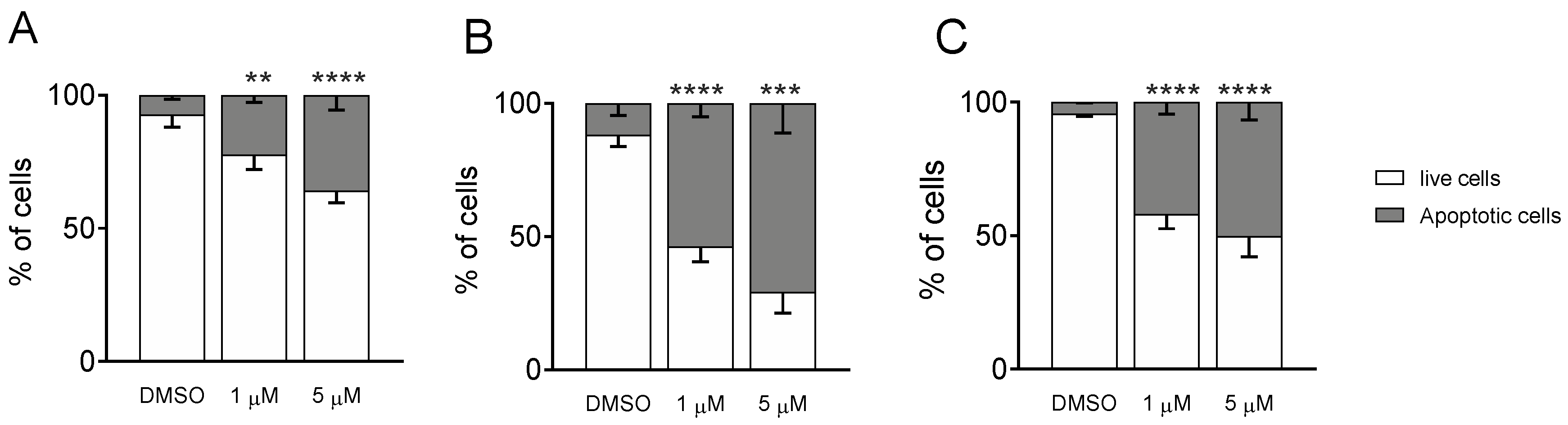
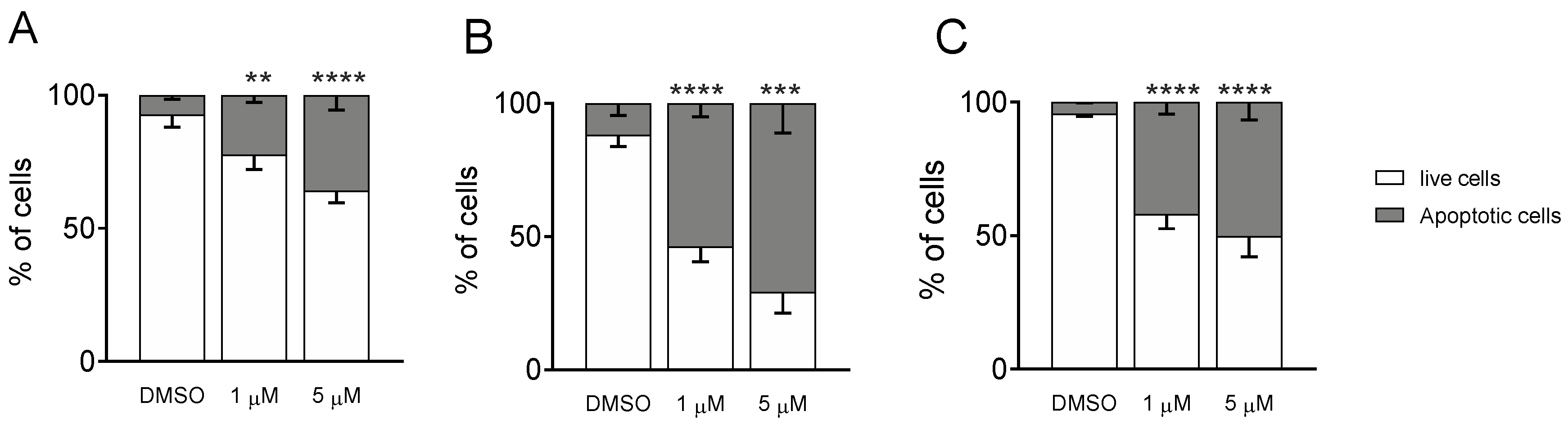
3.1.4. Measurement of Apoptosis by Flow Cytometry
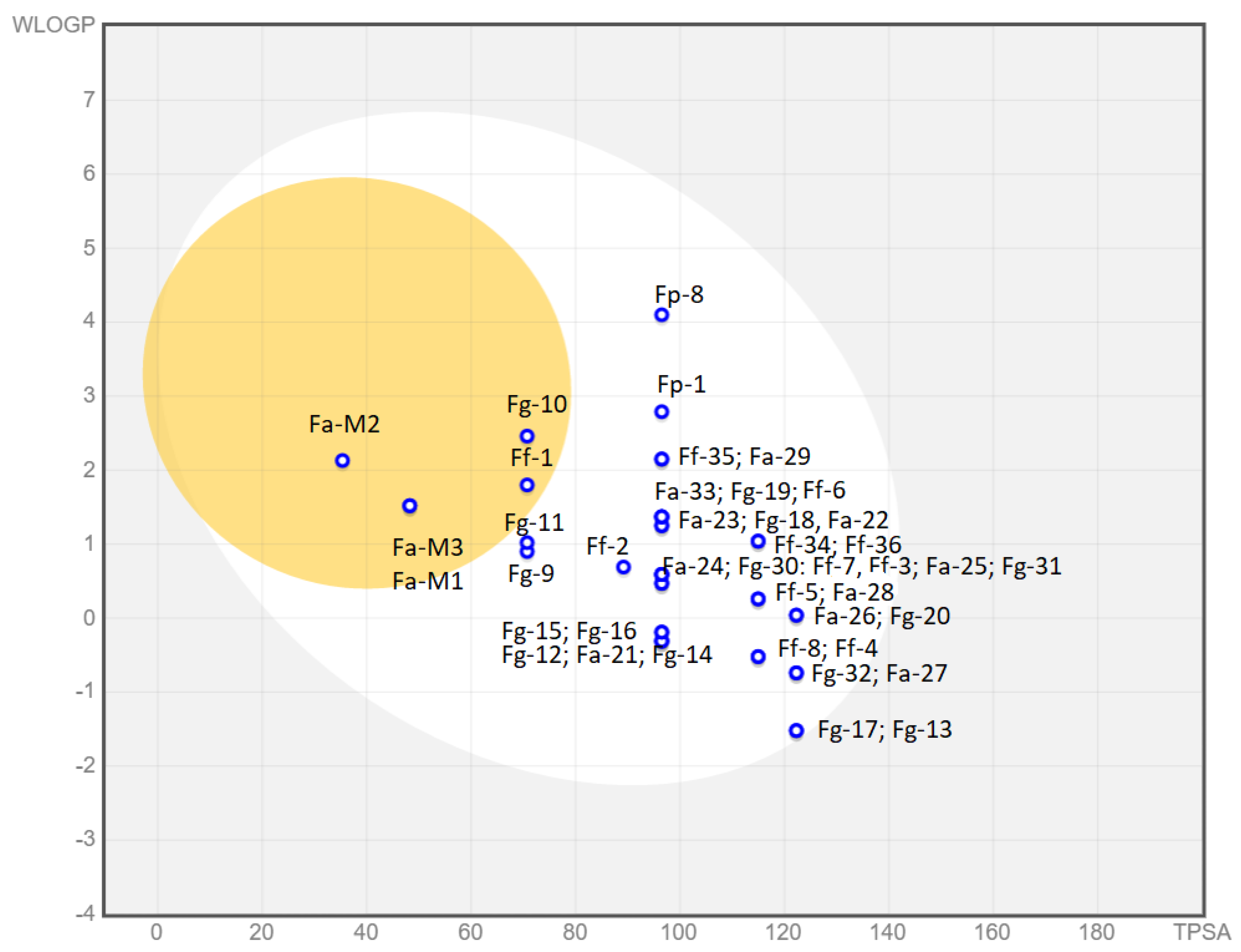
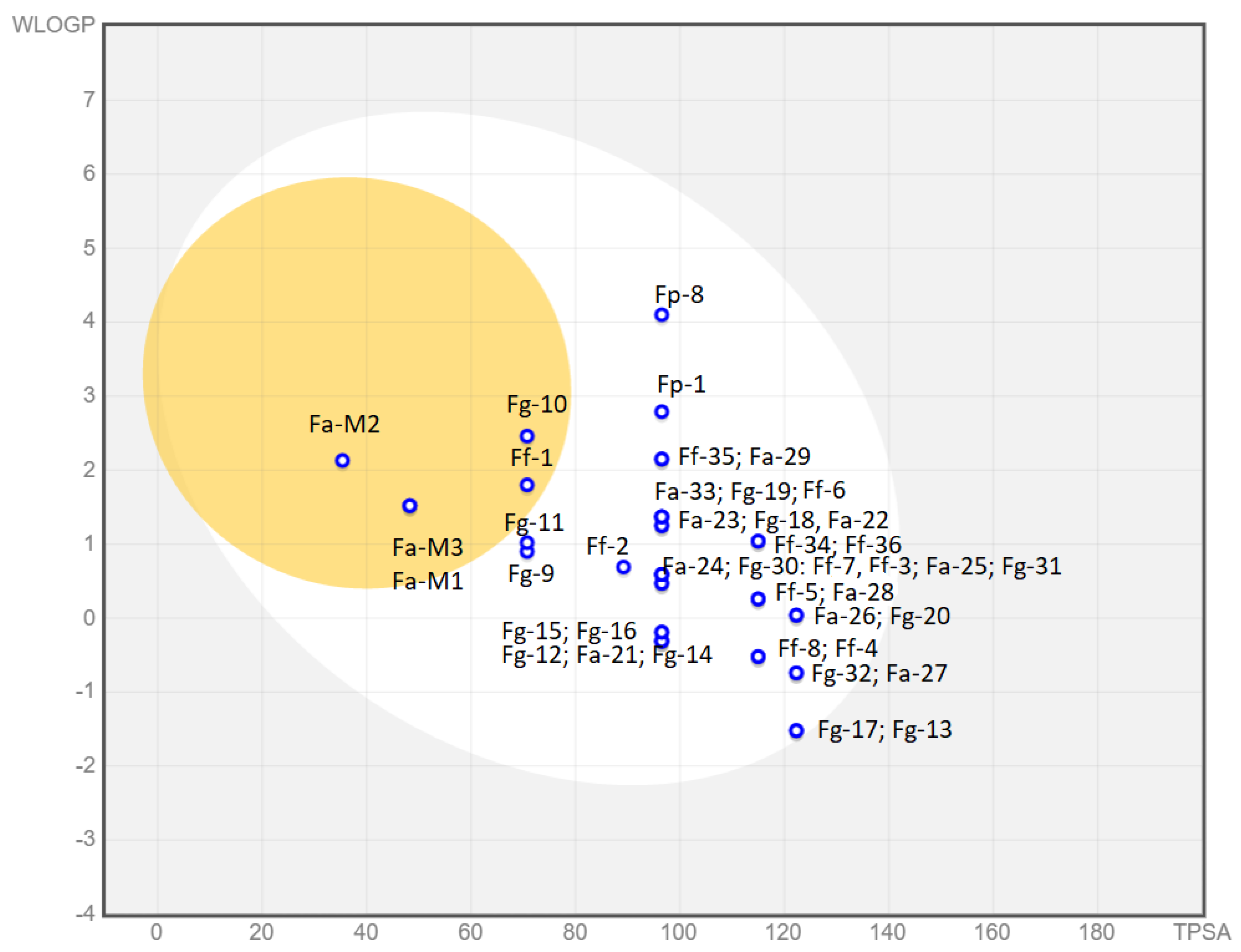
3.1.5. Predicted Parameters Related to the ADME (Absorption, Distribution, Metabolism, and Excretion) and PAINS (Pan Assay Interferences Structures)
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hernández-Alcoceba, R.; Saniger, L.; Campos, J.; Núñez, M.C.; Khaless, F.; Gallo, M.A.; Espinosa, A.; Lacal, J.C. Choline kinase inhibitors as a novel approach for antiproliferative drug design. Oncogene 1997, 15, 2289–2301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández-Alcoceba, R.; Fernández, F.; Lacal, J.C. In vivo antitumor activity of choline kinase inhibitors: A novel target for anticancer drug discovery. Cancer Res. 1999, 59, 3112–3118. [Google Scholar] [PubMed]
- Gibellini, F.; Smith, T.K. The Kennedy Pathway—De Novo Synthesis of Phosphatidylethanolamine and Phosphatidylcholine. IUBMB Life 2010, 62, 414–428. [Google Scholar] [CrossRef] [PubMed]
- De Molina, A.R.; Rodríguez-González, A.; Lacal, J.C. From Ras signalling to ChoK inhibitors: A further advance in anticancer drug design. Cancer Lett. 2004, 206, 137–148. [Google Scholar] [CrossRef]
- Katz-Brull, R.; Margalit, R.; Bendel, P.; Degani, H. Choline metabolism in breast cancer; 2H-, 13C- and 31P-NMR studies of cells and tumors. Magn. Reson. Mater. Phys. Biol. Med. 1998, 6, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Glunde, K.; Bhujwalla, Z.M.; Ronen, S.M. Choline metabolism in malignant transformation. Nat. Rev. Cancer 2011, 11, 835–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrán Aguilera, L.; Mariotto, E.; Rubbini, G.; Castro Navas, F.F.; Marco, C.; Carrasco-Jiménez, M.P.; Ballarotto, M.; Macchiarulo, A.; Hurtado-Guerrero, R.; Viola, G.; et al. Synthesis, biological evaluation, in silico modeling and crystallization of novel small monocationic molecules with potent antiproliferative activity by dual mechanism. Eur. J. Med. Chem. 2020, 207, 112797. [Google Scholar] [CrossRef]
- Conejo-García, A.; Campos, J.; Sánchez, R.M.; Rodríguez-González, A.; Lacal, J.C.; Gallo, M.A.; Espinosa, A. Choline kinase inhibitory effect and antiproliferative activity of new 1,1’,1’’-(benzene-1,3,5-triylmethylene)tris{4-[(disubstituted)amino]pyridinium} tribromides. Eur. J. Med. Chem. 2003, 38, 109–116. [Google Scholar] [CrossRef]
- Conejo-García, A.; Báñez-Coronel, M.; Sánchez-Martín, R.M.; Rodríguez-González, A.; Ramos, A.; de Molina, A.R.; Espinosa, A.; Gallo, M.A.; Campos, J.M.; Lacal, J.C. Influence of the linker in bispyridium compounds on the inhibition of human choline kinase. J. Med. Chem. 2004, 47, 5433–5440. [Google Scholar] [CrossRef]
- Sánchez-Martín, R.; Campos, J.M.; Conejo-García, A.; Cruz-López, O.; Báñez-Coronel, M.; Rodríguez-González, A.; Gallo, M.A.; Lacal, J.C.; Espinosa, A. Symmetrical bis-quinolinium compounds: New human choline kinase inhibitors with antiproliferative activity against the HT-29 cell line. J. Med. Chem. 2005, 48, 3354–3363. [Google Scholar] [CrossRef]
- TCD Pharma. Study of Intravenous TCD-717 in Patients With Advanced Solid Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT01215864 (accessed on 1 November 2021).
- Rodríguez-González, A.; Ramirez de Molina, A.; Fernández, F.; Lacal, J.C. Choline kinase inhibition induces the increase in ceramides resulting in a highly specific and selective cytotoxic antitumoral strategy as a potential mechanism of action. Oncogene 2004, 23, 8247–8259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trousil, S.; Kaliszczak, M.; Schug, Z.; Nguyen, Q.D.; Tomasi, G.; Favicchio, R.; Brickute, D.; Fortt, R.; Twyman, F.J.; Carroll, L.; et al. The novel choline kinase inhibitor ICL-CCIC-0019 reprograms cellular metabolism and inhibits cancer cell growth. Oncotarget 2016, 7, 37103–37120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arlauckas, S.P.; Popov, A.V.; Delikatny, E.J. Direct inhibition of choline kinase by a near-infrared fluorescent carbocyanine. Mol Cancer Ther. 2014, 13, 2149–2158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, B.S.; Allali-Hassani, A.; Tempel, W.; Finerty, P.J., Jr.; Mackenzie, F.; Dimov, S.; Vedadi, M.; Park, H.W. Crystal Structures of Human Choline Kinase Isoforms in Complex with Hemicholinium-3 Single Amino Acid near the Active Site Influences Inhibitor Sensitivity. J. Biol. Chem. 2010, 285, 16330–16340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trousil, S.; Carroll, L.; Kalusa, A.; Aberg, O.; Kaliszczak, M.; Aboagye, E.O. Design of symmetrical and nonsymmetrical N,N-dimethylaminopyridine derivatives as highly potent choline kinase alpha inhibitors. MedChemCommun 2013, 4, 693–696. [Google Scholar] [CrossRef] [Green Version]
- Jabalera, Y.; Sola-Leyva, A.; Peigneux, A.; Vurro, F.; Iglesias, G.R.; Vilchez-Garcia, J.; Pérez-Prieto, I.; Aguilar-Troyano, F.J.; López-Cara, L.C.; Carrasco-Jiménez, M.P.; et al. Biomimetic magnetic nanocarriers drive choline kinase alpha inhibitor inside cancer cells for combined chemo-hyperthermia therapy. Pharmaceutics 2019, 11, 408. [Google Scholar] [CrossRef] [Green Version]
- Castro-Navas, F.F.; Schiaffino-Ortega, S.; Carrasco-Jiménez, M.P.; Ríos-Marco, P.; Marco, C.; Espinosa, A.; Gallo, M.A.; Mariotto, E.; Basso, G.; Viola, G.; et al. New more polar symmetrical bipyridinic compounds: New strategy for the inhibition of choline kinase alpha 1. Future Med. Chem. 2015, 7, 417–436. [Google Scholar] [CrossRef]
- Schiaffino-Ortega, S.; Baglioni, E.; Mariotto, E.; Bortolozzi, R.; Serrán-Aguilera, L.; Ríos-Marco, P.; Carrasco-Jimenez, M.P.; Gallo, M.A.; Hurtado-Guerrero, R.; Marco, C.; et al. Design, synthesis, crystallization and biological evaluation of new symmetrical biscationic compounds as selective inhibitors of human Choline Kinase α1 (ChoKα1). Sci. Rep. 2016, 6, 23793. [Google Scholar] [CrossRef] [Green Version]
- Inazu, M.; Yamada, T.; Kubota, N.; Yamanaka, T. Functional expression of choline transporter-like protein 1 (CTL1) in small cell lung carcinoma cells: A target molecule for lung cancer therapy. Pharmacol. Res. 2013, 76, 119–131. [Google Scholar] [CrossRef] [Green Version]
- Sola-Leyva, A.; López-Cara, L.C.; Ríos-Marco, P.; Ríos, A.; Marco, C.; Carrasco-Jiménez, M.P. Choline kinase inhibitors EB-3D and EB-3P interferes with lipid homeostasis in HepG2 cells. Sci. Rep. 2019, 9, 5109. [Google Scholar] [CrossRef] [Green Version]
- Snaebjornsson, M.T.; Schulze, A. Tumours use a metabolic twist to make lipids. Nature 2019, 566, 333–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kall, S.L.; Delikatny, E.J.; Lavie, A. Identification of a Unique Inhibitor-Binding Site on Choline Kinase α. Biochemistry 2018, 57, 1316–1325. [Google Scholar] [CrossRef] [PubMed]
- Miyake, T.; Parsons, S.J. Functional interactions between Choline kinase α, epidermal growth factor receptor and c-Src in breast cancer cell proliferation. Oncogene 2012, 1431, 1441. [Google Scholar] [CrossRef] [Green Version]
- Falcon, S.C.; Hudson, C.S.; Huang, Y.; Mortimore, M.; Golec, J.M.; Charlton, P.A.; Weber, P.; Sundaram, H. A non-catalytic role of choline kinase alpha is important in promoting cancer cell survival. Oncogenesis. 2013, 2, e38. [Google Scholar] [CrossRef] [Green Version]
- Kall, S.; Whitlatch, K.; Smithgall, T.E.; Lavie, A. Molecular basis for the interaction between human choline kinase alpha and the SH3 domain of the c-Src tyrosine kinase. Sci. Rep. 2019, 9, 17121. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Troyano, F.J.; Torretta, A.; Rubbini, G.; Fasiolo, A.; Luque-Navarro, P.M.; Carrasco-Jimenez, M.P.; Pérez-Moreno, G.; Bosch-Navarrete, C.; González-Pacanowska, D.; Parisini, E.; et al. New Compounds with Bioisosteric Replacement of Classic Choline Kinase Inhibitors Show Potent Antiplasmodial Activity. Pharmaceutics 2021, 13, 1842. [Google Scholar] [CrossRef]
- Schiaffino-Ortega, S.; Mariotto, E.; Luque-Navarro, P.M.; Kimatrai-Salvador, M.; Rios-Marco, P.; Hurtado-Guerrero, R.; Marco, C.; Carrasco-Jimenez, M.P.; Viola, G.; López-Cara, L.C. Anticancer and structure activity relationship of non-symmetrical choline kinase inhibitors. Pharmaceutics 2021, 13, 1360. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 17391749. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Friesner, R.A.; Xiang, Z.; Honig, B. On the Role of the Crystal Environment in Determining Protein Side-chain Conformations. J. Mol. Biol. 2002, 320, 597–608. [Google Scholar] [CrossRef]
- Bochevarov, A.D.; Harder, E.; Hughes, T.F.; Greenwood, J.R.; Braden, D.A.; Philipp, D.M.; Rinaldo, D.; Halls, M.D.; Zhang, J.; Friesner, R.A.; et al. Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum. Chem. 2013, 113, 2110. [Google Scholar] [CrossRef]
- Brenk, R.; Schipani, A.; James, D.; Krasowski, A.; Gilbert, I.H.; Frearson, J.; Wyatt, P.G. Lessons learnt from assembling screening libraries for drug discovery for neglected diseases. ChemMedChem 2008, 3, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peisach, D.; Gee, P.; Kent, C.; Xu, Z. The Crystal Structure of Choline Kinase Reveals a Eukaryotic Protein Kinase Fold. Structure 2003, 11, 703–713. [Google Scholar] [CrossRef] [Green Version]
- Hunter, C.A.; Low, C.M.R.; Rotger, C.; Vinter, J.G.; Zonta, C. Substituent effects on cation- interactions: A quantitative study. Proc. Natl. Acad. Sci. USA 2002, 99, 4873–4876. [Google Scholar] [CrossRef] [Green Version]
- Mariotto, E.; Bortolozzi, R.; Volpin, I.; Carta, D.; Serafin, V.; Accordi, B.; Basso, G.; Navarro, P.L.; López-Cara, L.C.; Viola, G. EB-3D a novel choline kinase inhibitor induces deregulation of the AMPK-mTOR pathway and apoptosis in leukemia T-cells. Biochem Pharmacol. 2018, 155, 213–223. [Google Scholar] [CrossRef]
- Mariotto, E.; Viola, G.; Ronca, R.; Persano, L.; Aveic, S.; Bhujwalla, Z.M.; Mori, N.; Accordi, B.; Serafin, V.; López-Cara, L.C.; et al. T Choline Kinase Alpha Inhibition by EB-3D Triggers Cellular Senescence, Reduces Tumor Growth and Metastatic Dissemination in Breast Cancer. Cancers 2018, 10, 391. [Google Scholar] [CrossRef] [Green Version]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Zoete, V.A. BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [Green Version]
- Wildman, S.A.; Crippen, G.M. Prediction of physicochemical parameters by atomic contributions. J. Chem. Inf. Comput. Sci. 1999, 39, 868–873. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Family | Linker (a–f) | Bioisosteric Cationic Head | 7 or 4 Substituent | |
|---|---|---|---|---|---|
| Fa-M2 | Monocationic | A | Biphenyl | thieno[3,2-b]pyridin-1-ium | Pyrrolidinyl |
| Fa-M1 | thieno[3,2-d]pyrimidin-1-ium | ||||
| Fa-M3 | thieno[2,3-d]pyrimidin-1-ium | ||||
| Fg-9 | Biscationic | B | Biphenyl | thieno[3,2-b]pyridin-1-ium | Pyrrolidinyl |
| Fa-21 | thieno[3,2-d]pyrimidin-1-ium | ||||
| Fg-14 | thieno[2,3-d]pyrimidin-1-ium | ||||
| Fa-24 | thieno[3,2-d]pyrimidin-1-ium | Piperidinyl | |||
| Fg-30 | thieno[2,3-d]pyrimidin-1-ium | ||||
| Fg-10 | thieno[3,2-b]pyridin-1-ium | Azepanyl | |||
| Fa-22 | thieno[3,2-d]pyrimidin-1-ium | ||||
| Fg-18 | thieno[2,3-d]pyrimidin-1-ium | ||||
| Fp-1 | thieno[3,2-d]pyrimidin-1-ium | N-methyl-aniline | |||
| Fp-8 | thieno[2,3-d]pyrimidin-1-ium | p-Chloro-N-methylaniline | |||
| Fg-12 | C | Bipyridinyl | thieno[3,2-b]pyridin-1-ium | Pyrrolidinyl | |
| Fg-17 | thieno[3,2-d]pyrimidin-1-ium | ||||
| Fg-13 | thieno[2,3-d]pyrimidin-1-ium | ||||
| Fa-27 | thieno[3,2-d]pyrimidin-1-ium | Piperidinyl | |||
| Fg-32 | thieno[2,3-d]pyrimidin-1-ium | ||||
| Fa-26 | thieno[3,2-d]pyrimidin-1-ium | Azepanyl | |||
| Fg-20 | thieno[2,3-d]pyrimidin-1-ium | ||||
| Fg-11 | D | Bibenzyl | thieno[3,2-b]pyridin-1-ium | Pyrrolidinyl | |
| Fg-16 | thieno[3,2-d]pyrimidin-1-ium | ||||
| Fg-15 | thieno[2,3-d]pyrimidin-1-ium | ||||
| Fa-25 | thieno[3,2-d]pyrimidin-1-ium | Piperidinyl | |||
| Fg-31 | thieno[2,3-d]pyrimidin-1-ium | ||||
| Fa-23 | thieno[3,2-d]pyrimidin-1-ium | Azepanyl | |||
| Fg-19 | thieno[2,3-d]pyrimidin-1-ium | ||||
| Ff-1 | E | Biphenethyl | thieno[3,2-b]pyridin-1-ium | Pyrrolidinyl | |
| Ff-7 | thieno[3,2-d]pyrimidin-1-ium | ||||
| Ff-3 | thieno[2,3-d]pyrimidin-1-ium | ||||
| Fa-33 | thieno[3,2-d]pyrimidin-1-ium | Piperidinyl | |||
| Ff-6 | thieno[2,3-d]pyrimidin-1-ium | ||||
| Fa-29 | thieno[3,2-d]pyrimidin-1-ium | Azepanyl | |||
| Ff-35 | thieno[2,3-d]pyrimidin-1-ium | ||||
| Ff-2 | F | Diphenoxiethane | thieno[3,2-b]pyridin-1-ium | Pyrrolidinyl | |
| Ff-8 | thieno[3,2-d]pyrimidin-1-ium | ||||
| Ff-4 | thieno[2,3-d]pyrimidin-1-ium | ||||
| Fa-28 | thieno[3,2-d]pyrimidin-1-ium | Piperidinyl | |||
| Ff-5 | thieno[2,3-d]pyrimidin-1-ium | ||||
| Ff-34 | thieno[3,2-d]pyrimidin-1-ium | Azepanyl | |||
| Ff-36 | thieno[2,3-d]pyrimidin-1-ium | ||||
| Compound | % Inhibition at 10 µM | % Inhibition at 30 µM | 4BR3 | 4CG8 | ||
|---|---|---|---|---|---|---|
| g-score (kcal/mol) | ΔGbinding (kcal/mol) | g-score (kcal/mol) | ΔGbinding (kcal/mol) | |||
| Fa-29 | 100.00 a | 100.00 a | −7.429 | −90.93 | −8.740 | −116.31 |
| Fa-33 | 98.90 | 97.80 | −6.780 | −70.10 | −7.703 | −86.74 |
| Ff-34 | 94.31 | 99.02 | −7.502 | −107.36 | −6.838 | −108.16 |
| Fg-11 | 93.07 | 94.25 | −8.776 | −114.95 | −7.126 | −97.43 |
| Ff-35 | 90.55 | 97.80 | −6.483 | −95.67 | −7.907 | −96.44 |
| Ff-7 | 89.95 | 95.88 | −6.721 | −87.32 | −6.814 | −92.68 |
| Fa-22 | 89.59 | 97.15 | −7.105 | −112.67 | −5.921 | −88.89 |
| Fa-28 10 | 87.69 | 92.19 | −7.208 | −103.95 | −5.700 | −93.52 |
| Fa-24 | 84.64 | 98.40 | −7.571 | −109.75 | −6.298 | −106.62 |
| Ff-36 | 84.47 | 96.07 | −6.546 | −108.85 | −7.681 | −87.98 |
| Fg-12 | 83.33 | 98.61 | −6.890 | −89.43 | −5.406 | −82.50 |
| Ff-1 | 82.15 | 90.83 | −8.994 | −103.45 | −7.069 | −81.64 |
| Fg-10 | 80.76 | 96.98 | −6.645 | −97.26 | −6.409 | −116.55 |
| Fg-14 | 80.28 | 94.14 | −8.465 | −119.33 | −7.259 | −98.01 |
| Fg-16 | 80.28 | 85.87 | −7.542 | −122.68 | −5.556 | −83.41 |
| Ff-8 | 79.23 | 92.88 | −8.873 | −113.72 | −6.027 | −83.02 |
| Ff-2 | 77.41 | 82.77 | −9.403 | −117.89 | −7.085 | −104.19 |
| Fg-9 | 75.81 | 94.95 | −7.128 | −103.77 | −4.454 | −109.32 |
| Fg-30 | 75.46 | 85.71 | −7.379 | −118.12 | −5.856 | −79.10 |
| Fa-21 | 74.57 | 86.58 | −7.516 | −119.75 | −5.619 | −68.03 |
| Ff-3 | 74.17 | 80.09 | −7.833 | −99.93 | −6.937 | −93.24 |
| Fa-26 | 73.33 | 85.09 | −8.061 | −114.66 | −5.976 | −91.59 |
| Fg-20 | 71.88 | 80.83 | −7.632 | −111.57 | −5.544 | −73.41 |
| Fg-15 | 71.11 | 81.29 | −10.412 | −131.90 | −7.619 | −102.47 |
| Fa-27 | 70.28 | 81.49 | −6.060 | −72.32 | −5.137 | −75.34 |
| Fg-32 | 65.38 | 67.57 | −6.880 | −78.31 | −5.460 | −69.69 |
| Ff-5 | 64.21 | 85.71 | −7.810 | −114.51 | −5.318 | −55.38 |
| Ff-4 | 55.23 | 77.28 | −8.907 | −124.18 | −6.883 | −97.60 |
| Fg-17 | 54.90 | 60.95 | −7.405 | −113.16 | −6.103 | −77.09 |
| Fg-31 | 51.54 | 74.39 | −8.194 | −118.41 | −7.201 | −95.92 |
| Ff-6 | 46.43 | 73.19 | −6.992 | −76.19 | −7.657 | −109.86 |
| Fa-M1 | 34.59 | 31.68 | -6.978 | −82.49 | −5.331 | −51.72 |
| Fa-M3 | 34.42 | 37.94 | −5.922 | −82.47 | −5.357 | −68.05 |
| Fa-M2 | 30.24 | 35.06 | −6.308 | −75.23 | −5.755 | −62.55 |
| Fg-13 | 28.25 | 50.41 | −6.513 | −78.44 | −5.294 | −74.69 |
| Fa-23 | - b | - b | −7.346 | −127.89 | −7.105 | −102.87 |
| Fa-25 | - b | - b | −8.089 | −114.86 | −7.912 | −98.88 |
| Fg-18 | - b | - b | −7.376 | −95.92 | −8.146 | −90.75 |
| Fg-19 | - b | - b | −8.567 | −93.84 | −5.755 | −82.99 |
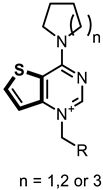 | 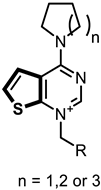 | ||
|---|---|---|---|
| Compound | % Inhibition at 10 µM | Compound | % Inhibition at 10 µM |
| Fa-29 | 100.00 | Ff-35 | 90.55 |
| Fa-33 | 98.9 | Ff-6 | 46.43 |
| Fa-28 | 87.69 | Ff-5 | 64.21 |
| Ff-34 | 94.31 | Ff-36 | 84.47 |
| GI50 c (µM) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Comp. | Family | IC50 CKα a | cLog P b | HL-60 | Jurkat | RS 4;11 | SEM | A549 | MDA-MB-231 | HeLa | HT29 | % Inhibition Choline Uptake d |
| Fa-M2 | A | ≥50 | 3.47 | 0.13 ± 0.01 | 2.40 ± 0.22 | 0.56 ± 0.03 | 0.96 ± 0.01 | 3.5 ± 0.3 | 0.81 ± 0.01 | 0.38 ± 0.005 | 0.40 ± 0.02 | |
| Fa-M1 | ≥50 | 3.00 | 0.15 ± 0.01 | 4.24 ± 0.32 | 0.68 ± 0.05 | 1.47± 0.2 | 0.39 ± 0.01 | 0.62 ± 0.03 | 0.41 ± 0.01 | 0.58 ± 0.02 | −1 μM: 49.93 ± 1.72 −10 μM: 85.12 ± 1.35 | |
| Fa-M3 | ≥50 | 3.14 | nd | Nd | nd | nd | nd | nd | nd | nd | ||
| Fg-9 | B | ≥5 | 3.54 | 9.12 ± 1.1 | >10 | >10 | >10 | 0.25 ± 0.02 | 2.02 ± 0.2 | >10 | >10 | |
| Fa-21 | ≥5 | 2.75 | 3.40 ± 0.7 | >10 | >10 | >10 | 0.22 ± 0.01 | 1.71 ± 0.4 | >10 | >10 | ||
| Fg-14 | 4.87 ± 0.57 | 2.81 | 1.35 ± 0.1 | >10 | >10 | >10 | 0.28 ± 0.02 | 0.94 ± 0.09 | 2.53 ± 0.8 | 4.16 ± 1.0 | ||
| Fa-24 | 2.33 ± 0.21 | 2.93 | 7.28 ± 1.1 | >10 | >10 | >10 | 1.17 ± 0.2 | 0.70 ± 0.07 | 7.08 ± 1.3 | >10 | ||
| Fg-30 | ≥5 | 3.07 | 2,78 ± 0.5 | >10 | >10 | >10 | 0.60 ± 0.02 | 0.80 ± 0.02 | 2.89 ± 0.3 | 8.32 ± 0.9 | ||
| Fg-10 | ≥10 μM | 4.33 | 0.44 ± 0.05 | 6.33 ± 0.6 | 2.77 ± 0.9 | 3.51 ± 0.8 | 0.31 ± 0.01 | 1.46 ± 0.2 | 1.59 ± 0.1 | 1.76 ± 0.2 | ||
| Fa-22 | 2.87 ± 0.19 | 3.49 | 3.29 ± 0.5 | >10 | 4.29 ± 0.7 | >10 | 0.97 ± 0.02 | 1.42 ± 0.01 | 6.55 ± 0.4 | 6.51 ± 0.6 | −0.1 μM: -1.15 ± 1.2 −0.5 μM: 60.6 ± 0.02 −2 μM: 90.08 ± 0.24 | |
| Fg-18 | --- | 3.72 | 0.79 ± 0.02 | 7.04 ± 0.8 | 2.65 ± 0.04 | >10 | 0.21 ± 0.01 | 0.61 ± 0.03 | 1.81 ± 0.02 | 2.32 ± 0.02 | ||
| Fp-1 | 1.06 ± 0.22 | 3.78 | 0.51 ± 0.05 | >10 | 2.12 ± 0.2 | 4.55 ± 0.5 | 0.21 ± 0.02 | 0.90 ± 0.02 | 1.66 ± 0.01 | 1.43 ± 0.03 | ||
| Fp-8 | --- | 5.12 | 0.90 ± 0.08 | >10 | 3.38 ± 0.05 | >10 | 0.16 ± 0.01 | 0.83 ± 0.04 | 1.77 ± 0.1 | 3.93 ± 0.5 | ||
| Fg-12 | C | --- | 2.23 | >10 | >10 | >10 | >10 | 0.19 ± 0.01 | 0.80 ± 0.02 | >10 | >10 | |
| Fg-17 | ≥25 | 1.32 | >10 | >10 | >10 | >10 | 0.14 ± 0.01 | >10 | >10 | >10 | ||
| Fg-13 | ≥25 | 1.27 | 8.71 ± 1.2 | >10 | >10 | >10 | 0.14 ± 0.01 | >10 | >10 | >10 | ||
| Fa-27 | ≥5 | 1.54 | >10 | >10 | >10 | >10 | 1.06 ± 0.02 | >10 | >10 | >10 | ||
| Fg-32 | ≥5 | 1.66 | >10 | >10 | >10 | >10 | 0.29 ± 0.02 | >10 | >10 | >10 | ||
| Fa-26 | ≥5 | 1.97 | >10 | >10 | >10 | >10 | 0.23 | >10 | >10 | >10 | ||
| Fg-20 | ≥5 | 2.18 | 1.22± | >10 | >10 | >10 | >10 | >10 | >10 | >10 | ||
| Fg-11 | D | 2.33 ± 0.20 | 3.76 | 0.47 ± 0.03 | 8.53 ± 1.1 | 5.72 ± 0.5 | >10 | 0.39 ± 0.04 | 0.68 ± 0.07 | 2.19 ± 0.3 | 3.65 ± 0.5 | |
| Fg-16 | 1.81 ± 0.17 | 2.93 | 0.21 ± 0.03 | >10 | 7.27 ± 0.8 | >10 | 0.25 ± 0.02 | 0.38 ± 0.01 | 0.67 ± 0.02 | 2.64 ± 0.06 | ||
| Fg-15 | ≥5 | 2.99 | 0.13 ± 0.01 | >10 | 3.71 ± 0.04 | >10 | 0.22 ± 0.03 | 0.26 ± 0.02 | 1.00 ± 0.1 | 2.01 ± 0.03 | ||
| Fa-25 | --- | 3.35 | 0.14 ± 0.01 | 5.48 ± 0.5 | 1.84 ± 0.1 | 2.53 ± 0.03 | 0.28 ± 0.02 | 0.33 ± 0.02 | 0.62 ± 0.02 | 1.47 ± 0.06 | ||
| Fg-31 | ≥10 | 3.43 | 0.76 ± 0.05 | >10 | 7.20 ± 1.0 | >10 | 1.17 ± 0.01 | 1.09 ± 0.1 | 1.40 ± 0.02 | 4.50 ± 0.4 | ||
| Fa-23 | --- | 3.95 | 0.51 ± 0.05 | 6.15 ± 0.6 | 2.08 ± 0.03 | 2.17 ± 0.02 | 0.60 ± 0.06 | 1.13 ± 0.2 | 1.17 ± 0.1 | 1.56 ± 0.2 | ||
| Fg-19 | --- | 4.15 | 0.15 ± 0.01 | 4.72 ± 0.6 | 1.51 ± 0.2 | 2.59 ± 0.03 | 0.31 ± 0.04 | 0.50 ± 0.02 | 0.78 ± 0.02 | 1.42 ± 0.1 | ||
| Ff-1 | E | 1.54 ± 0.17 | 4.42 | 0.79 ± 0.05 | 4.13 ± 0.4 | 7.90 ± 1.3 | 3.07 ± 0.3 | 0.97 ± 0.02 | 1.23 ± 0.01 | 1.90 ± 0.01 | 3.32 ± 0.03 | |
| Ff-7 | 8.57 ± 2.80 | 3.59 | 0.40± 0.05 | 8.61 ± 1.3 | 2.33 ± 0.3 | 2.93 ± 0.2 | 0.21 ± 0.03 | 0.47 ± 0.04 | 0.87 ± 0.05 | 1.48 ± 0.04 | ||
| Ff-3 | ≥5 | 3.80 | 0.21 ± 0.03 | 7.54 ± 1.5 | 2.05 ± 0.1 | 3.70 ± 0.4 | 0.21 ± 0.02 | 0.36 ± 0.03 | 0.63 ± 0.02 | 1.33 ± 0.01 | ||
| Fa-33 | 1.67 ± 0.08 | 4.09 | 0.14 ± 0.01 | 1.46 ± 0.1 | 0.76 ± 0.1 | 1.37 ± 0.07 | 0.16 ± 0.01 | 0.41 ± 0.02 | 0.28 ± 0.02 | 1.07 ± 0.1 | ||
| Ff-6 | ≥10 | 4.21 | 0.19 ± 0.01 | 6.7 ± 0.8 | 1.50 ± 0.2 | 2.52 ± 0.2 | 0.19 ± 0.03 | 0.50 ± 0.02 | 0.42 ± 0.05 | 0.93 ± 0.04 | ||
| Fa-29 | 1.08 ± 0.07 | 4.81 | 0.55± 0.02 | 5.23 ± 0.8 | 1.60 ± 0.1 | 1.77 ± 0.2 | 0.14 ± 0.02 | 0.50 ± 0.02 | 0.84 ± 0.04 | 1.09 ± 0.2 | ||
| Ff-35 | 0.46 ± 0.08 | 4.91 | 0.15 ± 0.01 | 1.54 ± 0.02 | 0.95 ± 0.05 | 0.76 ± 0.03 | 0.14 ± 0.02 | 0.097 ± 0.001 | 0.30 ± 0.01 | 0.78 ± 0.03 | −0.1 μM: 35.64 ± 0.7 −0.5 μM: 84.99 ± 1.3 −2 μM: 96.62 ± 0.316 | |
| Ff-2 | F | nd | 3.14 | 2.14 ± 0.1 | >10 | >10 | >10 | 1.06 ± 0.1 | 1.43 ± 0.2 | 3.26 ± 0.4 | >10 | |
| Ff-8 | nd | 2.28 | 0.55 ± 0.02 | >10 | 3.47 ± 0.3 | >10 | 0.29 ± 0.3 | 0.32 ± 0.05 | 0.91 ± 0.09 | 1.89 ± 0.2 | ||
| Ff-4 | ≥10 | 2.50 | 0.79 ± 0.05 | >10 | >10 | >10 | 0.23 ± 0.03 | 0.72 ± 0.03 | >10 | >10 | ||
| Fa-28 | 3.25 ± 0.54 | 2.78 | 0.35 ± 0.01 | >10 | 2.32 ± 0.03 | 2.41 ± 0.04 | 0.29 ± 0.02 | 0.19 ± 0.01 | 0.59 ± 0.02 | >10 | ||
| Ff-5 | ≥5 | 2.94 | 0.20 ± 0.01 | >10 | 2.45 ± 0.02 | 3.27 ± 0.03 | 0.34 ±0.06 | 1.10 ± 0.2 | 1.03 ± 0.1 | 1.73 ± 0.3 | ||
| Ff-34 | 0.70 ± 0.03 | 3.48 | 0.27 ± 0.01 | 6.1 ± 1.1 | 1.8 ± 0.1 | 2.5 ± 0.1 | 0.29 ± 0.03 | 0.91 ± 0.08 | 0.93 ± 0.01 | 1.63 ± 0.1 | ||
| Ff-36 | 0.35 ± 0.05 | 3.69 | 0.44 ± 0.02 | 6.2 ± 0.9 | 2.26 ± 0.3 | 2.54 ± 0.2 | 1.43 ± 0.1 | 2.26 ± 0.3 | 3.02 ± 0.4 | >10 | −0.5 μM: 71.78 ± 0.1 −2 μM: 95.32 ± 0.29 | |
| MN-48b d | 0.78 ± 0.03 | 1.27 | 0.32 ± 0.03 | 0.35 ± 0.1 | 1.0 ± 0.3 | nd | 0.54 ± 0.2 | 0.31 ± 0.12 | 1.9 ± 0.1 | 1.9 ± 0.4 | ||
| RSM-932A d | 1.92 ± 0.06 | 4.13 | 0.93 ± 0.1 | 0.41 ± 0.1 | 0.17 ± 0.04 | nd | 0.45 ± 0.09 | 0.17 ± 0.05 | 0.83 ± 0.1 | 0.4 ± 0.2 | ||
| Compound | cLog P | PAINS | Mw | Structural Alert | Lipinski Rules | Suitability | |
|---|---|---|---|---|---|---|---|
| Monocationic Biphenyl | Fa-M2 | 3.47 | NO | 451.42 | Quaternary N | Yes | No |
| Fa-M1 | 3.00 | NO | 452.41 | Quaternary N | Yes | No | |
| Fa-M3 | 3.14 | NO | 452.41 | Quaternary N | Yes | No | |
| Biscationic | Fg-9 | 3.54 | NO | 748.64 | Quaternary N | No | Moderate |
| Biphenyl | Fa-21 | 2.75 | NO | 750.62 | Quaternary N | No | Yes |
| Fg-14 | 2.81 | NO | 750.62 | Quaternary N | No | Yes | |
| Fa-24 | 2.93 | NO | 778.67 | Quaternary N | No | Yes | |
| Fg-30 | 3.07 | NO | 778.67 | Quaternary N | No | Yes | |
| Fg-10 | 4.33 | NO | 804.75 | Quaternary N | No | No | |
| Fa-22 | 3.49 | NO | 806.72 | Quaternary N | No | Yes | |
| Fg-18 | 3.72 | NO | 806.72 | Quaternary N | No | Yes | |
| Fp-1 | 3.78 | NO | 822.68 | Quaternary N | No | Yes | |
| Fp-8 | 5.12 | NO | 891.57 | Quaternary N | No | Yes | |
| Bipyridinyl | Fg-12 | 2.23 | NO | 750.62 | Quaternary N | No | Yes |
| Fg-17 | 1.32 | NO | 752.59 | Quaternary N | Yes | No | |
| Fg-13 | 1.27 | NO | 752.59 | Quaternary N | Yes | No | |
| Fa-27 | 1.54 | NO | 780.65 | Quaternary N | No | Yes | |
| Fg-32 | 1.66 | NO | 780.65 | Quaternary N | Yes | Yes | |
| Fa-26 | 1.97 | NO | 808.7 | Quaternary N | No | Yes | |
| Fg-20 | 2.18 | NO | 808.7 | Quaternary N | No | Yes | |
| Bibenzyl | Fg-11 | 3.76 | NO | 776.69 | Quaternary N | No | Moderate |
| Fg-16 | 2.93 | NO | 778.67 | Quaternary N | No | Yes | |
| Fg-15 | 2.99 | NO | 778.67 | Quaternary N | No | Yes | |
| Fa-25 | 3.35 | NO | 806.72 | Quaternary N | No | Yes | |
| Fg-31 | 3.43 | NO | 806.72 | Quaternary N | No | Yes | |
| Fa-23 | 3.95 | NO | 834.78 | Quaternary N | No | Yes | |
| Fg-19 | 4.15 | NO | 834.78 | No | Yes | ||
| Biphenethyl | Ff-1 | 4.42 | NO | 804.75 | Quaternary Quaternary NN | No | No |
| Ff-7 | 3.59 | NO | 806.72 | Quaternary N | No | Yes | |
| Ff-3 | 3.80 | NO | 806.72 | Quaternary N | No | Yes | |
| Fa-33 | 4.09 | NO | 834.78 | Quaternary N | No | Yes | |
| Ff-6 | 4.21 | NO | 834.78 | Quaternary N | No | Yes | |
| Fa-29 | 4.81 | NO | 862.83 | Quaternary N | No | Yes | |
| Ff-35 | 4.91 | NO | 862.83 | Quaternary N | No | Yes | |
| Diphenoxiethane | Ff-2 | 3.14 | NO | 808.69 | Quaternary N | No | Yes |
| Ff-8 | 2.28 | NO | 810.67 | Quaternary N | No | Yes | |
| Ff-4 | 2.50 | NO | 810.67 | Quaternary N | No | Yes | |
| Fa-28 | 2.78 | NO | 838.72 | Quaternary N | No | Yes | |
| Ff-5 | 2.94 | NO | 838.72 | Quaternary N | No | Yes | |
| Ff-34 | 3.48 | NO | 866.78 | Quaternary N | No | Yes | |
| Ff-36 | 3.69 | NO | 866.78 | Quaternary N | No | Yes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luque-Navarro, P.M.; Mariotto, E.; Ballarotto, M.; Rubbini, G.; Aguilar-Troyano, F.J.; Fasiolo, A.; Torretta, A.; Parisini, E.; Macchiarulo, A.; Laso, A.; et al. Biological Evaluation of New Thienopyridinium and Thienopyrimidinium Derivatives as Human Choline Kinase Inhibitors. Pharmaceutics 2022, 14, 715. https://doi.org/10.3390/pharmaceutics14040715
Luque-Navarro PM, Mariotto E, Ballarotto M, Rubbini G, Aguilar-Troyano FJ, Fasiolo A, Torretta A, Parisini E, Macchiarulo A, Laso A, et al. Biological Evaluation of New Thienopyridinium and Thienopyrimidinium Derivatives as Human Choline Kinase Inhibitors. Pharmaceutics. 2022; 14(4):715. https://doi.org/10.3390/pharmaceutics14040715
Chicago/Turabian StyleLuque-Navarro, Pilar María, Elena Mariotto, Marco Ballarotto, Gianluca Rubbini, Francisco José Aguilar-Troyano, Alberto Fasiolo, Archimede Torretta, Emilio Parisini, Antonio Macchiarulo, Alejandro Laso, and et al. 2022. "Biological Evaluation of New Thienopyridinium and Thienopyrimidinium Derivatives as Human Choline Kinase Inhibitors" Pharmaceutics 14, no. 4: 715. https://doi.org/10.3390/pharmaceutics14040715
APA StyleLuque-Navarro, P. M., Mariotto, E., Ballarotto, M., Rubbini, G., Aguilar-Troyano, F. J., Fasiolo, A., Torretta, A., Parisini, E., Macchiarulo, A., Laso, A., Marco, C., Viola, G., Carrasco-Jimenez, M. P., & López-Cara, L. C. (2022). Biological Evaluation of New Thienopyridinium and Thienopyrimidinium Derivatives as Human Choline Kinase Inhibitors. Pharmaceutics, 14(4), 715. https://doi.org/10.3390/pharmaceutics14040715