
Manipulating Macrophage/Microglia Polarization to Treat Glioblastoma or Multiple Sclerosis



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Different Polarization States of Macrophages
2.1. The M1 Polarization
2.2. The M2 Polarization
2.3. The New Concepts about “Polarization”
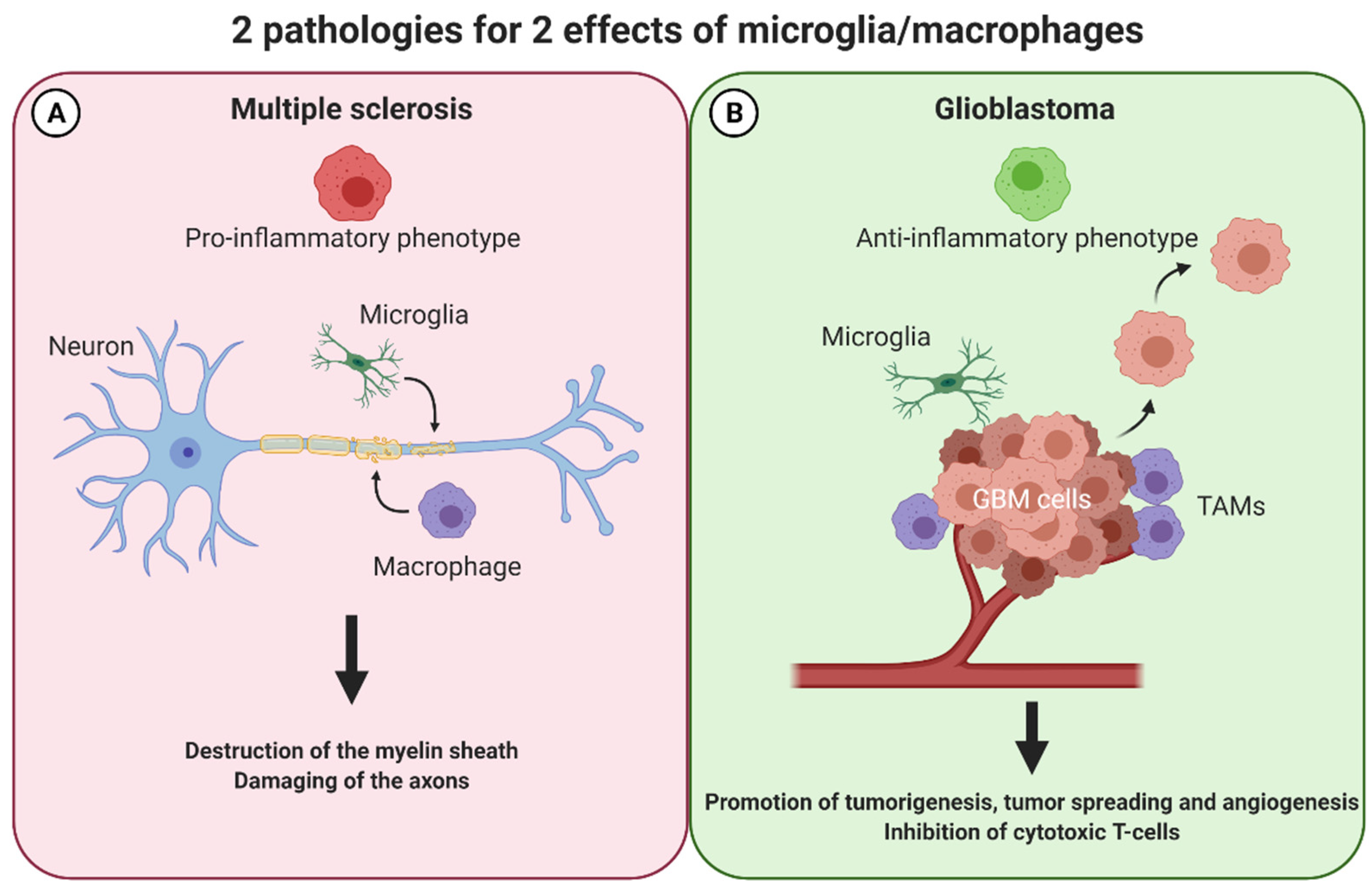
3. Macrophages/Microglia in MS
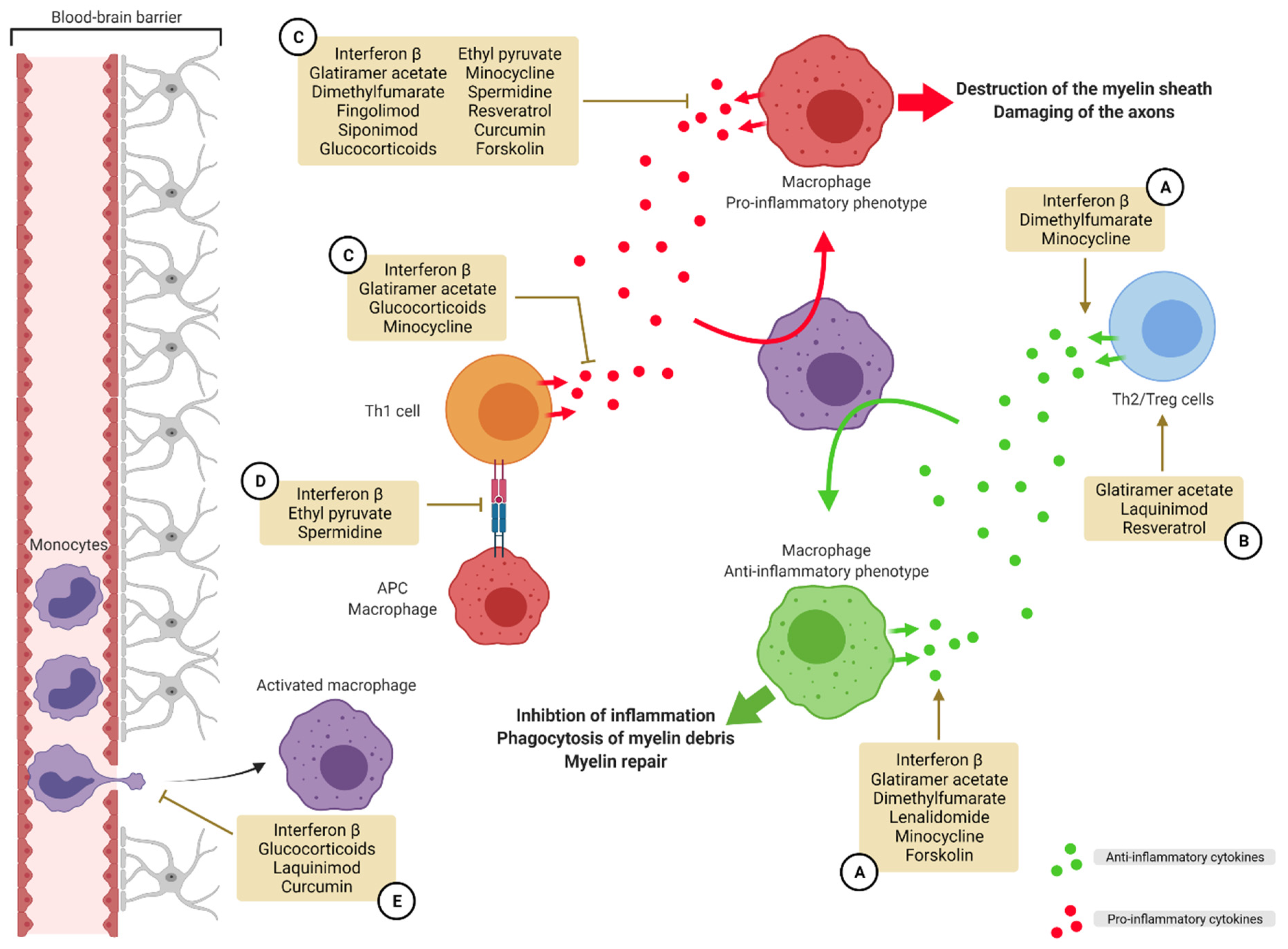
3.1. The M1-M2 Balance Consequences on Multiple Sclerosis
3.2. Treatments Influencing Polarization for MS Care Already on the Market
3.2.1. Proteins/Peptides
3.2.2. Small Molecules
3.3. Compounds Influencing Polarization for MS Care: New Perspectives
3.3.1. Synthetic Small Molecules
3.3.2. Natural Occurring Compounds
4. Macrophages/Microglia in GBM
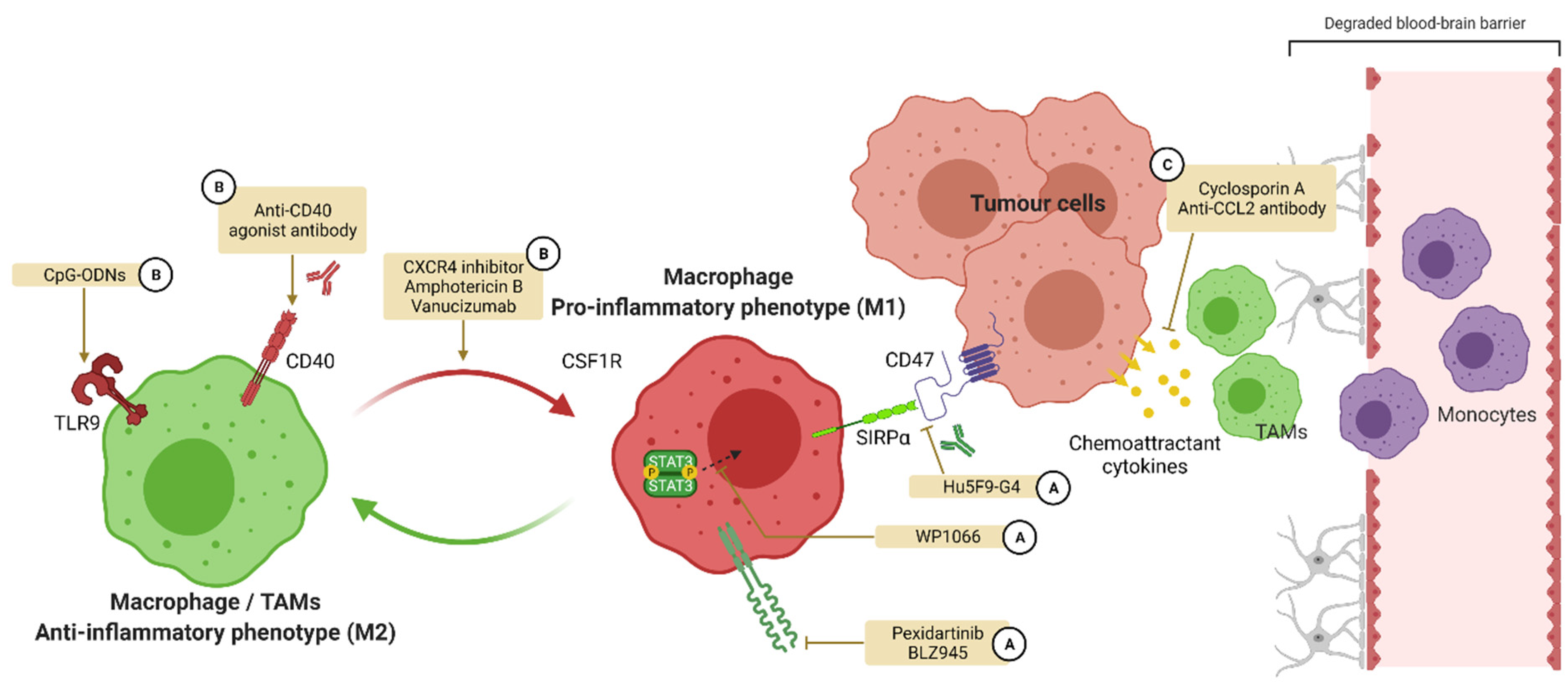
4.1. TAMs and M1-M2 Balance
4.2. Treatments Influencing Polarization for GBM Care
4.2.1. Colony Stimulating Factor 1 and Its Receptor
4.2.2. CD47
4.2.3. CD40
4.2.4. Toll-like Receptors (TLRs)
4.2.5. Signal Transducers and Activators of Transcription (STATs)
4.2.6. Other Targets
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shrivastava, R.; Shukla, N. Attributes of alternatively activated (M2) macrophages. Life Sci. 2019, 224, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Parisi, L.; Gini, E.; Baci, D.; Tremolati, M.; Fanuli, M.; Bassani, B.; Farronato, G.; Bruno, A.; Mortara, L. Macrophage Polarization in Chronic Inflammatory Diseases: Killers or Builders? J. Immunol. Res. 2018, 2018, 8917804. [Google Scholar] [CrossRef] [PubMed]
- Chu, F.; Shi, M.; Zheng, C.; Shen, D.; Zhu, J.; Zheng, X.; Cui, L. The roles of macrophages and microglia in multiple sclerosis and experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2018, 318, 1–7. [Google Scholar] [CrossRef]
- Omuro, A.; DeAngelis, L.M. Glioblastoma and Other Malignant Gliomas: A Clinical Review. JAMA 2013, 310, 1842. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef]
- Reich, D.S.; Lucchinetti, C.F.; Calabresi, P.A. Multiple Sclerosis. N. Engl. J. Med. 2018, 378, 169–180. [Google Scholar] [CrossRef]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 Macrophages and the Th1/Th2 Paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef]
- Arora, S.; Dev, K.; Agarwal, B.; Das, P.; Syed, M.A. Macrophages: Their role, activation and polarization in pulmonary diseases. Immunobiology 2018, 223, 383–396. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Gabrusiewicz, K.; Heimberger, A. The Controversial Role of Microglia in Malignant Gliomas. Clin. Dev. Immunol. 2013, 2013, 285246. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Rőszer, T. Understanding the Mysterious M2 Macrophage through Activation Markers and Effector Mechanisms. Mediat. Inflamm. 2015, 2015, 816460. [Google Scholar] [CrossRef]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.-A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Vogel, D.Y.; Vereyken, E.J.; Glim, J.E.; Heijnen, P.D.; Moeton, M.; van der Valk, P.; Amor, S.; Teunissen, C.E.; van Horssen, J.; Dijkstra, C.D. Macrophages in inflammatory multiple sclerosis lesions have an intermediate activation status. J. Neuroinflamm. 2013, 10, 809. [Google Scholar] [CrossRef]
- Li, C.; Menoret, A.; Farragher, C.; Ouyang, Z.; Bonin, C.; Holvoet, P.; Vella, A.T.; Zhou, B. Single-cell transcriptomics–based MacSpectrum reveals macrophage activation signatures in diseases. JCI Insight 2019, 4, e126453. [Google Scholar] [CrossRef]
- Saliba, A.-E.; Li, L.; Westermann, A.J.; Appenzeller, S.; Stapels, D.A.C.; Schulte, L.N.; Helaine, S.; Vogel, J. Single-cell RNA-seq ties macrophage polarization to growth rate of intracellular Salmonella. Nat. Microbiol. 2017, 2, 16206. [Google Scholar] [CrossRef]
- Hume, D.A. The Many Alternative Faces of Macrophage Activation. Front. Immunol. 2015, 6, 370. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Luo, C.; Jian, C.; Liao, Y.; Huang, Q.; Wu, Y.; Liu, X.; Zou, D.; Wu, Y. The role of microglia in multiple sclerosis. Neuropsychiatr. Dis. Treat. 2017, 13, 1661–1667. [Google Scholar] [CrossRef] [PubMed]
- Peferoen, L.A.N.; Vogel, D.Y.S.; Ummenthum, K.; Breur, M.; Heijnen, P.D.A.M.; Gerritsen, W.H.; Peferoen-Baert, R.M.B.; van der Valk, P.; Dijkstra, C.D.; Amor, S. Activation Status of Human Microglia Is Dependent on Lesion Formation Stage and Remyelination in Multiple Sclerosis. J. Neuropathol. Exp. Neurol. 2015, 74, 48–63. [Google Scholar] [CrossRef] [PubMed]
- Kieseier, B.C. The Mechanism of Action of Interferon-b in Relapsing Multiple Sclerosis. CNS Drugs 2011, 25, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.K.; Yong, V.W. Myeloid cells—Targets of medication in multiple sclerosis. Nat. Rev. Neurol. 2016, 12, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Nally, F.K.; De Santi, C.; McCoy, C.E. Nanomodulation of Macrophages in Multiple Sclerosis. Cells 2019, 8, 543. [Google Scholar] [CrossRef] [PubMed]
- Molnarfi, N.; Prod’homme, T.; Schulze-Topphoff, U.; Spencer, C.M.; Weber, M.S.; Patarroyo, J.C.; Lalive, P.H.; Zamvil, S.S. Glatiramer acetate treatment negatively regulates type I interferon signaling. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e179. [Google Scholar] [CrossRef]
- Schilling, S.; Goelz, S.; Linker, R.; Luehder, F.; Gold, R. Fumaric acid esters are effective in chronic experimental autoimmune encephalomyelitis and suppress macrophage infiltration. Clin. Exp. Immunol. 2006, 145, 101–107. [Google Scholar] [CrossRef]
- Wierinckx, A.; Brevé, J.; Mercier, D.; Schultzberg, M.; Drukarch, B.; Van Dam, A.-M. Detoxication enzyme inducers modify cytokine production in rat mixed glial cells. J. Neuroimmunol. 2005, 166, 132–143. [Google Scholar] [CrossRef]
- Di Dario, M.; Colombo, E.; Govi, C.; De Feo, D.; Messina, M.J.; Romeo, M.; Sangalli, F.; Moiola, L.; Rodegher, M.; Martino, G.; et al. Myeloid cells as target of fingolimod action in multiple sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e157. [Google Scholar] [CrossRef]
- Jackson, S.J.; Giovannoni, G.; Baker, D. Fingolimod modulates microglial activation to augment markers of remyelination. J. Neuroinflamm. 2011, 8, 76. [Google Scholar] [CrossRef]
- Aoki, M.; Kondo, A.; Matsunaga, N.; Honda, A.; Okubo, Y.; Takabe, K.; Ogawa, R. The Immunosuppressant Fingolimod (FTY720) for the Treatment of Mechanical Force-Induced Abnormal Scars. J. Immunol. Res. 2020, 2020, 7057195. [Google Scholar] [CrossRef] [PubMed]
- Behrangi, N.; Fischbach, F.; Kipp, M. Mechanism of Siponimod: Anti-Inflammatory and Neuroprotective Mode of Action. Cells 2019, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Gentile, A.; Musella, A.; Bullitta, S.; Fresegna, D.; De Vito, F.; Fantozzi, R.; Piras, E.; Gargano, F.; Borsellino, G.; Battistini, L.; et al. Siponimod (BAF312) prevents synaptic neurodegeneration in experimental multiple sclerosis. J. Neuroinflamm. 2016, 13, 207. [Google Scholar] [CrossRef] [PubMed]
- Kipp, M. Does Siponimod Exert Direct Effects in the Central Nervous System? Cells 2020, 9, 1771. [Google Scholar] [CrossRef]
- Fischer, H.J.; Finck, T.L.K.; Pellkofer, H.L.; Reichardt, H.M.; Lühder, F. Glucocorticoid Therapy of Multiple Sclerosis Patients Induces Anti-inflammatory Polarization and Increased Chemotaxis of Monocytes. Front. Immunol. 2019, 10, 1200. [Google Scholar] [CrossRef]
- Weng, Q.; Wang, J.; Wang, J.; Wang, J.; Sattar, F.; Zhang, Z.; Zheng, J.; Xu, Z.; Zhao, M.; Liu, X.; et al. Lenalidomide regulates CNS autoimmunity by promoting M2 macrophages polarization. Cell Death Dis. 2018, 9, 251. [Google Scholar] [CrossRef]
- Miljković, D.; Blaževski, J.; Petković, F.; Djedović, N.; Momčilović, M.; Stanisavljević, S.; Jevtić, B.; Mostarica Stojković, M.; Spasojević, I. A Comparative Analysis of Multiple Sclerosis–Relevant Anti-Inflammatory Properties of Ethyl Pyruvate and Dimethyl Fumarate. J. Immunol. 2015, 194, 2493–2503. [Google Scholar] [CrossRef]
- He, Y.; An, J.; Yin, J.-J.; Sui, R.-X.; Miao, Q.; Ding, Z.-B.; Han, Q.-X.; Wang, Q.; Ma, C.-G.; Xiao, B.-G. Ethyl pyruvate enhances spontaneous remyelination by targeting microglia phagocytosis. Int. Immunopharmacol. 2019, 77, 105929. [Google Scholar] [CrossRef]
- Brück, W.; Wegner, C. Insight into the mechanism of laquinimod action. J. Neurol. Sci. 2011, 306, 173–179. [Google Scholar] [CrossRef]
- Kieseier, B.C. Defining a role for laquinimod in multiple sclerosis. Ther. Adv. Neurol. Disord. 2014, 7, 195–205. [Google Scholar] [CrossRef]
- Mishra, M.K.; Wang, J.; Keough, M.B.; Fan, Y.; Silva, C.; Sloka, S.; Hayardeny, L.; Brück, W.; Yong, V.W. Laquinimod reduces neuroaxonal injury through inhibiting microglial activation. Ann. Clin. Transl. Neurol. 2014, 1, 409–422. [Google Scholar] [CrossRef] [PubMed]
- Comi, G.; Jeffery, D.; Kappos, L.; Montalban, X.; Boyko, A.; Rocca, M.A.; Filippi, M. Placebo-Controlled Trial of Oral Laquinimod for Multiple Sclerosis. N. Engl. J. Med. 2012, 366, 1000–1009. [Google Scholar] [CrossRef] [PubMed]
- On behalf of the BRAVO Study Group; Vollmer, T.L.; Sorensen, P.S.; Selmaj, K.; Zipp, F.; Havrdova, E.; Cohen, J.A.; Sasson, N.; Gilgun-Sherki, Y.; Arnold, D.L. A randomized placebo-controlled phase III trial of oral laquinimod for multiple sclerosis. J. Neurol. 2014, 261, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Brundula, V.; Rewcastle, N.B.; Metz, L.M.; Bernard, C.C.; Yong, V.W. Targeting leukocyte MMPs and transmigration. Brain 2002, 125, 1297–1308. [Google Scholar] [CrossRef]
- Giuliani, F. Minocycline attenuates T cell and microglia activity to impair cytokine production in T cell-microglia interaction. J. Leukoc. Biol. 2005, 78, 135–143. [Google Scholar] [CrossRef]
- Nikodemova, M.; Watters, J.J.; Jackson, S.J.; Yang, S.K.; Duncan, I.D. Minocycline Down-regulates MHC II Expression in Microglia and Macrophages through Inhibition of IRF-1 and Protein Kinase C (PKC)α/βII. J. Biol. Chem. 2007, 282, 15208–15216. [Google Scholar] [CrossRef]
- Kobayashi, K.; Imagama, S.; Ohgomori, T.; Hirano, K.; Uchimura, K.; Sakamoto, K.; Hirakawa, A.; Takeuchi, H.; Suzumura, A.; Ishiguro, N.; et al. Minocycline selectively inhibits M1 polarization of microglia. Cell Death Dis. 2013, 4, e525. [Google Scholar] [CrossRef]
- Metz, L.M.; Li, D.K.B.; Traboulsee, A.L.; Duquette, P.; Eliasziw, M.; Cerchiaro, G.; Greenfield, J.; Riddehough, A.; Yeung, M.; Kremenchutzky, M.; et al. Trial of Minocycline in a Clinically Isolated Syndrome of Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 2122–2133. [Google Scholar] [CrossRef]
- Marchand, D.K.; Butcher, R. Minocycline for Relapsing-Remitting Multiple Sclerosis and Clinically Isolated Syndrome: A Review of Clinical Effectiveness and Guidelines; CADTH Rapid Response Reports; Canadian Agency for Drugs and Technologies in Health: Ottawa, ON, USA, 2019. [Google Scholar]
- Sørensen, P.S.; Sellebjerg, F.; Lycke, J.; Färkkilä, M.; Créange, A.; Lund, C.G.; Schluep, M.; Frederiksen, J.L.; Stenager, E.; Pfleger, C.; et al. Minocycline added to subcutaneous interferon β-1a in multiple sclerosis: Randomized RECYCLINE study. Eur. J. Neurol. 2016, 23, 861–870. [Google Scholar] [CrossRef]
- Saqib, U.; Sarkar, S.; Suk, K.; Mohammad, O.; Baig, M.S.; Savai, R. Phytochemicals as modulators of M1-M2 macrophages in inflammation. Oncotarget 2018, 9, 17937–17950. [Google Scholar] [CrossRef]
- Wang, Y.; Smith, W.; Hao, D.; He, B.; Kong, L. M1 and M2 macrophage polarization and potentially therapeutic naturally occurring compounds. Int. Immunopharmacol. 2019, 70, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, K.; Barthelmes, J.; Stolz, L.; Beyer, S.; Diehl, O.; Tegeder, I. “Disease modifying nutricals” for multiple sclerosis. Pharmacol. Ther. 2015, 148, 85–113. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Park, H. Anti-inflammatory effects of spermidine in lipopolysaccharide-stimulated BV2 microglial cells. J. Biomed. Sci. 2012, 19, 31. [Google Scholar] [CrossRef]
- Yang, Q.; Zheng, C.; Cao, J.; Cao, G.; Shou, P.; Lin, L.; Velletri, T.; Jiang, M.; Chen, Q.; Han, Y.; et al. Spermidine alleviates experimental autoimmune encephalomyelitis through inducing inhibitory macrophages. Cell Death Differ. 2016, 23, 1850–1861. [Google Scholar] [CrossRef]
- Guo, X.; Harada, C.; Namekata, K.; Kimura, A.; Mitamura, Y.; Yoshida, H.; Matsumoto, Y.; Harada, T. Spermidine Alleviates Severity of Murine Experimental Autoimmune Encephalomyelitis. Investig. Opthalmology Vis. Sci. 2011, 52, 2696. [Google Scholar] [CrossRef] [PubMed]
- Ghaiad, H.R.; Nooh, M.M.; El-Sawalhi, M.M.; Shaheen, A.A. Resveratrol Promotes Remyelination in Cuprizone Model of Multiple Sclerosis: Biochemical and Histological Study. Mol. Neurobiol. 2017, 54, 3219–3229. [Google Scholar] [CrossRef]
- Schwager, J.; Richard, N.; Widmer, F.; Raederstorff, D. Resveratrol distinctively modulates the inflammatory profiles of immune and endothelial cells. BMC Complement. Altern. Med. 2017, 17, 309. [Google Scholar] [CrossRef]
- Buttari, B.; Profumo, E.; Segoni, L.; D’Arcangelo, D.; Rossi, S.; Facchiano, F.; Saso, L.; Businaro, R.; Iuliano, L.; Riganò, R. Resveratrol Counteracts Inflammation in Human M1 and M2 Macrophages upon Challenge with 7-Oxo-Cholesterol: Potential Therapeutic Implications in Atherosclerosis. Oxid. Med. Cell. Longev. 2014, 2014, 257543. [Google Scholar] [CrossRef]
- Imler, T.J.; Petro, T.M. Decreased severity of experimental autoimmune encephalomyelitis during resveratrol administration is associated with increased IL-17+IL-10+ T cells, CD4− IFN-γ+ cells, and decreased macrophage IL-6 expression. Int. Immunopharmacol. 2009, 9, 134–143. [Google Scholar] [CrossRef]
- Sato, F.; Martinez, N.E.; Shahid, M.; Rose, J.W.; Carlson, N.G.; Tsunoda, I. Resveratrol Exacerbates Both Autoimmune and Viral Models of Multiple Sclerosis. Am. J. Pathol. 2013, 183, 1390–1396. [Google Scholar] [CrossRef]
- Qureshi, M.; Al-Suhaimi, E.A.; Wahid, F.; Shehzad, O.; Shehzad, A. Therapeutic potential of curcumin for multiple sclerosis. Neurol. Sci. 2018, 39, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Ghanaatian, N.; Lashgari, N.; Abdolghaffari, A.H.; Rajaee, S.M.; Panahi, Y.; Barreto, G.E.; Butler, A.E.; Sahebkar, A. Curcumin as a therapeutic candidate for multiple sclerosis: Molecular mechanisms and targets. J. Cell. Physiol. 2019, 234, 12237–12248. [Google Scholar] [CrossRef] [PubMed]
- Esmaeilzadeh, E.; Soleimani, M.; Zare-Abdollahi, D.; Jameie, B.; Khorram Kohrshid, H.R. Curcumin ameliorates experimental autoimmune encephalomyelitis in a C57BL/6 mouse model. Drug Dev. Res. 2019, 80, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, F.; Bagheri, H.; Barreto, G.E.; Read, M.I.; Sahebkar, A. Effects of Curcumin on Microglial Cells. Neurotox. Res. 2019, 36, 12–26. [Google Scholar] [CrossRef]
- Lu, L.; Qi, S.; Chen, Y.; Luo, H.; Huang, S.; Yu, X.; Luo, Q.; Zhang, Z. Targeted immunomodulation of inflammatory monocytes across the blood-brain barrier by curcumin-loaded nanoparticles delays the progression of experimental autoimmune encephalomyelitis. Biomaterials 2020, 245, 119987. [Google Scholar] [CrossRef]
- Dolati, S.; Ahmadi, M.; Aghebti-Maleki, L.; Nikmaram, A.; Marofi, F.; Rikhtegar, R.; Ayromlou, H.; Yousefi, M. Nanocurcumin is a potential novel therapy for multiple sclerosis by influencing inflammatory mediators. Pharmacol. Rep. 2018, 70, 1158–1167. [Google Scholar] [CrossRef]
- Veremeyko, T.; Yung, A.W.Y.; Dukhinova, M.; Kuznetsova, I.S.; Pomytkin, I.; Lyundup, A.; Strekalova, T.; Barteneva, N.S.; Ponomarev, E.D. Cyclic AMP Pathway Suppress Autoimmune Neuroinflammation by Inhibiting Functions of Encephalitogenic CD4 T Cells and Enhancing M2 Macrophage Polarization at the Site of Inflammation. Front. Immunol. 2018, 9, 50. [Google Scholar] [CrossRef]
- Rolle, C.E.; Sengupta, S.; Lesniak, M.S. Mechanisms of Immune Evasion by Gliomas. In Glioma; Yamanaka, R., Ed.; Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2012; Volume 746, pp. 53–76. ISBN 978-1-4614-3145-9. [Google Scholar]
- Hui, L.; Chen, Y. Tumor microenvironment: Sanctuary of the devil. Cancer Lett. 2015, 368, 7–13. [Google Scholar] [CrossRef]
- Ruffell, B.; Affara, N.I.; Coussens, L.M. Differential macrophage programming in the tumor microenvironment. Trends Immunol. 2012, 33, 119–126. [Google Scholar] [CrossRef]
- Solinas, G.; Germano, G.; Mantovani, A.; Allavena, P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J. Leukoc. Biol. 2009, 86, 1065–1073. [Google Scholar] [CrossRef]
- Ao, J.-Y.; Zhu, X.-D.; Chai, Z.-T.; Cai, H.; Zhang, Y.-Y.; Zhang, K.-Z.; Kong, L.-Q.; Zhang, N.; Ye, B.-G.; Ma, D.-N.; et al. Colony-Stimulating Factor 1 Receptor Blockade Inhibits Tumor Growth by Altering the Polarization of Tumor-Associated Macrophages in Hepatocellular Carcinoma. Mol. Cancer Ther. 2017, 16, 1544–1554. [Google Scholar] [CrossRef] [PubMed]
- Cannarile, M.A.; Weisser, M.; Jacob, W.; Jegg, A.-M.; Ries, C.H.; Rüttinger, D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J. Immunother. Cancer 2017, 5, 53. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Knolhoff, B.L.; Meyer, M.A.; Nywening, T.M.; West, B.L.; Luo, J.; Wang-Gillam, A.; Goedegebuure, S.P.; Linehan, D.C.; DeNardo, D.G. CSF1/CSF1R Blockade Reprograms Tumor-Infiltrating Macrophages and Improves Response to T-cell Checkpoint Immunotherapy in Pancreatic Cancer Models. Cancer Res. 2014, 74, 5057–5069. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Kowal, J.; Akkari, L.; Schuhmacher, A.J.; Huse, J.T.; West, B.L.; Joyce, J.A. Inhibition of colony stimulating factor-1 receptor abrogates microenvironment-mediated therapeutic resistance in gliomas. Oncogene 2017, 36, 6049–6058. [Google Scholar] [CrossRef] [PubMed]
- Coniglio, S.J.; Eugenin, E.; Dobrenis, K.; Stanley, E.R.; West, B.L.; Symons, M.H.; Segall, J.E. Microglial Stimulation of Glioblastoma Invasion Involves Epidermal Growth Factor Receptor (EGFR) and Colony Stimulating Factor 1 Receptor (CSF-1R) Signaling. Mol. Med. 2012, 18, 519–527. [Google Scholar] [CrossRef]
- Butowski, N.; Colman, H.; De Groot, J.F.; Omuro, A.M.; Nayak, L.; Wen, P.Y.; Cloughesy, T.F.; Marimuthu, A.; Haidar, S.; Perry, A.; et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: An Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro-Oncology 2016, 18, 557–564. [Google Scholar] [CrossRef]
- A Phase 1b/2 Study of PLX3397 + Radiation Therapy + Temozolomide in Patients With Newly Diagnosed Glioblastoma—Study Results—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/results/NCT01790503 (accessed on 11 June 2021).
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef]
- Quail, D.F.; Bowman, R.L.; Akkari, L.; Quick, M.L.; Schuhmacher, A.J.; Huse, J.T.; Holland, E.C.; Sutton, J.C.; Joyce, J.A. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science 2016, 352, aad3018. [Google Scholar] [CrossRef]
- Zhang, M.; Hutter, G.; Kahn, S.A.; Azad, T.D.; Gholamin, S.; Xu, C.Y.; Liu, J.; Achrol, A.S.; Richard, C.; Sommerkamp, P.; et al. Anti-CD47 Treatment Stimulates Phagocytosis of Glioblastoma by M1 and M2 Polarized Macrophages and Promotes M1 Polarized Macrophages In Vivo. PLoS ONE 2016, 11, e0153550. [Google Scholar] [CrossRef]
- Zhu, H.; Leiss, L.; Yang, N.; Rygh, C.B.; Mitra, S.S.; Cheshier, S.H.; Weissman, I.L.; Huang, B.; Miletic, H.; Bjerkvig, R.; et al. Surgical debulking promotes recruitment of macrophages and triggers glioblastoma phagocytosis in combination with CD47 blocking immunotherapy. Oncotarget 2017, 8, 12145–12157. [Google Scholar] [CrossRef]
- Li, F.; Lv, B.; Liu, Y.; Hua, T.; Han, J.; Sun, C.; Xu, L.; Zhang, Z.; Feng, Z.; Cai, Y.; et al. Blocking the CD47-SIRPα axis by delivery of anti-CD47 antibody induces antitumor effects in glioma and glioma stem cells. OncoImmunology 2018, 7, e1391973. [Google Scholar] [CrossRef] [PubMed]
- Chonan, M.; Saito, R.; Shoji, T.; Shibahara, I.; Kanamori, M.; Sonoda, Y.; Watanabe, M.; Kikuchi, T.; Ishii, N.; Tominaga, T. CD40/CD40L expression correlates with the survival of patients with glioblastomas and an augmentation in CD40 signaling enhances the efficacy of vaccinations against glioma models. Neuro-Oncology 2015, 17, 1453–1462. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Chiorean, E.G.; Fishman, M.P.; Saboury, B.; Teitelbaum, U.R.; Sun, W.; Huhn, R.D.; Song, W.; Li, D.; Sharp, L.L.; et al. CD40 Agonists Alter Tumor Stroma and Show Efficacy Against Pancreatic Carcinoma in Mice and Humans. Science 2011, 331, 1612–1616. [Google Scholar] [CrossRef] [PubMed]
- Walker, P.R.; Migliorini, D. The CD40/CD40L axis in glioma progression and therapy. Neuro-Oncology 2015, 17, 1428–1430. [Google Scholar] [CrossRef] [PubMed]
- Kosaka, A.; Ohkuri, T.; Okada, H. Combination of an agonistic anti-CD40 monoclonal antibody and the COX-2 inhibitor celecoxib induces anti-glioma effects by promotion of type-1 immunity in myeloid cells and T-cells. Cancer Immunol. Immunother. 2014, 63, 847–857. [Google Scholar] [CrossRef] [PubMed]
- Abarca-Merlin, D.M.; Maldonado-Bernal, C.; Alvarez-Arellano, L. Toll-Like Receptors as Therapeutic Targets in Central Nervous System Tumors. BioMed Res. Int. 2019, 2019, 5286358. [Google Scholar] [CrossRef]
- Grauer, O.M.; Molling, J.W.; Bennink, E.; Toonen, L.W.J.; Sutmuller, R.P.M.; Nierkens, S.; Adema, G.J. TLR Ligands in the Local Treatment of Established Intracerebral Murine Gliomas. J. Immunol. 2008, 181, 6720–6729. [Google Scholar] [CrossRef]
- Carpentier, A.F.; Xie, J.; Mokhtari, K.; Delattre, J.Y. Successful treatment of intracranial gliomas in rat by oligodeoxynucleotides containing CpG motifs. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2000, 6, 2469–2473. [Google Scholar]
- Ursu, R.; Taillibert, S.; Banissi, C.; Vicaut, E.; Bailon, O.; Le Rhun, E.; Guillamo, J.-S.; Psimaras, D.; Tibi, A.; Sacko, A.; et al. Immunotherapy with CpG-ODN in neoplastic meningitis: A phase I trial. Cancer Sci. 2015, 106, 1212–1218. [Google Scholar] [CrossRef]
- Carpentier, A.; Laigle-Donadey, F.; Zohar, S.; Capelle, L.; Behin, A.; Tibi, A.; Martin-Duverneuil, N.; Sanson, M.; Lacomblez, L.; Taillibert, S.; et al. Phase 1 trial of a CpG oligodeoxynucleotide for patients with recurrent glioblastoma1. Neuro-Oncology 2006, 8, 60–66. [Google Scholar] [CrossRef]
- Carpentier, A.; Metellus, P.; Ursu, R.; Zohar, S.; Lafitte, F.; Barrie, M.; Meng, Y.; Richard, M.; Parizot, C.; Laigle-Donadey, F.; et al. Intracerebral administration of CpG oligonucleotide for patients with recurrent glioblastoma: A phase II study. Neuro-Oncology 2010, 12, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Schön, M.P.; Schön, M. TLR7 and TLR8 as targets in cancer therapy. Oncogene 2008, 27, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Iwamaru, A.; Szymanski, S.; Iwado, E.; Aoki, H.; Yokoyama, T.; Fokt, I.; Hess, K.; Conrad, C.; Madden, T.; Sawaya, R.; et al. A novel inhibitor of the STAT3 pathway induces apoptosis in malignant glioma cells both in vitro and in vivo. Oncogene 2007, 26, 2435–2444. [Google Scholar] [CrossRef] [PubMed]
- Carvalho da Fonseca, A.C.; Badie, B. Microglia and Macrophages in Malignant Gliomas: Recent Discoveries and Implications for Promising Therapies. Clin. Dev. Immunol. 2013, 2013, 264124. [Google Scholar] [CrossRef]
- Hussain, S.F.; Kong, L.-Y.; Jordan, J.; Conrad, C.; Madden, T.; Fokt, I.; Priebe, W.; Heimberger, A.B. A Novel Small Molecule Inhibitor of Signal Transducers and Activators of Transcription 3 Reverses Immune Tolerance in Malignant Glioma Patients. Cancer Res. 2007, 67, 9630–9636. [Google Scholar] [CrossRef]
- Mercurio, L.; Ajmone-Cat, M.A.; Cecchetti, S.; Ricci, A.; Bozzuto, G.; Molinari, A.; Manni, I.; Pollo, B.; Scala, S.; Carpinelli, G.; et al. Targeting CXCR4 by a selective peptide antagonist modulates tumor microenvironment and microglia reactivity in a human glioblastoma model. J. Exp. Clin. Cancer Res. 2016, 35, 55. [Google Scholar] [CrossRef]
- Sarkar, S.; Döring, A.; Zemp, F.J.; Silva, C.; Lun, X.; Wang, X.; Kelly, J.; Hader, W.; Hamilton, M.; Mercier, P.; et al. Therapeutic activation of macrophages and microglia to suppress brain tumor-initiating cells. Nat. Neurosci. 2014, 17, 46–55. [Google Scholar] [CrossRef]
- Kloepper, J.; Riedemann, L.; Amoozgar, Z.; Seano, G.; Susek, K.; Yu, V.; Dalvie, N.; Amelung, R.L.; Datta, M.; Song, J.W.; et al. Ang-2/VEGF bispecific antibody reprograms macrophages and resident microglia to anti-tumor phenotype and prolongs glioblastoma survival. Proc. Natl. Acad. Sci. USA 2016, 113, 4476–4481. [Google Scholar] [CrossRef]
- Russo, C.D.; Cappoli, N. Glioma associated microglia/macrophages, a potential pharmacological target to promote antitumor inflammatory immune response in the treatment of glioblastoma. Neuroimmunol. Neuroinflamm. 2018, 5, 36. [Google Scholar] [CrossRef][Green Version]
- Zhu, X.; Fujita, M.; Snyder, L.A.; Okada, H. Systemic delivery of neutralizing antibody targeting CCL2 for glioma therapy. J. Neurooncol. 2011, 104, 83–92. [Google Scholar] [CrossRef]
- Jacob, L.; Sawma, P.; Garnier, N.; Meyer, L.A.T.; Fritz, J.; Hussenet, T.; Spenlé, C.; Goetz, J.; Vermot, J.; Fernandez, A.; et al. Inhibition of PlexA1-mediated brain tumor growth and tumor-associated angiogenesis using a transmembrane domain targeting peptide. Oncotarget 2016, 7, 57851–57865. [Google Scholar] [CrossRef] [PubMed]
- Nasarre, C.; Roth, M.; Jacob, L.; Roth, L.; Koncina, E.; Thien, A.; Labourdette, G.; Poulet, P.; Hubert, P.; Crémel, G.; et al. Peptide-based interference of the transmembrane domain of neuropilin-1 inhibits glioma growth in vivo. Oncogene 2010, 29, 2381–2392. [Google Scholar] [CrossRef] [PubMed]
- Binamé, F.; Pham-Van, L.D.; Spenlé, C.; Jolivel, V.; Birmpili, D.; Meyer, L.A.; Jacob, L.; Meyer, L.; Mensah-Nyagan, A.G.; Po, C.; et al. Disruption of Sema3A/Plexin-A1 inhibitory signalling in oligodendrocytes as a therapeutic strategy to promote remyelination. EMBO Mol. Med. 2019, 11, e10378. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, C.; Appert-Collin, A.; Bagnard, D.; Blaise, S.; Romier-Crouzet, B.; Efremov, R.G.; Sartelet, H.; Duca, L.; Maurice, P.; Bennasroune, A. Transmembrane Peptides as Inhibitors of Protein-Protein Interactions: An Efficient Strategy to Target Cancer Cells? Front. Oncol. 2020, 10, 519. [Google Scholar] [CrossRef]
- Alves, D.S.; Westerfield, J.M.; Shi, X.; Nguyen, V.P.; Stefanski, K.M.; Booth, K.R.; Kim, S.; Morrell-Falvey, J.; Wang, B.-C.; Abel, S.M.; et al. A novel pH-dependent membrane peptide that binds to EphA2 and inhibits cell migration. eLife 2018, 7, e36645. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuntzel, T.; Bagnard, D. Manipulating Macrophage/Microglia Polarization to Treat Glioblastoma or Multiple Sclerosis. Pharmaceutics 2022, 14, 344. https://doi.org/10.3390/pharmaceutics14020344
Kuntzel T, Bagnard D. Manipulating Macrophage/Microglia Polarization to Treat Glioblastoma or Multiple Sclerosis. Pharmaceutics. 2022; 14(2):344. https://doi.org/10.3390/pharmaceutics14020344
Chicago/Turabian StyleKuntzel, Thomas, and Dominique Bagnard. 2022. "Manipulating Macrophage/Microglia Polarization to Treat Glioblastoma or Multiple Sclerosis" Pharmaceutics 14, no. 2: 344. https://doi.org/10.3390/pharmaceutics14020344
APA StyleKuntzel, T., & Bagnard, D. (2022). Manipulating Macrophage/Microglia Polarization to Treat Glioblastoma or Multiple Sclerosis. Pharmaceutics, 14(2), 344. https://doi.org/10.3390/pharmaceutics14020344