
Effect of Particle Carriers for Intraperitoneal Drug Delivery on the Course of Ovarian Cancer and Its Immune Microenvironment in a Mouse Model

 ,
,  , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Ovarian Cancer Tumor Model
2.2. Drug Carrier Administration in Mice
2.3. Chemotherapy Treatment in Mice
2.4. Experimental Design
2.5. Bioluminescence Imaging
2.6. Blood and Peritoneal Fluid Sampling for Immune Readout
2.7. Flow Cytometry
2.8. Luminex®
2.9. Statistical Analysis
3. Results
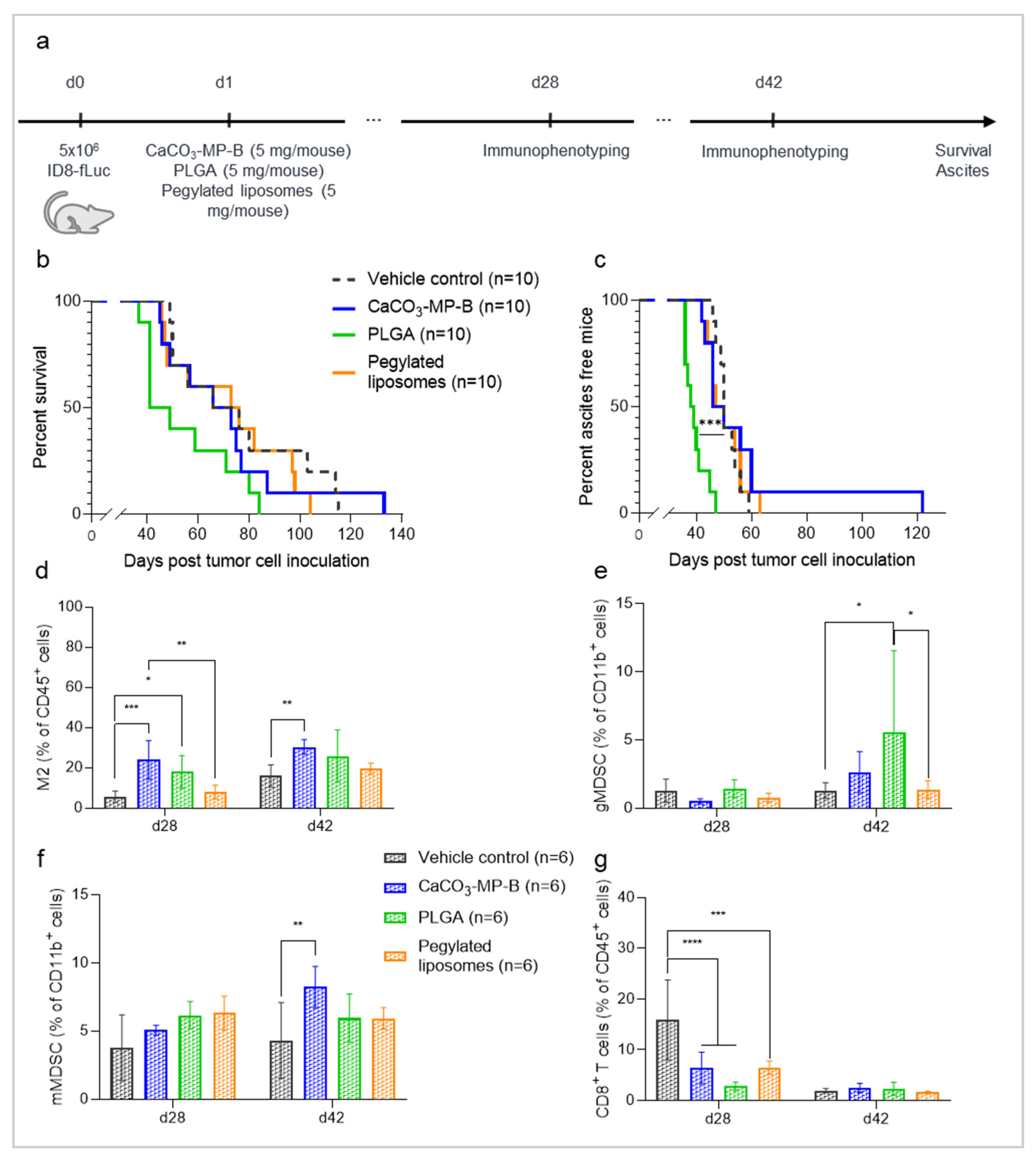
3.1. Different Types of Drug Carriers Worsen Disease Symptoms in an Ovarian Cancer Mouse Model
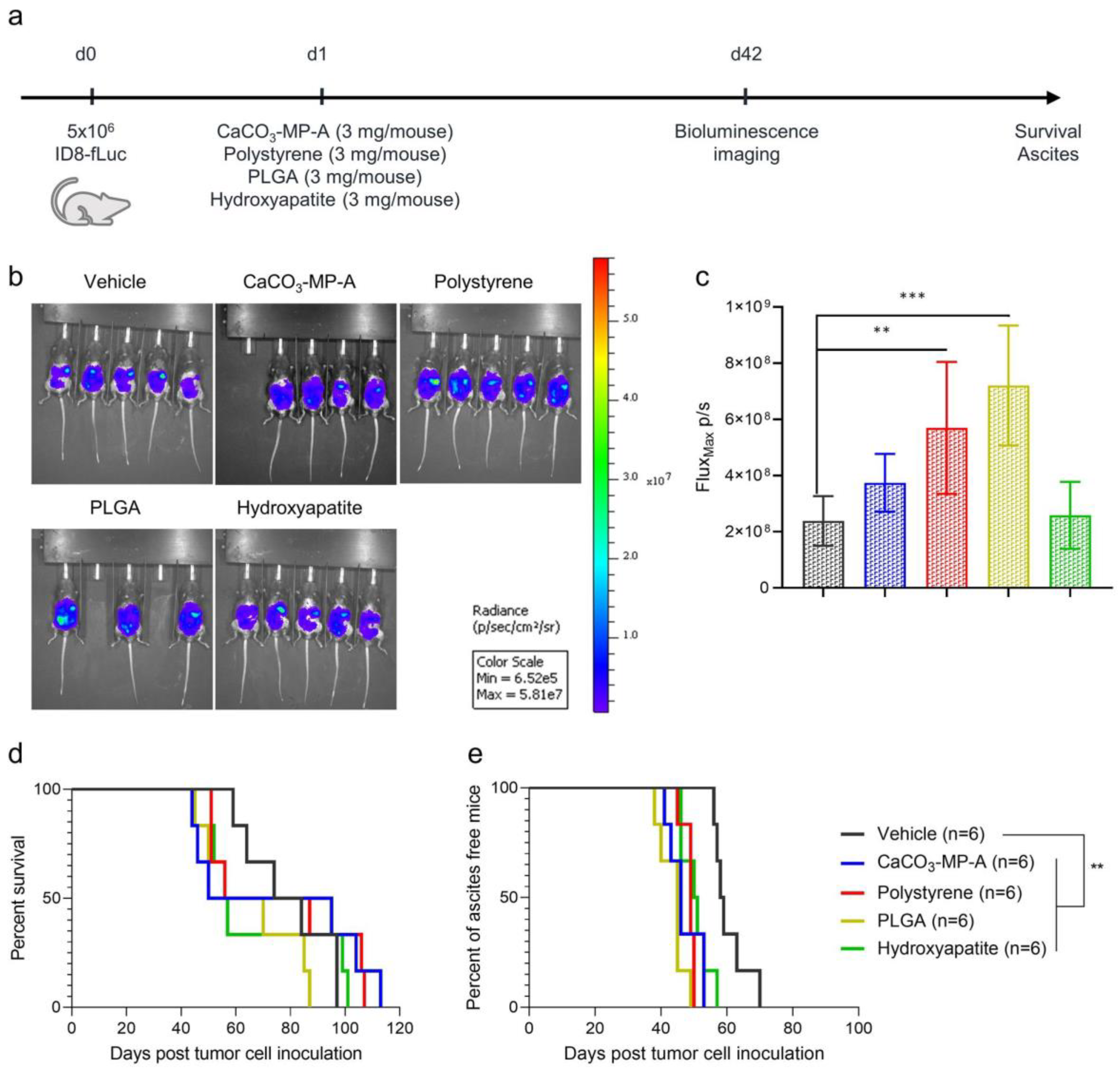
3.2. A Higher Microparticle Dose Can Result in a Hyperproliferation of the Tumor and an Increase in Innate Immune Suppression
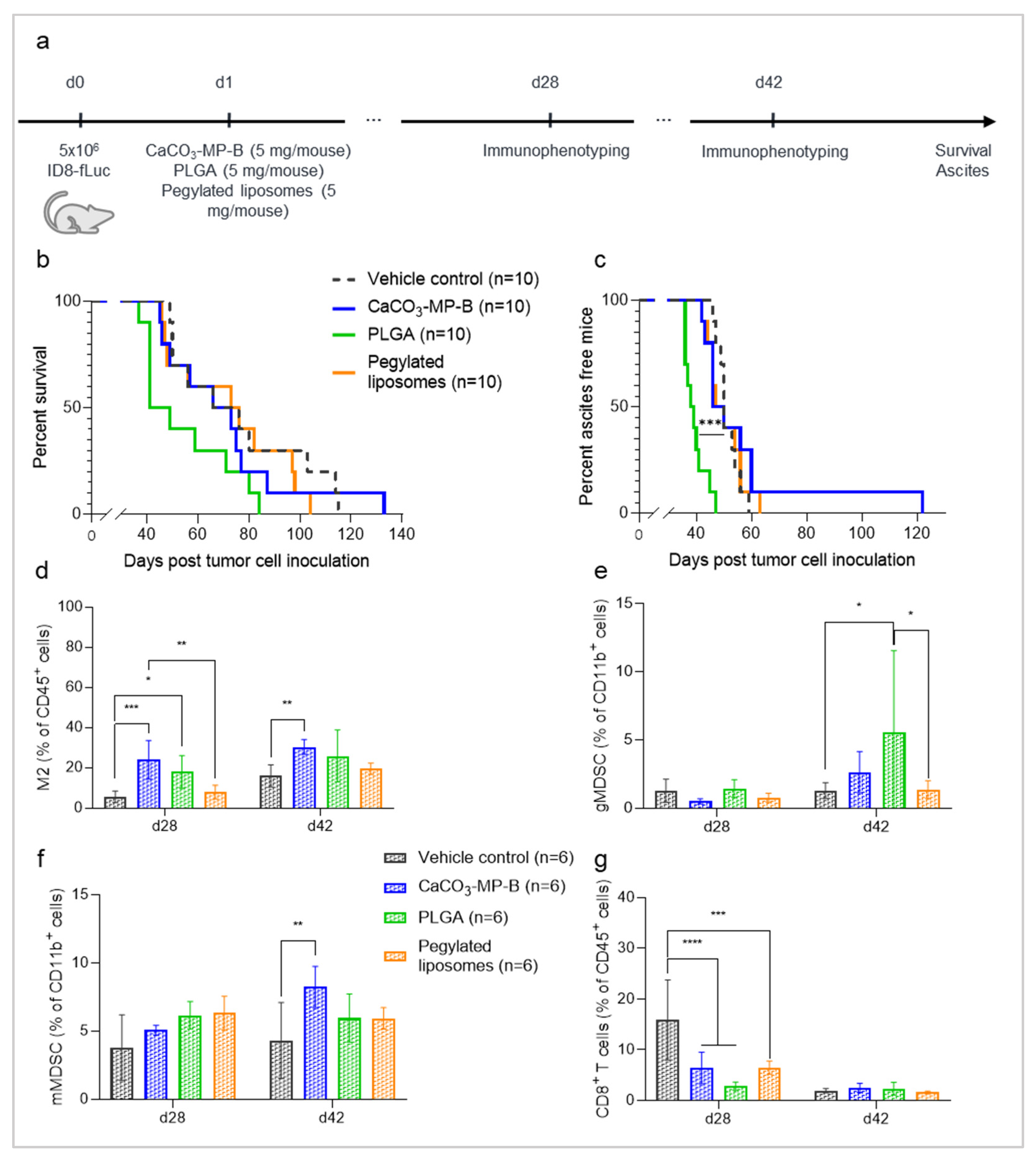
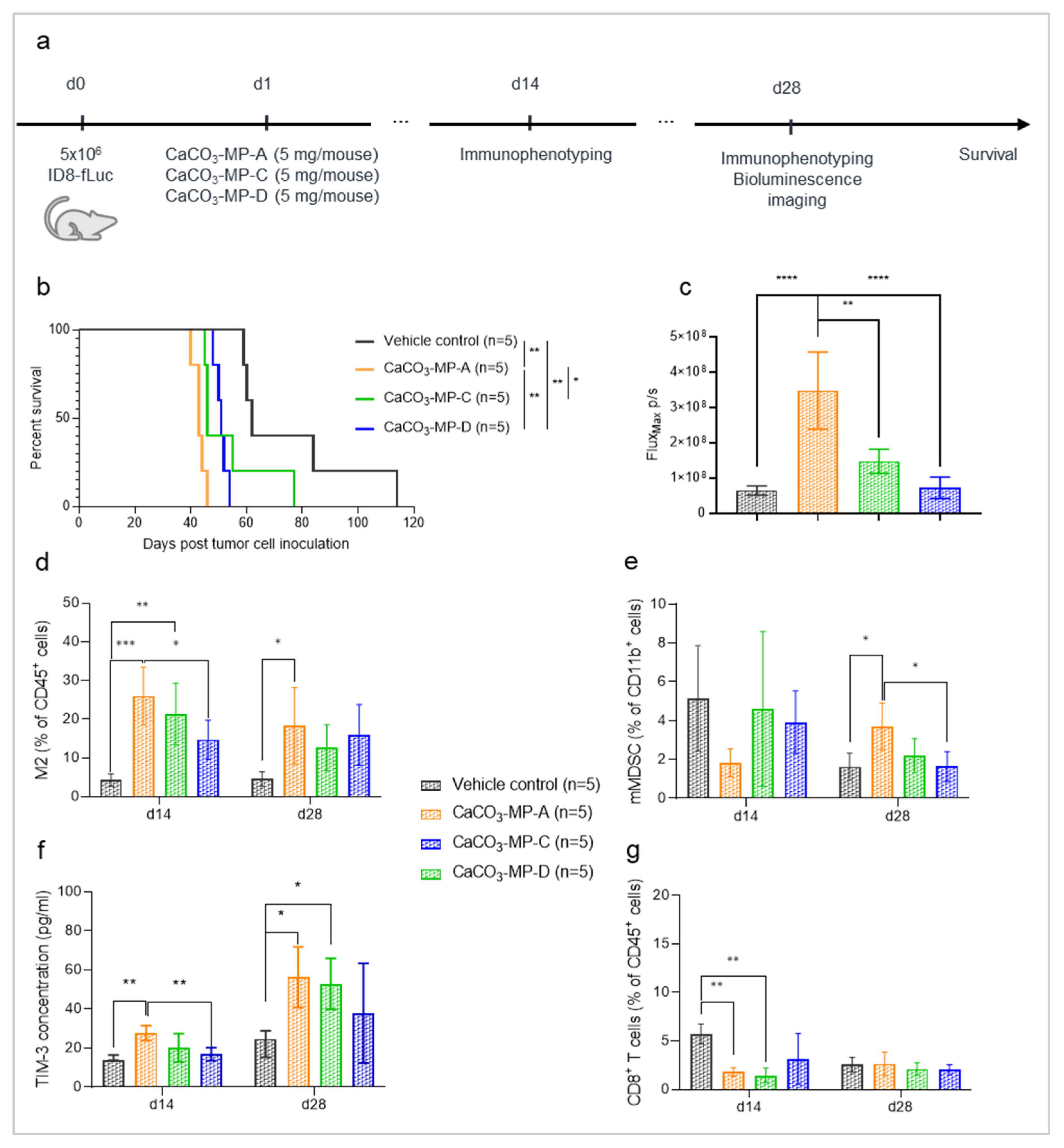
3.3. Different Product Formulations of CaCO3-Microparticles Have Different Effects on Tumor Progression and Innate Immune Status

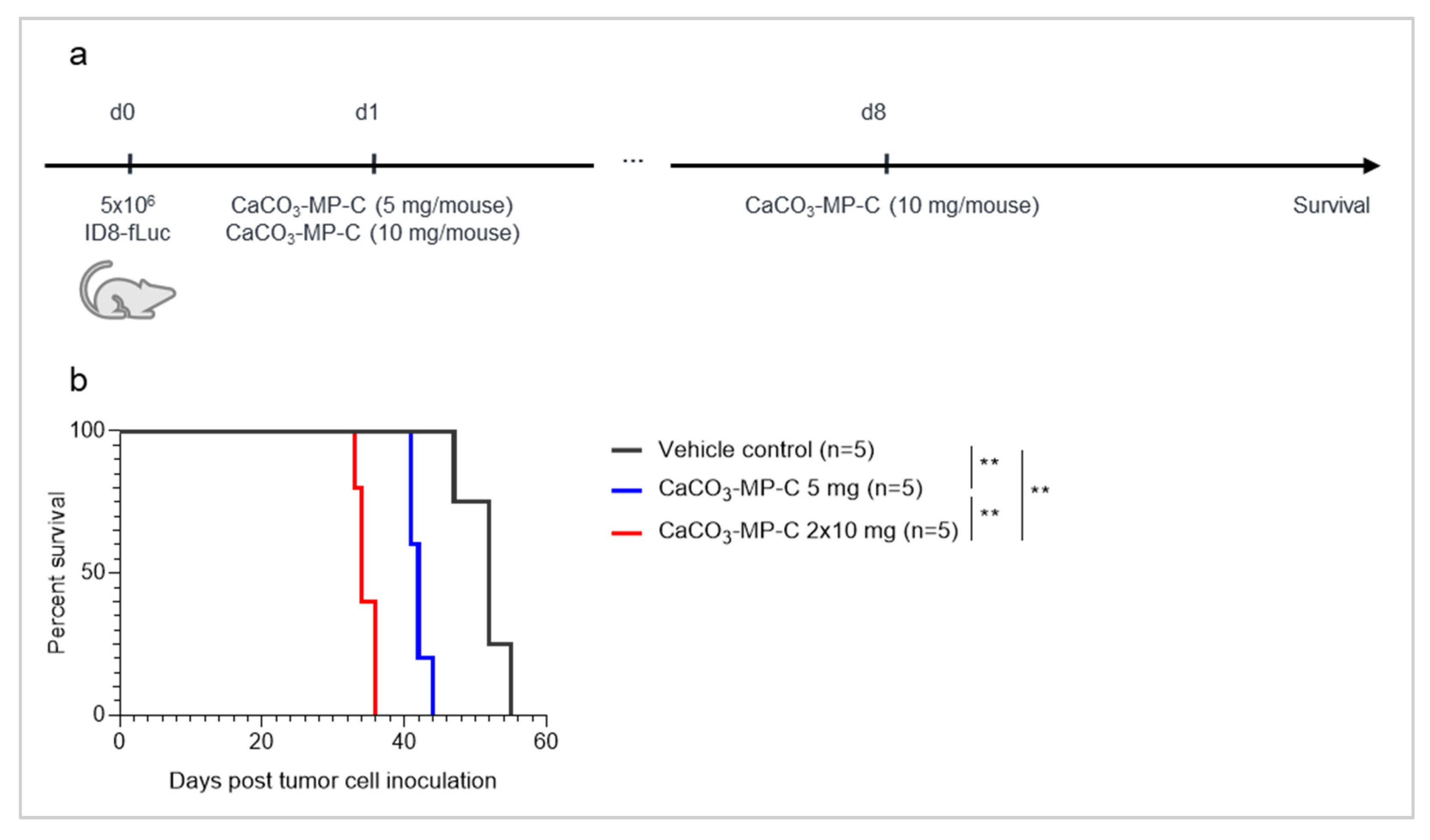
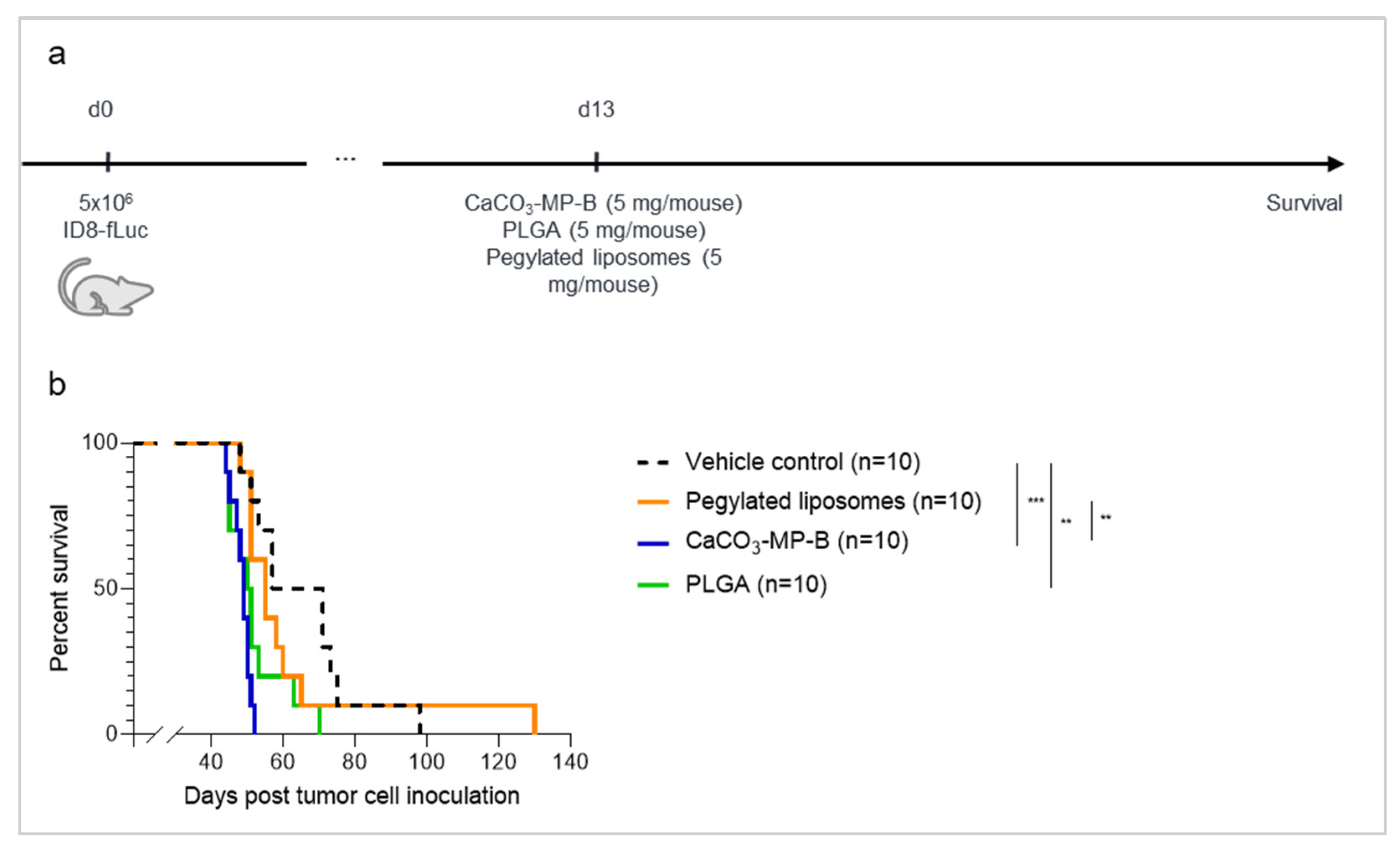
3.4. The Dose of CaCO3-Microparticles Influences the Degree of Hyperproliferation

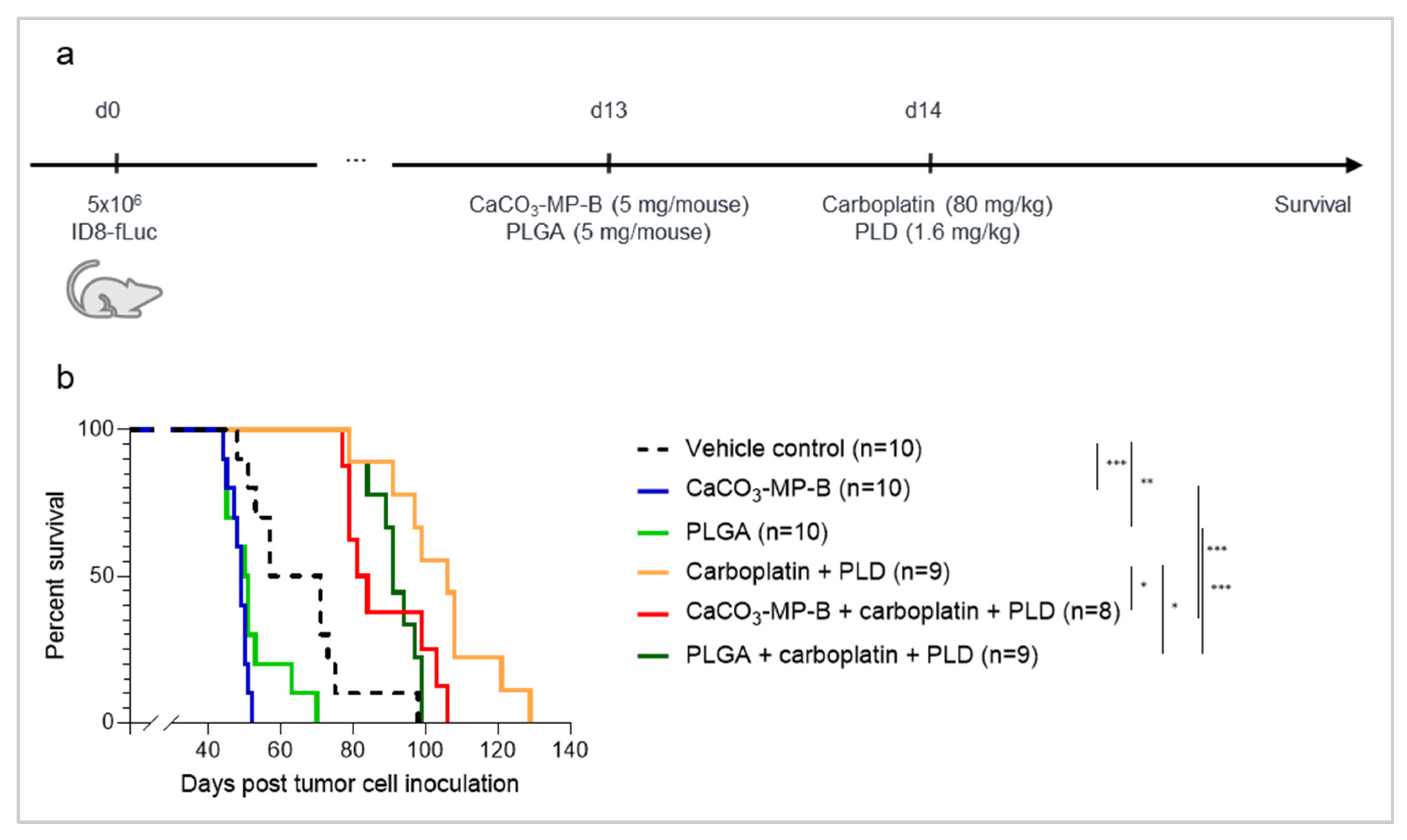
3.5. Chemotherapy Treatment Is Not Able to Sufficiently Reverse the Hyperproliferation Effect Seen with PLGA and CaCO3 Drug Carriers
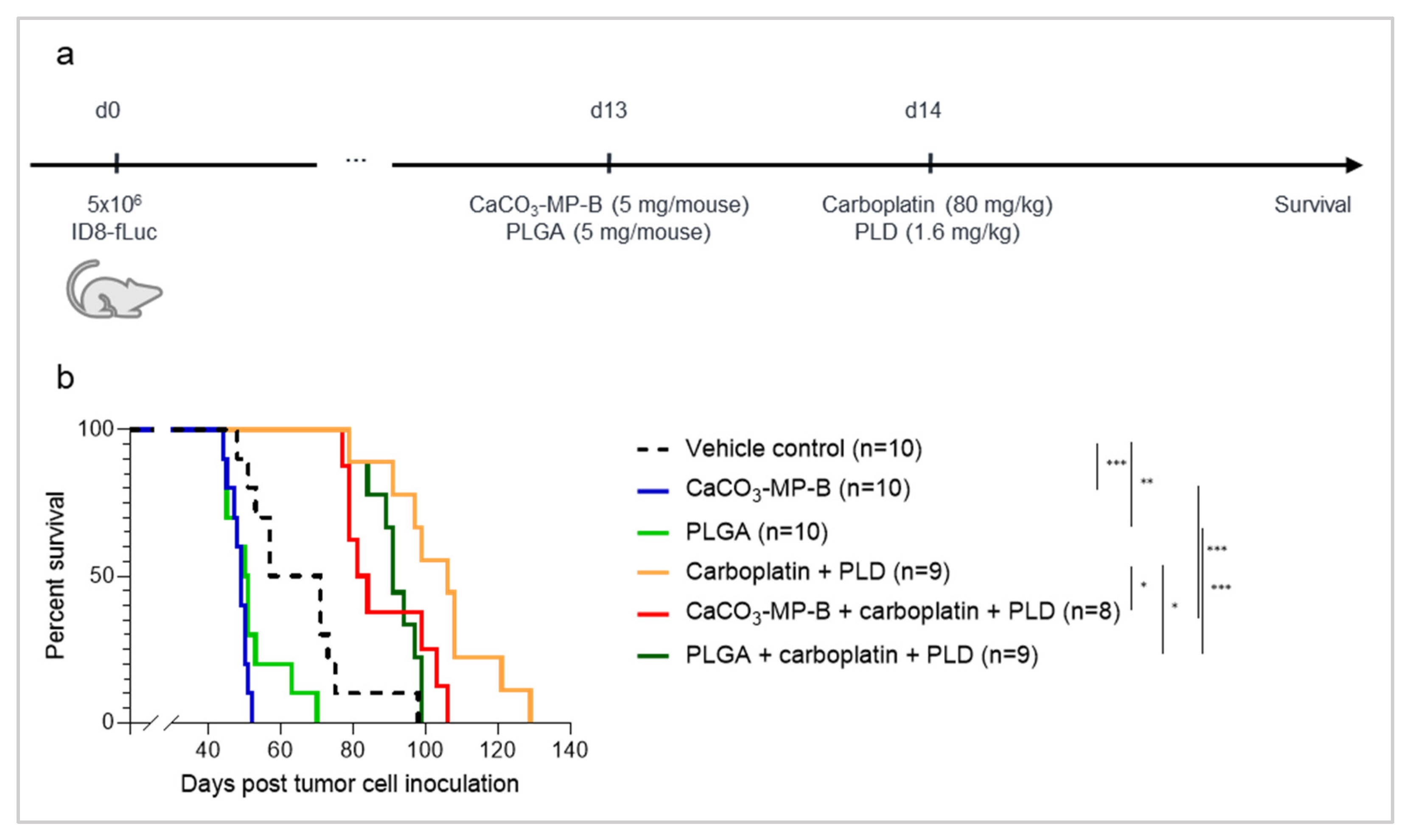
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA. Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prat, J. Staging classification for cancer of the ovary, fallopian tube, and peritoneum. Int. J. Gynecol. Obstet. 2014, 124, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Colombo, N.; Van Gorp, T.; Parma, G.; Amant, F.; Gatta, G.; Sessa, C.; Vergote, I. Ovarian cancer. Crit. Rev. Oncol. Hematol. 2006, 60, 159–179. [Google Scholar] [CrossRef]
- Vetter, M.H.; Hays, J.L. Use of Targeted Therapeutics in Epithelial Ovarian Cancer: A Review of Current Literature and Future Directions. Clin. Ther. 2018, 40, 361–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ledermann, J.A.; Colombo, N.; Oza, A.M.; Fujiwara, K.; Birrer, M.J.; Randall, L.M.; Poddubskaya, E.V.; Scambia, G.; Shparyk, Y.V.; Lim, M.C.; et al. Avelumab in combination with and/or following chemotherapy vs chemotherapy alone in patients with previously untreated epithelial ovarian cancer: Results from the phase 3 javelin ovarian 100 trial. Gynecol. Oncol. 2020, 159, 13–14. [Google Scholar] [CrossRef]
- Senapati, S.; Mahanta, A.K.; Kumar, S.; Maiti, P. Controlled drug delivery vehicles for cancer treatment and their performance. Signal Transduct. Target. Ther. 2018, 3, 7. [Google Scholar] [CrossRef] [Green Version]
- Dakwar, G.R.; Shariati, M.; Willaert, W.; Ceelen, W.; De Smedt, S.C.; Remaut, K. Nanomedicine-based intraperitoneal therapy for the treatment of peritoneal carcinomatosis—Mission possible? Adv. Drug Deliv. Rev. 2017, 108, 13–24. [Google Scholar] [CrossRef] [Green Version]
- Drummond, D.C.; Meyer, O.; Hong, K.; Kirpotin, D.B.; Papahadjopoulos, D. Optimizing Liposomes for Delivery of Chemotherapeutic agents to Solid Tumors. Farmcol. Rev. 1999, 51, 691–743. [Google Scholar]
- Amoozgar, Z.; Wang, L.; Brandstoetter, T.; Wallis, S.S.; Wilson, E.M.; Goldberg, M.S. Dual-Layer Surface Coating of PLGA-Based Nanoparticles Provides Slow-Release Drug Delivery To Achieve Metronomic Therapy in a Paclitaxel-Resistant Murine Ovarian Cancer Model. Biomacromolecules 2014, 15, 4187–4194. [Google Scholar] [CrossRef]
- Takeyama, H.; Mohri, N.; Mizuno, I.; Akamo, Y.; Sawai, H.; Manabe, T.; Yotsuyanagi, T.; Nakamura, S. Treatment of Peritoneal Carcinomatosis Using Carboplatin-loaded Hydroxyapatite Particles. Anticancer Res. 2006, 26, 4603–4606. [Google Scholar] [PubMed]
- Larina, I.V.; Evers, B.M.; Ashitkov, T.V.; Bartels, C.; Larin, K.V.; Esenaliev, R.O. Enhancement of drug delivery in tumors by using interaction of nanoparticles with ultrasound radiation. Technol. Cancer Res. Treat. 2005, 4, 217–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.; Wang, R.; Liu, Z.; Zhang, H. Preparation and characterization of calcium carbonate microspheres and their potential application as drug carriers. Mol. Med. Rep. 2018, 17, 8403–8408. [Google Scholar] [CrossRef] [PubMed]
- Williamson, S.K.; Johnson, G.A.; Maulhardt, H.A.; Moore, K.M.; McMeekin, D.S.; Schulz, T.K.; Reed, G.A.; Roby, K.F.; Mackay, C.B.; Smith, H.J.; et al. A phase I study of intraperitoneal nanoparticulate paclitaxel (Nanotax®) in patients with peritoneal malignancies. Cancer Chemother. Pharmacol. 2015, 75, 1075. [Google Scholar] [CrossRef] [Green Version]
- Coleman, R.L.; Brady, W.E.; McMeekin, D.S.; Rose, P.G.; Soper, J.T.; Lentz, S.S.; Hoffman, J.S.; Shahin, M.S. A phase II evaluation of nanoparticle, albumin-bound (nab) paclitaxel) in the treatment of recurrent or persistent platinum-resistant ovarian, fallopian tube, or primary peritoneal cancer: A Gynecologic Oncology Group Study. Gynecol. Oncol. 2011, 122, 111. [Google Scholar] [CrossRef] [Green Version]
- Pujade-Lauraine, E.; Wagner, U.; Aavall-Lundqvist, E.; Gebski, V.; Heywood, M.; Vasey, P.A.; Volgger, B.; Vergote, I.; Pignata, S.; Ferrero, A.; et al. Pegylated liposomal doxorubicin and carboplatin compared with paclitaxel and carboplatin for patients with platinum-sensitive ovarian cancer in late relapse. J. Clin. Oncol. 2010, 28, 3323–3329. [Google Scholar] [CrossRef]
- Wagner, U.; Marth, C.; Largillier, R.; Kaern, J.; Brown, C.; Heywood, M.; Bonaventura, T.; Vergote, I.; Piccirillo, M.C.; Fossati, R.; et al. Final overall survival results of phase III GCIG CALYPSO trial of pegylated liposomal doxorubicin and carboplatin vs paclitaxel and carboplatin in platinum-sensitive ovarian cancer patients. Br. J. Cancer 2012, 107, 588–591. [Google Scholar] [CrossRef] [Green Version]
- Li, R.G.; Lindland, K.; Tonstad, S.K.; Bønsdorff, T.B.; Juzeniene, A.; Westrøm, S.; Larsen, R.H. Improved formulation of 224Ra-labeled calcium carbonate microparticles by surface layer encapsulation and addition of EDTMP. Pharmaceutics 2021, 13, 634. [Google Scholar] [CrossRef]
- Westrøm, S.; Malenge, M.; Jorstad, I.S.; Napoli, E.; Bruland, Ø.S.; Bønsdorff, T.B.; Larsen, R.H. Ra-224 labeling of calcium carbonate microparticles for internal α-therapy: Preparation, stability, and biodistribution in mice. J. Label. Comp. Radiopharm. 2018, 61, 472–486. [Google Scholar] [CrossRef] [Green Version]
- Muslimov, A.R.; Antuganov, D.O.; Tarakanchikova, Y.V.; Zhukov, M.V.; Nadporojskii, M.A.; Zyuzin, M.V.; Timin, A.S. Calcium Carbonate Core—Shell Particles for Incorporation of 225Ac and Their Application in Local α-Radionuclide Therapy. ACS Appl. Mater. Interfaces 2021, 13, 25599–25610. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity′s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Conejo-Garcia, J.R.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N.; Rubin, S.C.; et al. Intratumoral T Cells, Recurrence, and Survival in Epithelial Ovarian Cancer. N. Engl. J. Med. 2003, 348, 203–213. [Google Scholar] [CrossRef] [Green Version]
- Vankerckhoven, A.; Wouters, R.; Mathivet, T.; Ceusters, J.; Baert, T.; Van Hoylandt, A.; Gerhardt, H.; Vergote, I.; Coosemans, A. Opposite Macrophage Polarization in Different Subsets of Ovarian Cancer: Observation from a Pilot Study. Cells 2020, 9, 305. [Google Scholar] [CrossRef] [Green Version]
- Coosemans, A.; Baert, T.; Ceusters, J.; Busschaert, P.; Landolfo, C.; Verschuere, T.; Van Rompuy, A.S.; Vanderstichele, A.; Froyman, W.; Neven, P.; et al. Myeloid-derived suppressor cells at diagnosis may discriminate between benign and malignant ovarian tumors. Int. J. Gynecol. Cancer 2019, 29, 1381–1388. [Google Scholar] [CrossRef] [PubMed]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving bioscience research reporting: The arrive guidelines for reporting animal research. PLoS Biol. 2010, 8, e1000412. [Google Scholar] [CrossRef] [PubMed]
- Council, N.R. Guide for the Care and Use of Laboratory Animals; National Academies Press: Washington, DC, USA, 2011. [Google Scholar]
- Moughon, D.L.; He, H.; Schokrpur, S.; Jiang, Z.K.; Yaqoob, M.; David, J.; Lin, C.; Luisa Iruela-Arispe, M.; Dorigo, O.; Wu, L. Microenvironment and Immunology Macrophage Blockade Using CSF1R Inhibitors Reverses the Vascular Leakage Underlying Malignant Ascites in Late-Stage Epithelial Ovarian Cancer. Cancer Res. 2015, 75, 4742–4752. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; He, Y.; Sun, X.; Li, Q.; Wang, W.; Zhao, A.; Di, W. A high M1/M2 ratio of tumor-associated macrophages is associated with extended survival in ovarian cancer patients. J. Ovarian Res. 2014, 7, 19. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Hu, R.; Zeng, Y.; Zhang, W.; Zhou, H.H. Tumour immune cell infiltration and survival after platinum-based chemotherapy in high-grade serous ovarian cancer subtypes: A gene expression-based computational study. EBioMedicine 2020, 51, 102602. [Google Scholar] [CrossRef] [Green Version]
- Camelliti, S.; Le Noci, V.; Bianchi, F.; Moscheni, C.; Arnaboldi, F.; Gagliano, N.; Balsari, A.; Garassino, M.C.; Tagliabue, E.; Sfondrini, L.; et al. Mechanisms of hyperprogressive disease after immune checkpoint inhibitor therapy: What we (don′t) know. J. Exp. Clin. Cancer Res. 2020, 39, 236. [Google Scholar] [CrossRef] [PubMed]
- Champiat, S.; Dercle, L.; Ammari, S.; Massard, C.; Hollebecque, A.; Postel-Vinay, S.; Chaput, N.; Eggermont, A.; Marabelle, A.; Soria, J.-C.; et al. Hyperprogressive Disease Is a New Pattern of Progression in Cancer Patients Treated by Anti-PD-1/PD-L1. Clin. Cancer Res. 2017, 23, 1920–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baert, T.; Vankerckhoven, A.; Riva, M.; Van Hoylandt, A.; Thirion, G.; Holger, G.; Mathivet, T.; Vergote, I.; Coosemans, A. Myeloid Derived Suppressor Cells: Key Drivers of Immunosuppression in Ovarian Cancer. Front. Immunol. 2019, 10, 1273. [Google Scholar] [CrossRef] [PubMed]
- Baek, H.; Lee, H.W.; Gangadaran, P.; Oh, J.M.; Zhu, L.; Rajendran, R.L.; Lee, J.; Ahn, B.-C. Role of M2-like macrophages in the progression of ovarian cancer. Exp. Cell Res. 2020, 395, 112211. [Google Scholar] [CrossRef] [PubMed]
- La-Beck, N.M.; Liu, X.; Wood, L.M. Harnessing liposome interactions with the immune system for the next breakthrough in cancer drug delivery. Front. Pharmacol. 2019, 10, 220. [Google Scholar] [CrossRef] [Green Version]
- Sabnani, M.K.; Rajan, R.; Rowland, B.; Mavinkurve, V.; Wood, L.M.; Gabizon, A.A.; La-Beck, N.M. Liposome promotion of tumor growth is associated with angiogenesis and inhibition of antitumor immune responses. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 259–262. [Google Scholar] [CrossRef]
- Lam, S.F.; Bishop, K.W.; Mintz, R.; Fang, L.; Achilefu, S. Calcium carbonate nanoparticles stimulate cancer cell reprogramming to suppress tumor growth and invasion in an organ-on-a-chip system. Sci. Rep. 2021, 11, 9246. [Google Scholar] [CrossRef]
- Kohane, D.S.; Tse, J.Y.; Yeo, Y.; Padera, R.; Shubina, M.; Langer, R. Biodegradable polymeric microspheres and nanospheres for drug delivery in the peritoneum. J. Biomed. Mater. Res. Part A 2006, 77, 351–361. [Google Scholar] [CrossRef]
- Lebre, F.; Sridharan, R.; Sawkins, M.J.; Kelly, D.J.; O′Brien, F.J.; Lavelle, E.C. The shape and size of hydroxyapatite particles dictate inflammatory responses following implantation. Sci. Rep. 2017, 7, 2922. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Locati, M. Tumor-associated macrophages as a paradigm of macrophage plasticity, diversity, and polarization lessons and open questions. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1478–1483. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wouters, R.; Westrøm, S.; Vankerckhoven, A.; Thirion, G.; Ceusters, J.; Claes, S.; Schols, D.; Bønsdorff, T.B.; Vergote, I.; Coosemans, A. Effect of Particle Carriers for Intraperitoneal Drug Delivery on the Course of Ovarian Cancer and Its Immune Microenvironment in a Mouse Model. Pharmaceutics 2022, 14, 687. https://doi.org/10.3390/pharmaceutics14040687
Wouters R, Westrøm S, Vankerckhoven A, Thirion G, Ceusters J, Claes S, Schols D, Bønsdorff TB, Vergote I, Coosemans A. Effect of Particle Carriers for Intraperitoneal Drug Delivery on the Course of Ovarian Cancer and Its Immune Microenvironment in a Mouse Model. Pharmaceutics. 2022; 14(4):687. https://doi.org/10.3390/pharmaceutics14040687
Chicago/Turabian StyleWouters, Roxanne, Sara Westrøm, Ann Vankerckhoven, Gitte Thirion, Jolien Ceusters, Sandra Claes, Dominique Schols, Tina B. Bønsdorff, Ignace Vergote, and An Coosemans. 2022. "Effect of Particle Carriers for Intraperitoneal Drug Delivery on the Course of Ovarian Cancer and Its Immune Microenvironment in a Mouse Model" Pharmaceutics 14, no. 4: 687. https://doi.org/10.3390/pharmaceutics14040687
APA StyleWouters, R., Westrøm, S., Vankerckhoven, A., Thirion, G., Ceusters, J., Claes, S., Schols, D., Bønsdorff, T. B., Vergote, I., & Coosemans, A. (2022). Effect of Particle Carriers for Intraperitoneal Drug Delivery on the Course of Ovarian Cancer and Its Immune Microenvironment in a Mouse Model. Pharmaceutics, 14(4), 687. https://doi.org/10.3390/pharmaceutics14040687