
Formulation and Evaluation of Self-Nanoemulsifying Drug Delivery System Derived Tablet Containing Sertraline

 ,
,  , , ,
, , ,  ,
,  ,
,  , and
, and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Quantification of Sertraline
2.3. Liquid Self-Nanoemulsifying Drug Delivery System (L-SNEDDS)
2.3.1. Preliminary Studies for Components of L-SNEDDS
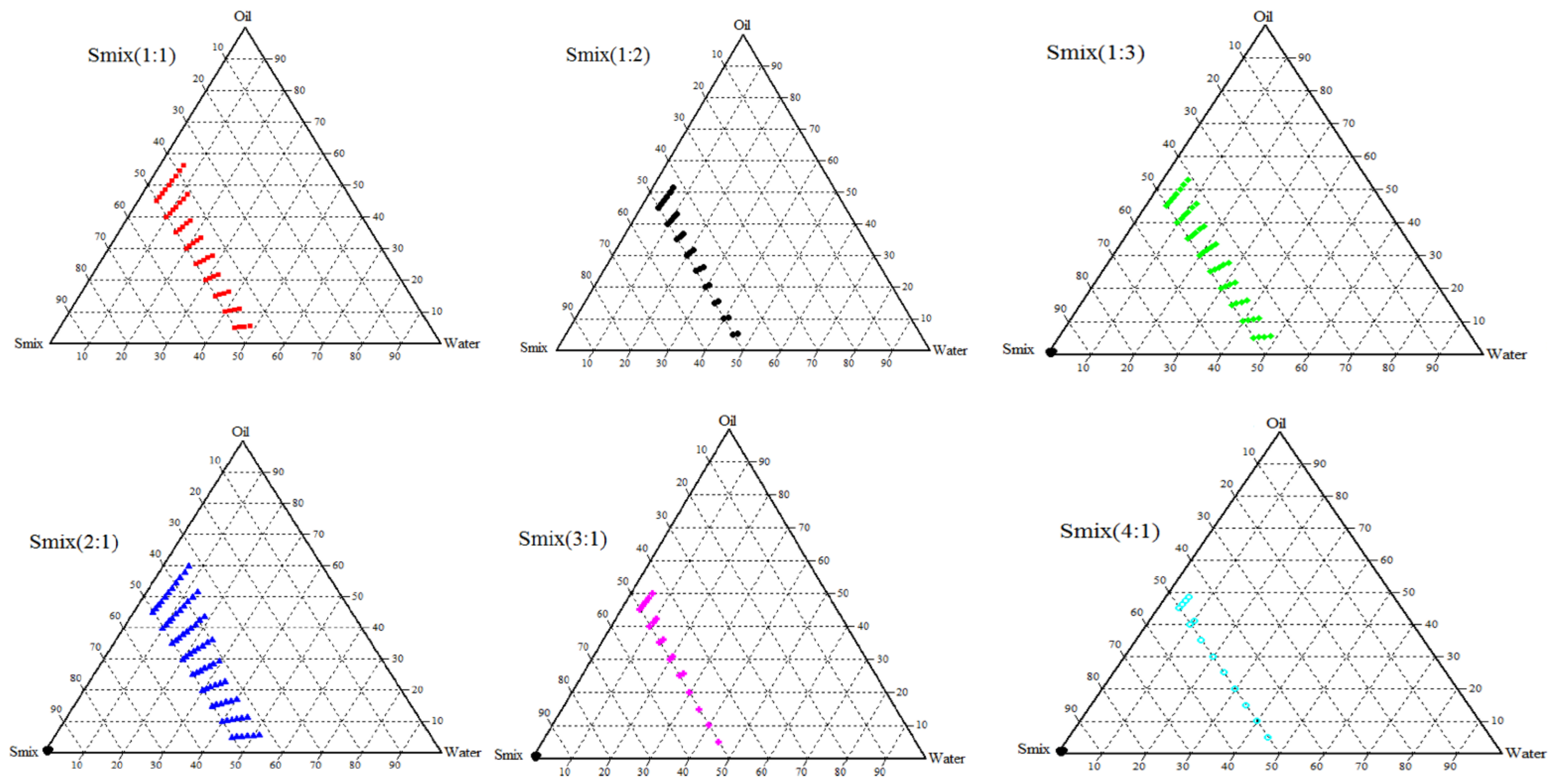
2.3.2. Creation of Ternary Phase Diagram
2.4. Thermodynamic Stability and Dispersibility Studies of L-SNEDDS Preparations
2.5. Screening of Formulations by Full Factorial Design
2.6. Characterization of Designed Batches of L-SNEDDS
2.6.1. Determination of Dissolution Efficiency
2.6.2. Globule Size
2.6.3. Determination of Self-Emulsification Time
2.6.4. Determination of Viscosity, Zeta Potential, Percentage Transmittance
2.7. Solidification of L-SNEDDS
Flowability and Compressibility
2.8. Tablet Preparation of S-SNEDDS
2.9. Characterization of S-SNEDDS Loaded Tablets
2.9.1. Thickness and Hardness
2.9.2. Friability
2.9.3. Disintegration
2.9.4. Drug Content
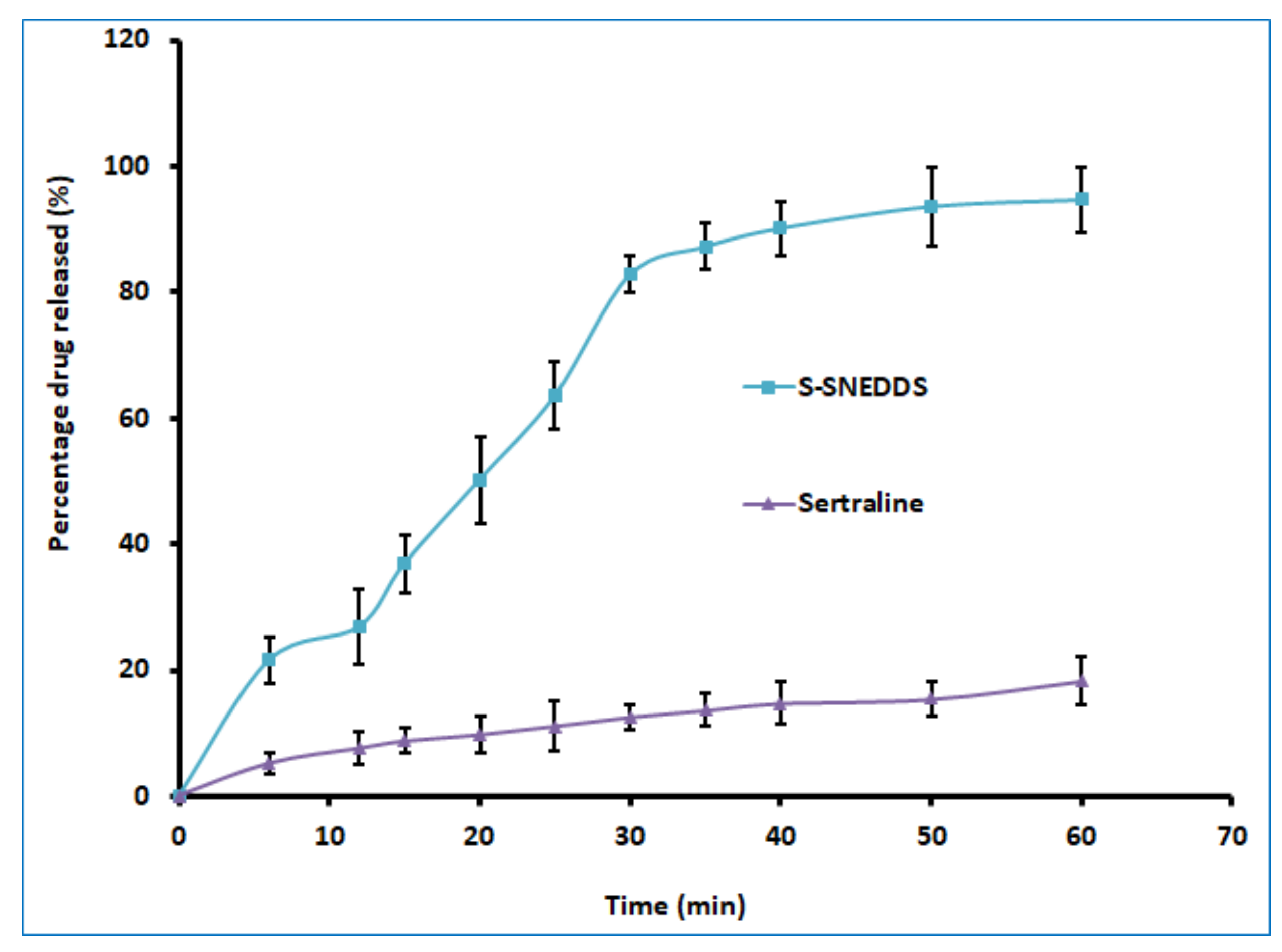
2.9.5. Drug Release
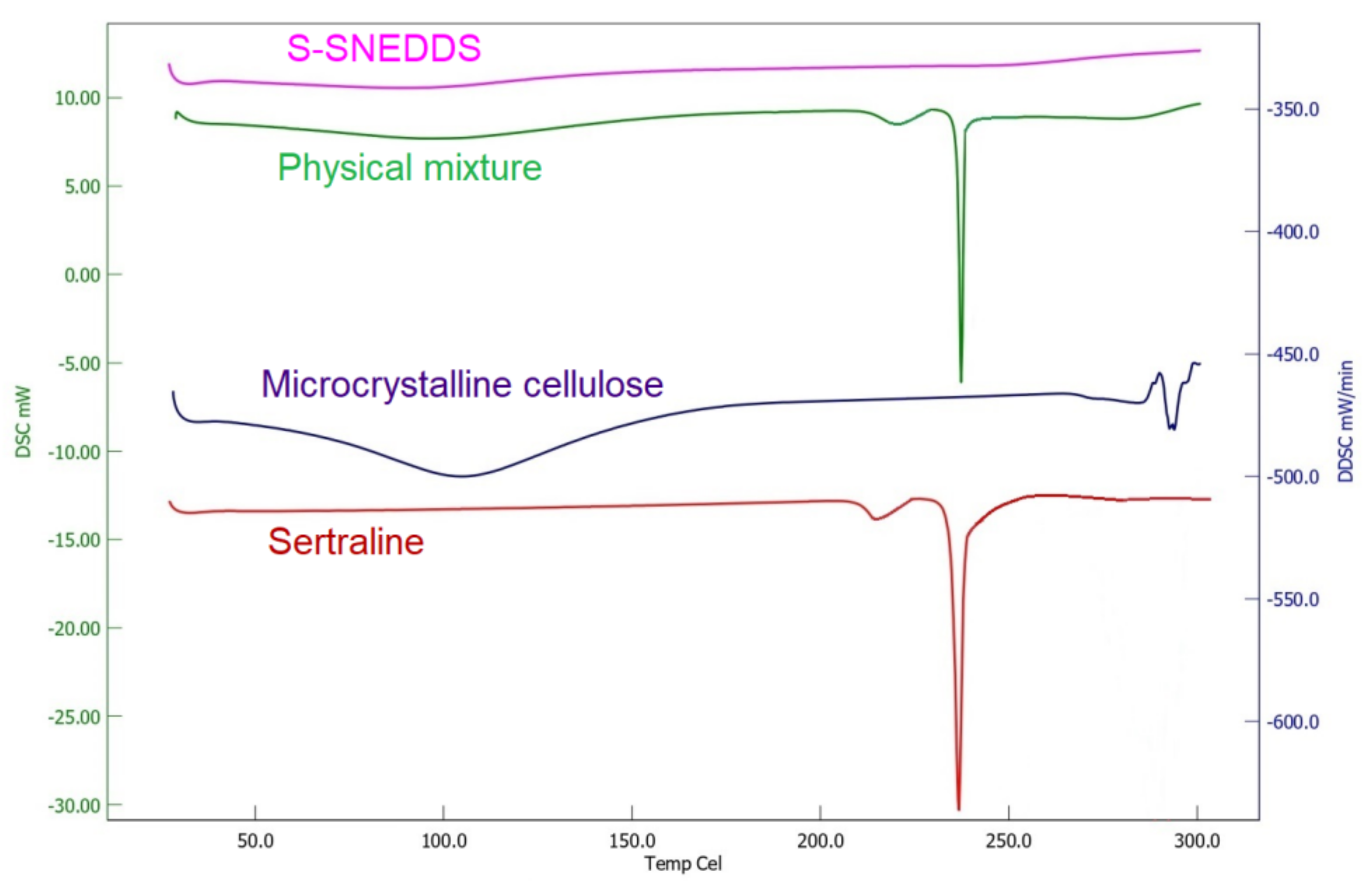
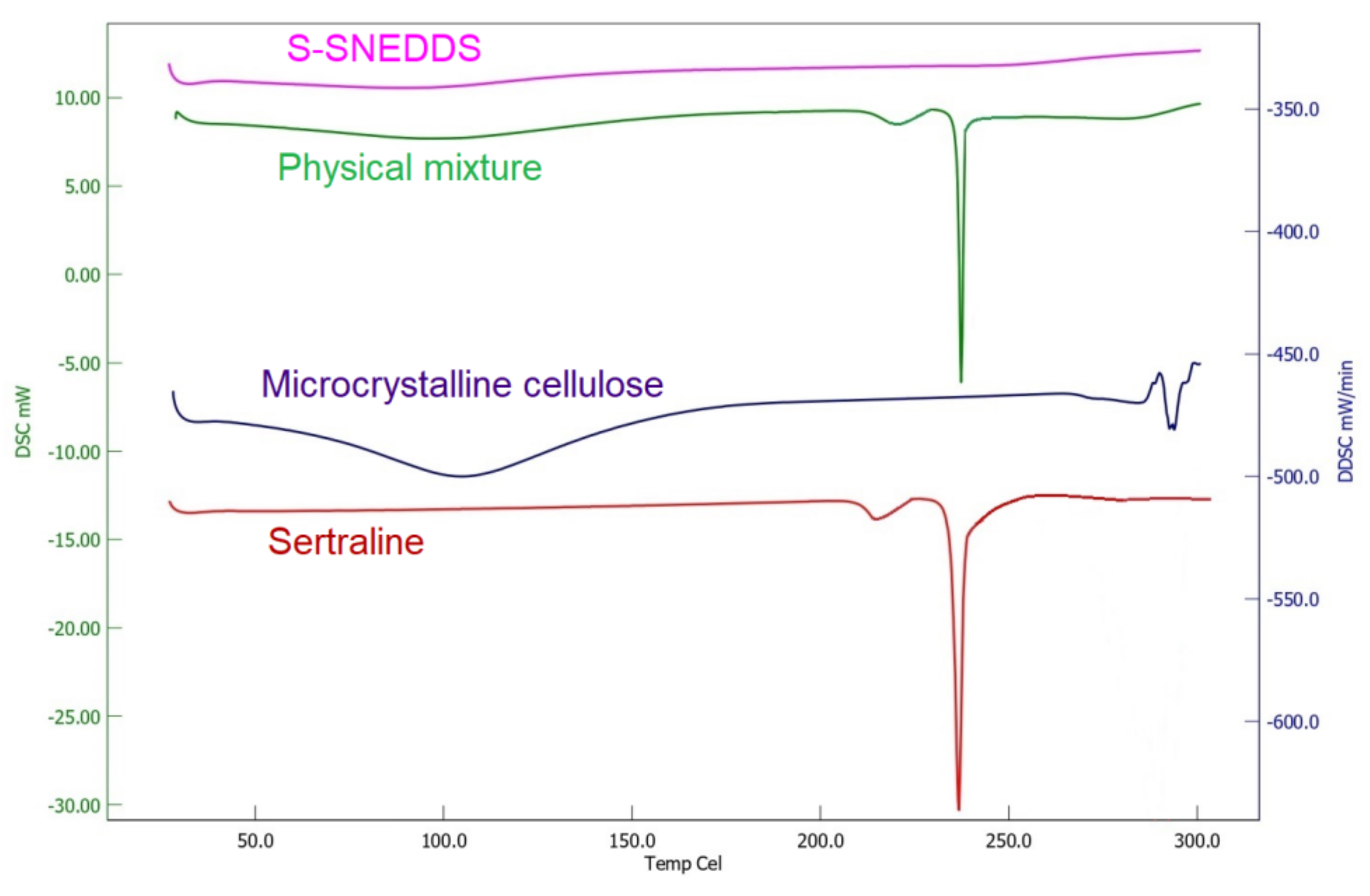
2.9.6. Differential Scanning Calorimetry (DSC)
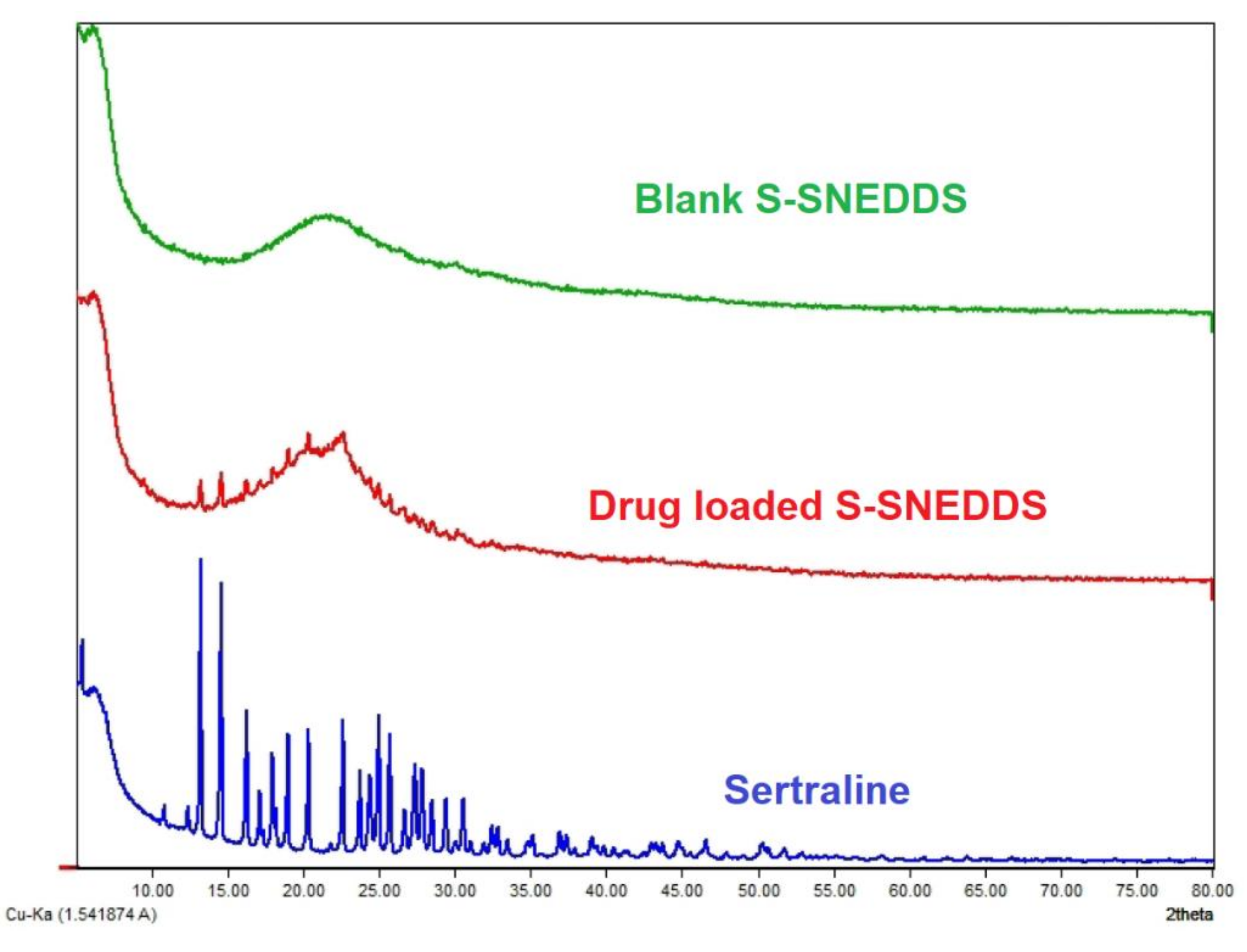
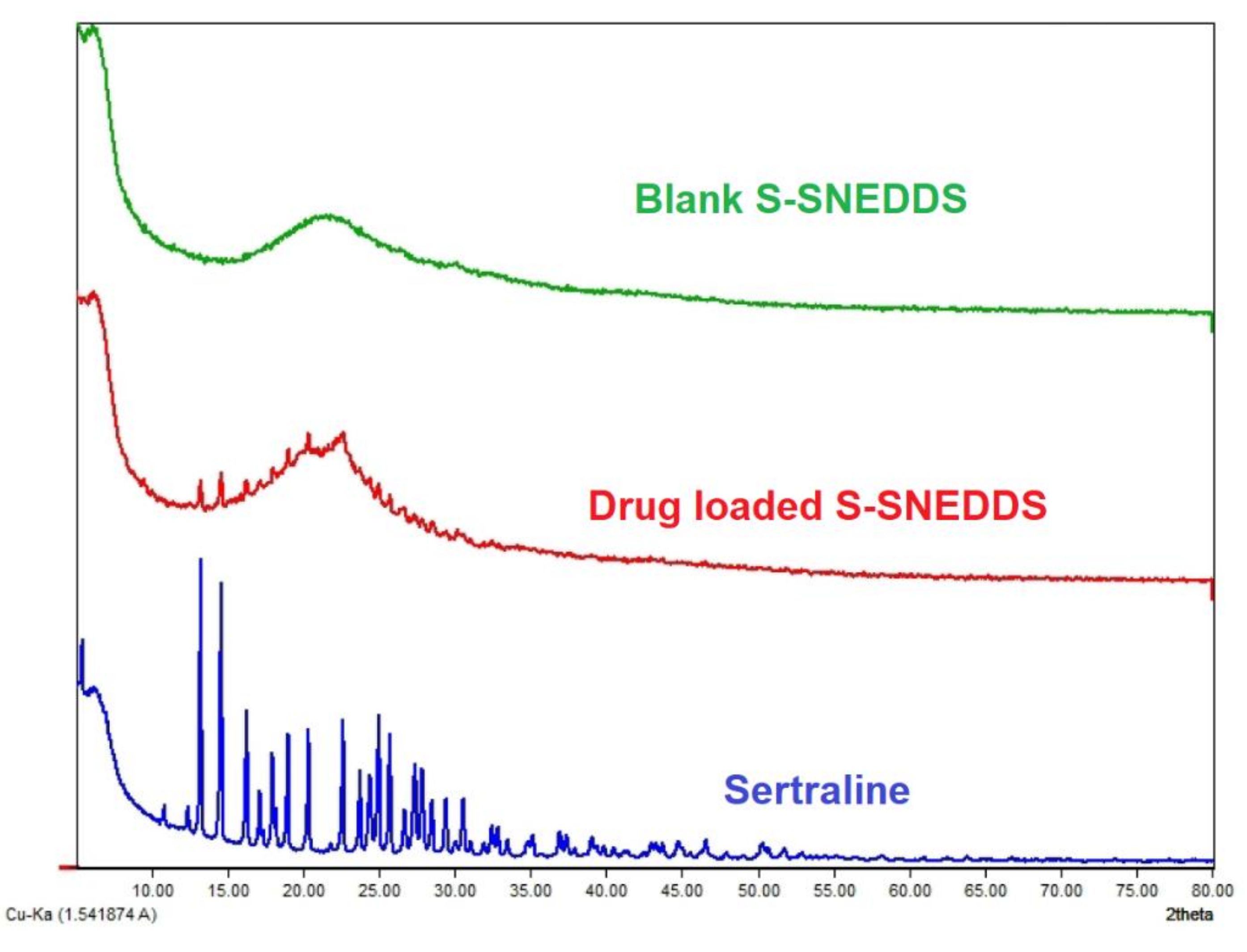
2.9.7. X-ray Diffraction (XRD)
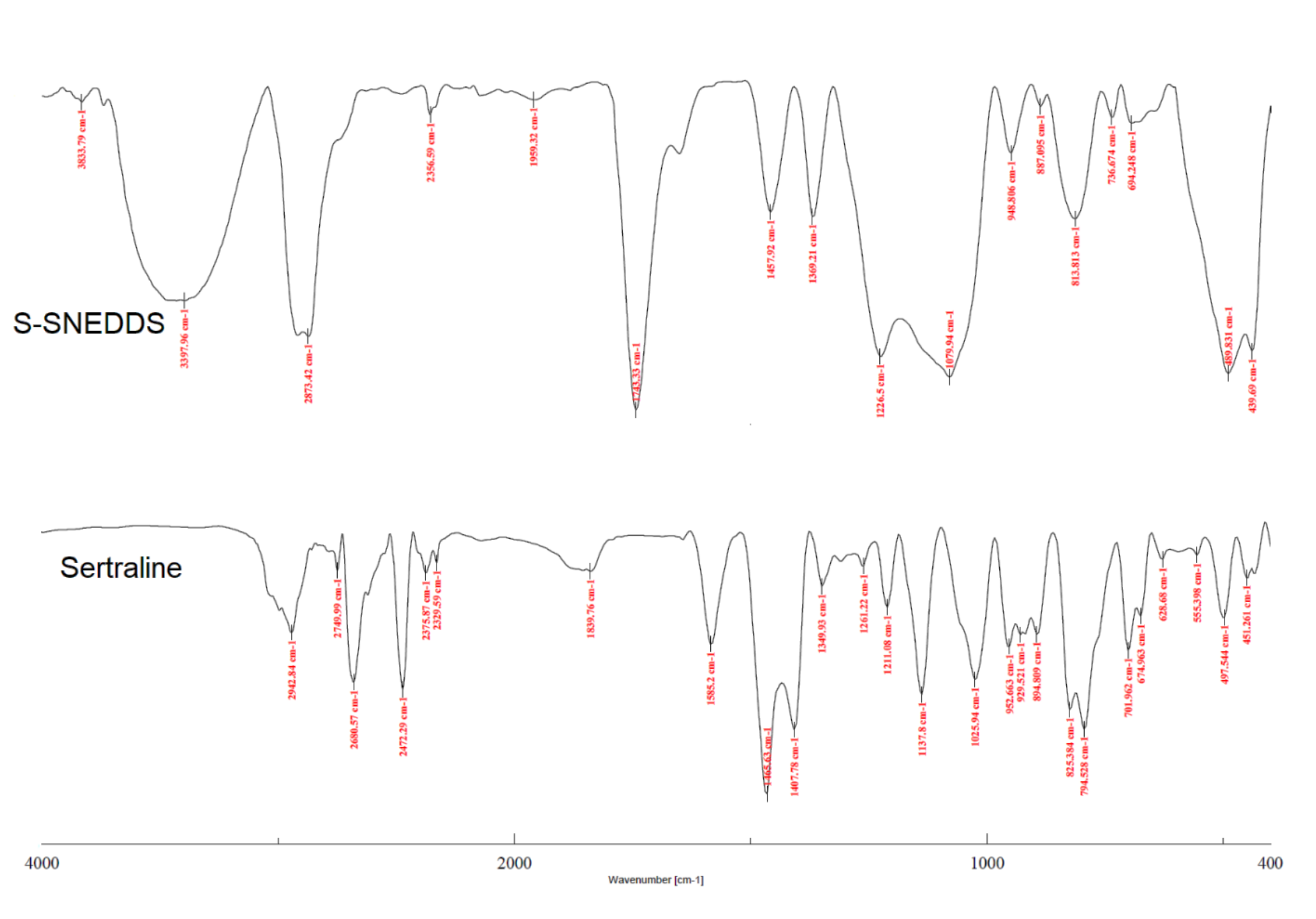
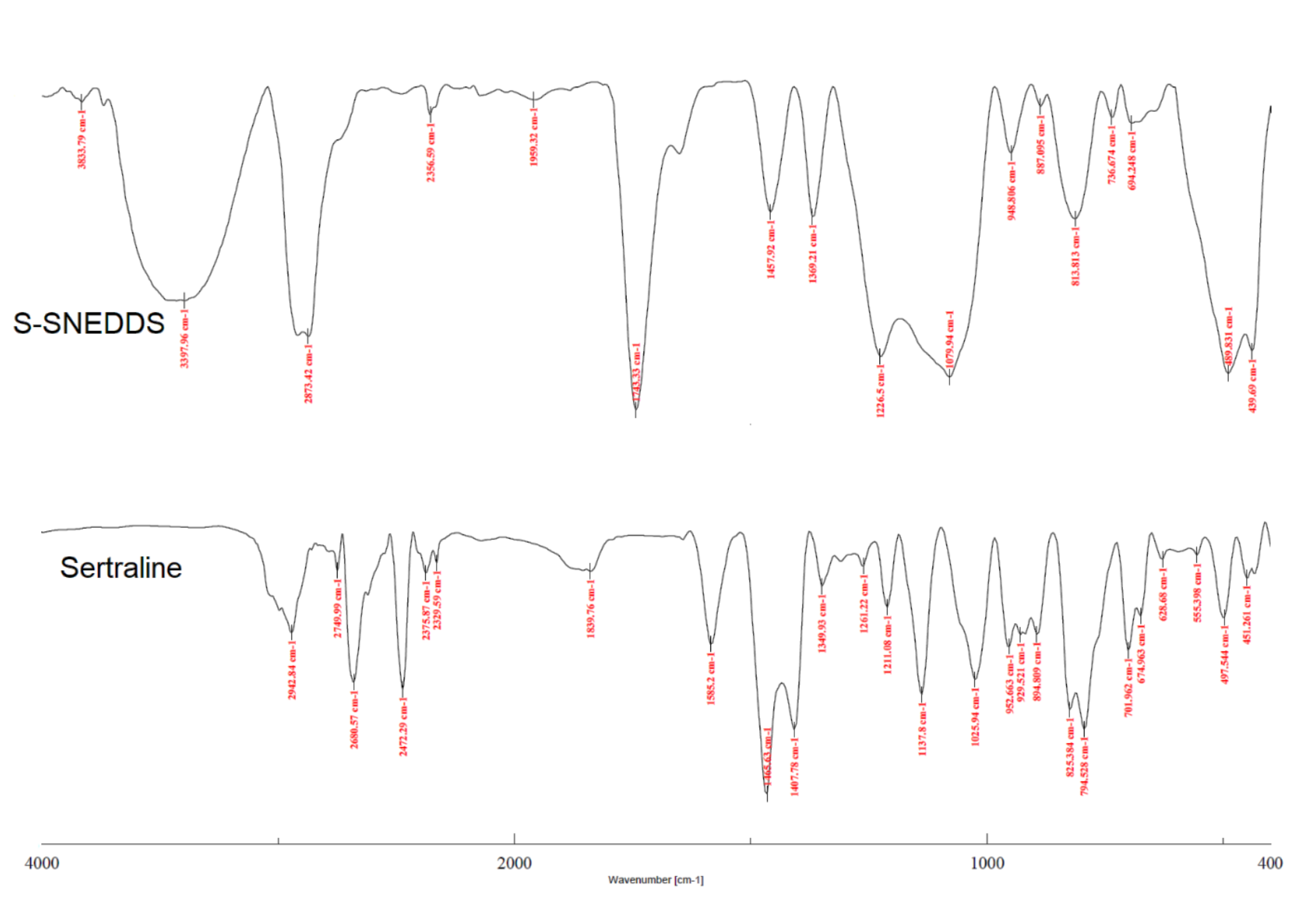
2.9.8. Fourier Transform Infrared (FTIR)
2.9.9. Scanning Electron Microscopy (SEM)
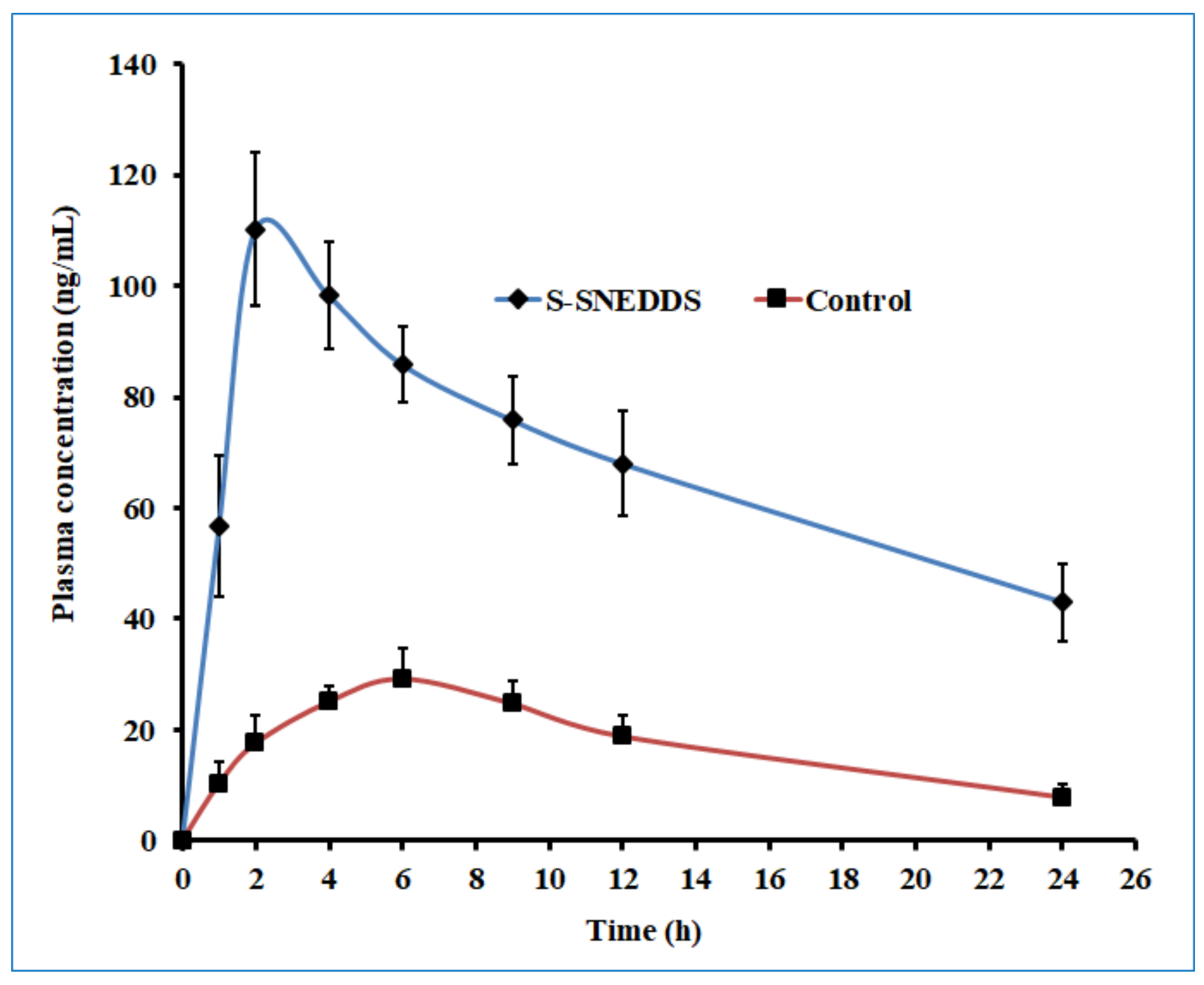
2.10. Oral Bioavailability Studies
3. Results and Discussion
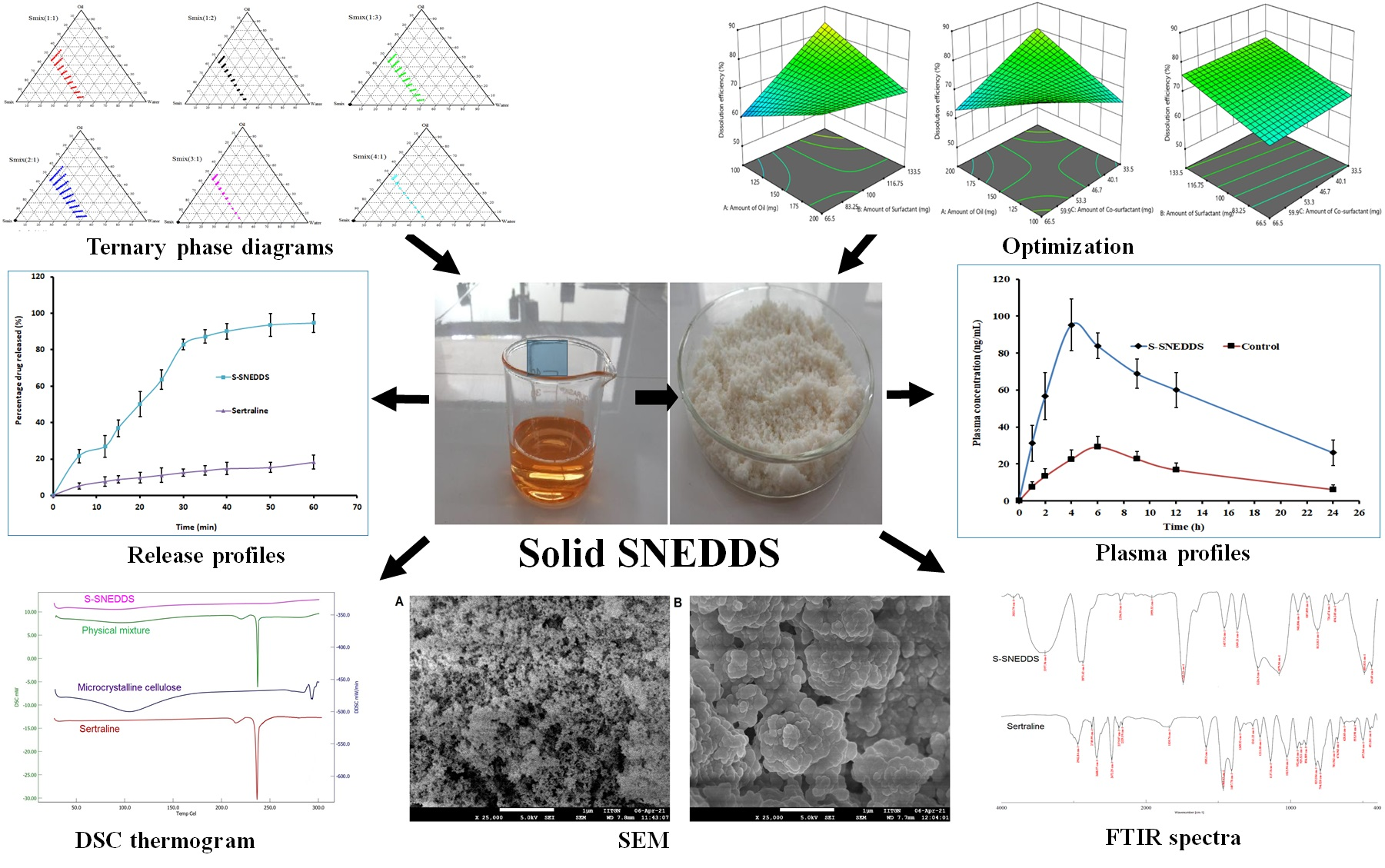
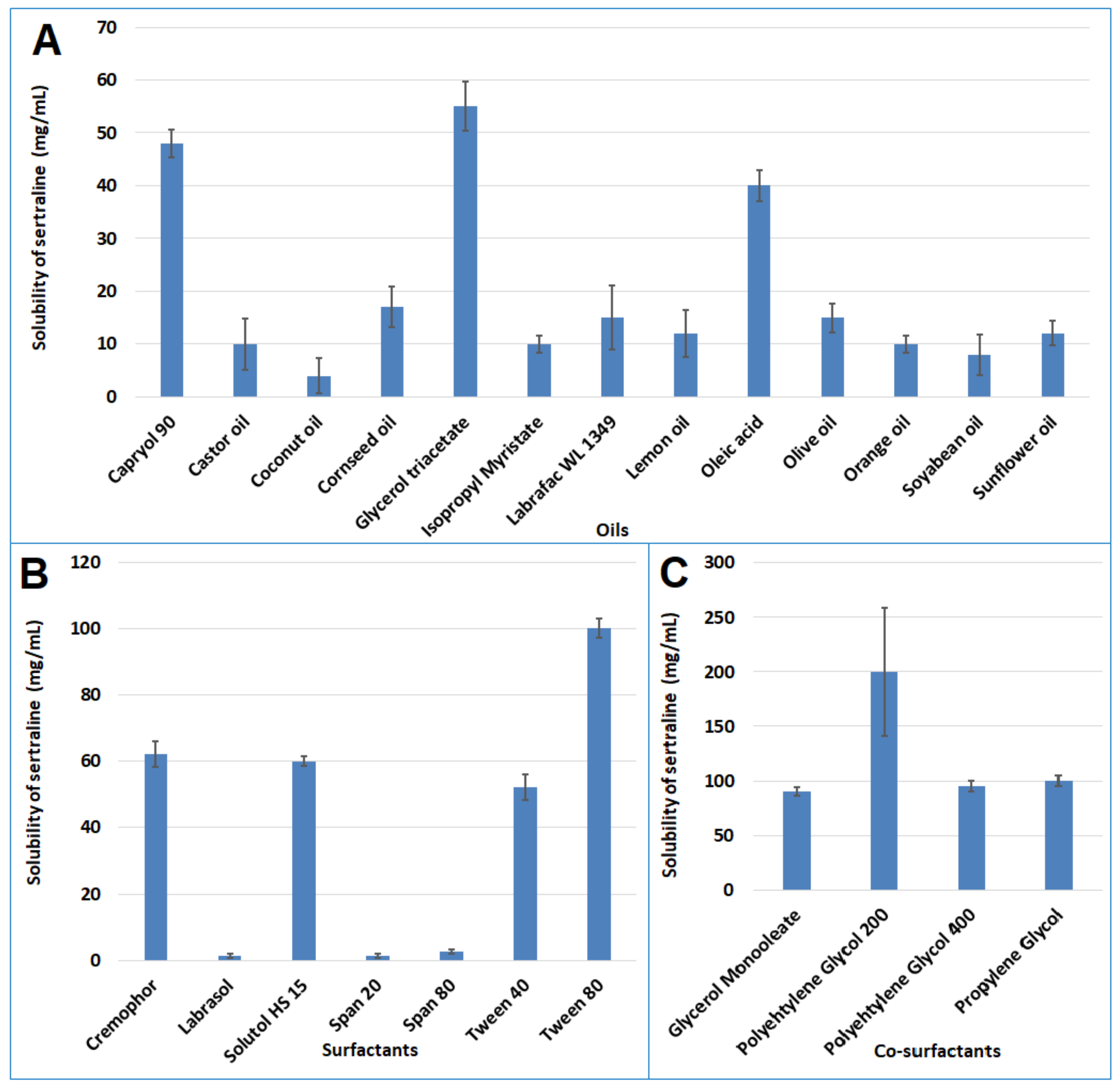
3.1. Preliminary Studies for Components of L-SNEDDS
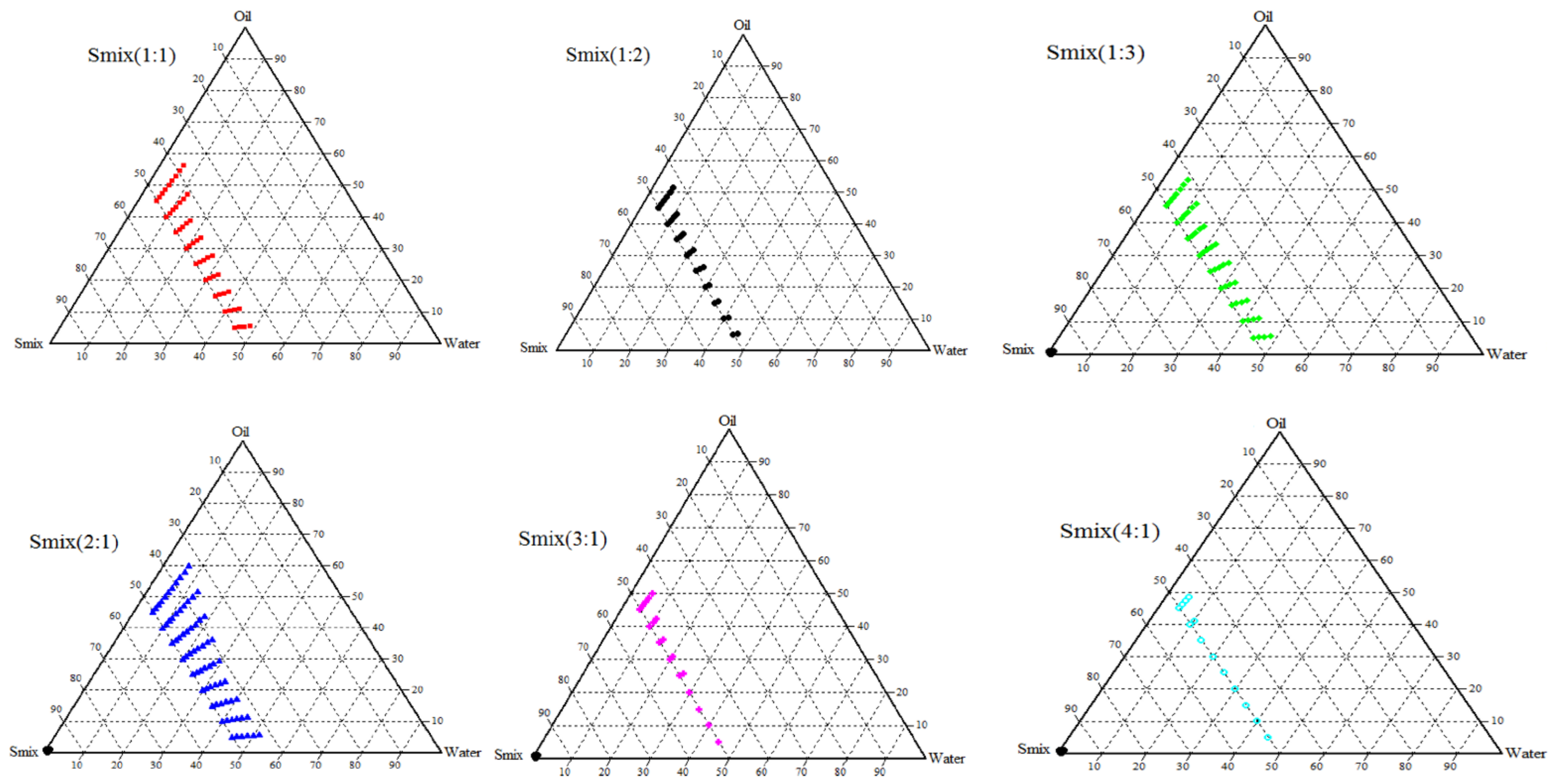
3.2. Construction of Ternary Phase Diagram
3.3. Thermodynamic Stability and Dispersibility Studies
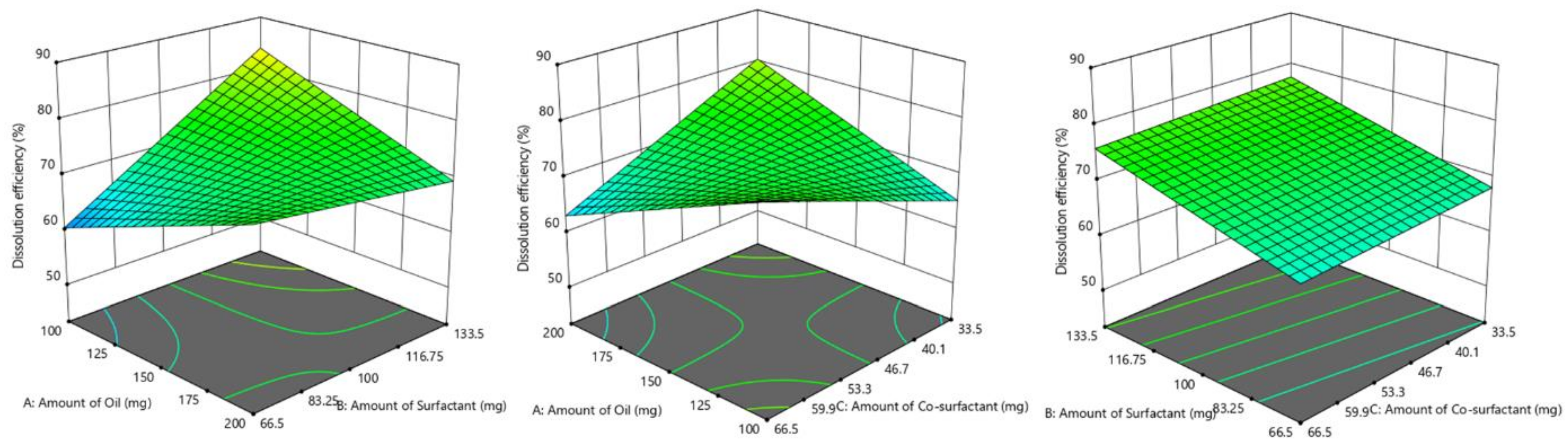
3.4. Screening of Formulations by Full Factorial Design
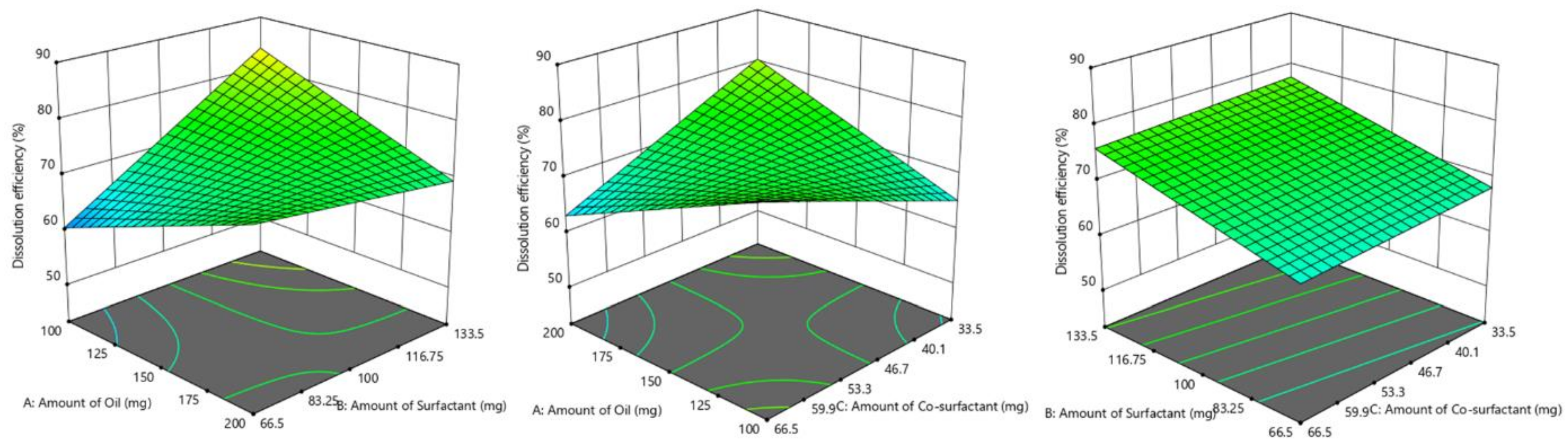
3.4.1. Influence of Formulation Factors on Y1 (Dissolution Efficiency %)
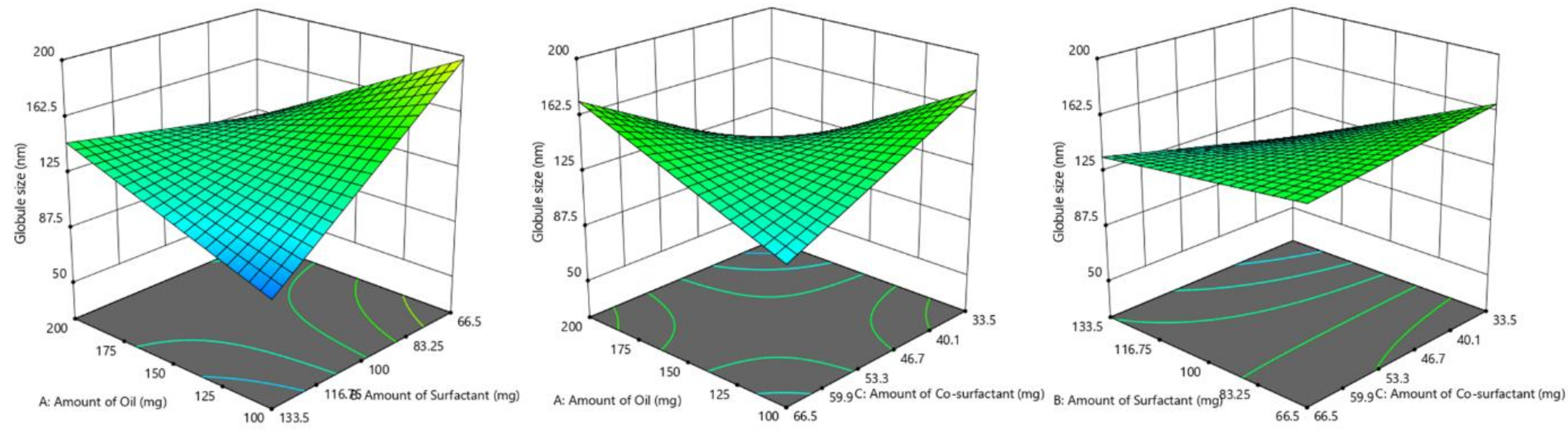
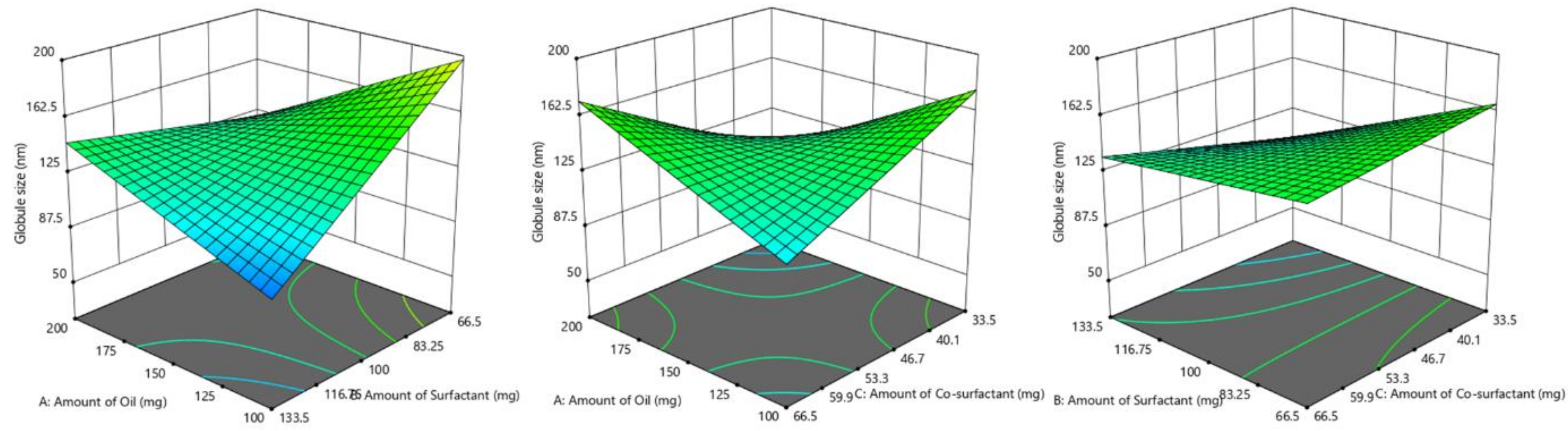
3.4.2. Influence of Formulation Factors on Y2 (Globule Size)
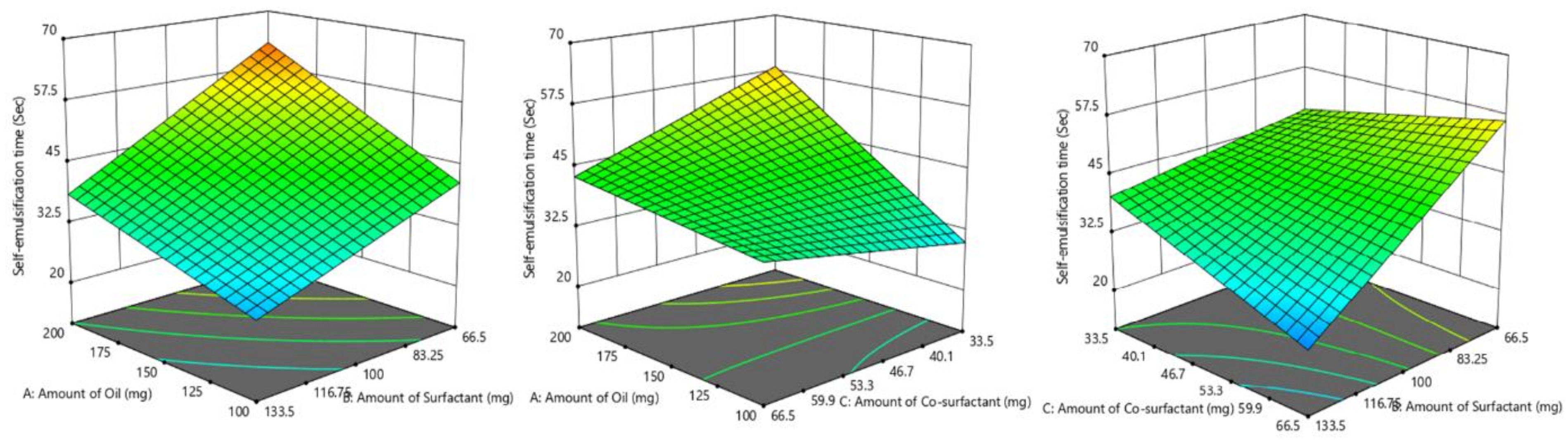
3.4.3. Influence of Formulation Factors on Y3 (Self Emulsification Time)
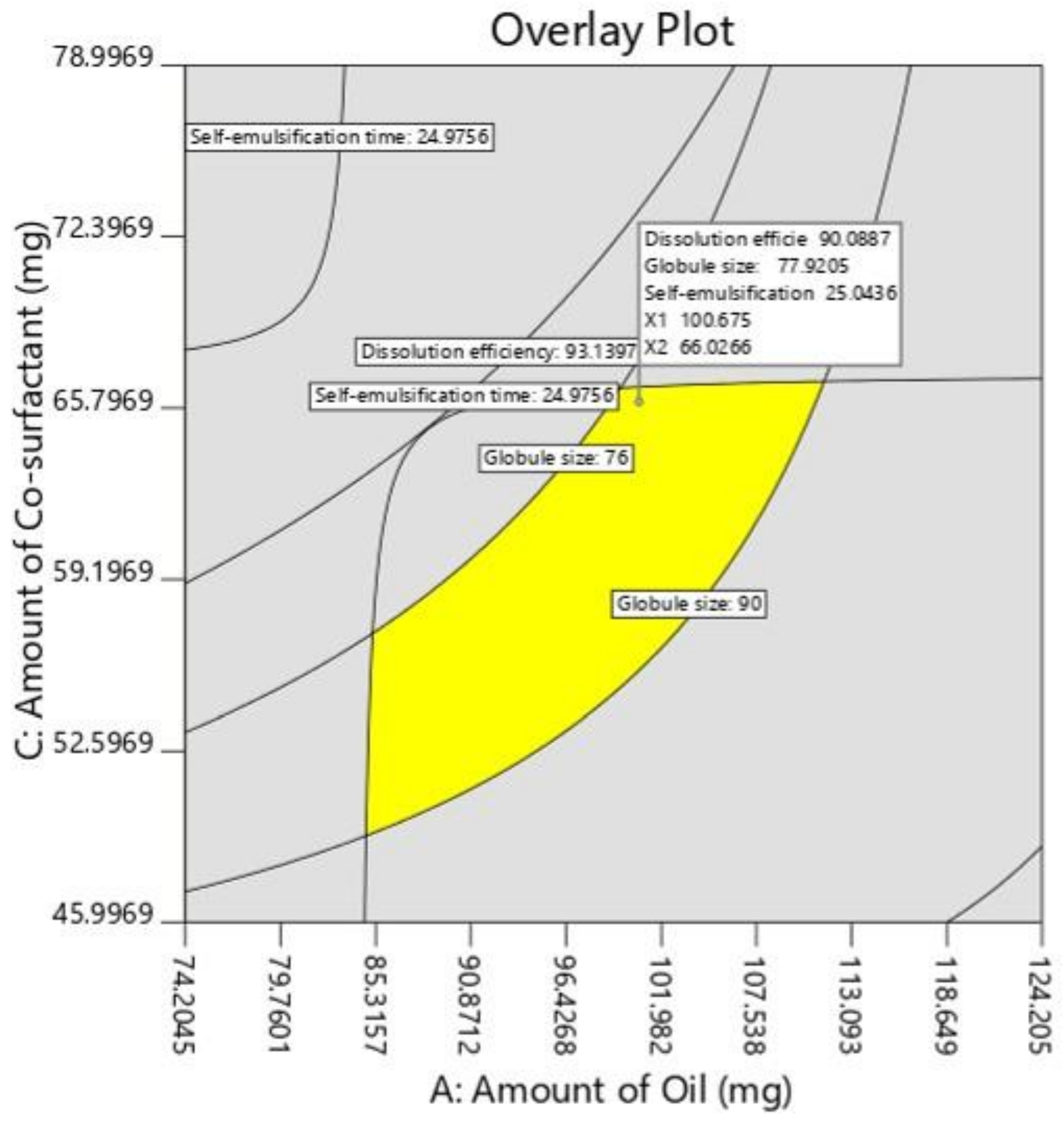
3.4.4. Characteristics of Selected Nanoemulsion
3.5. Solidification of L-SNEDDS
Flowability and Compressibility
3.6. Characterization of S-SNEDDS Loaded Tablets
3.6.1. Thickness and Hardness
3.6.2. Friability
3.6.3. Disintegration
3.6.4. Drug Content
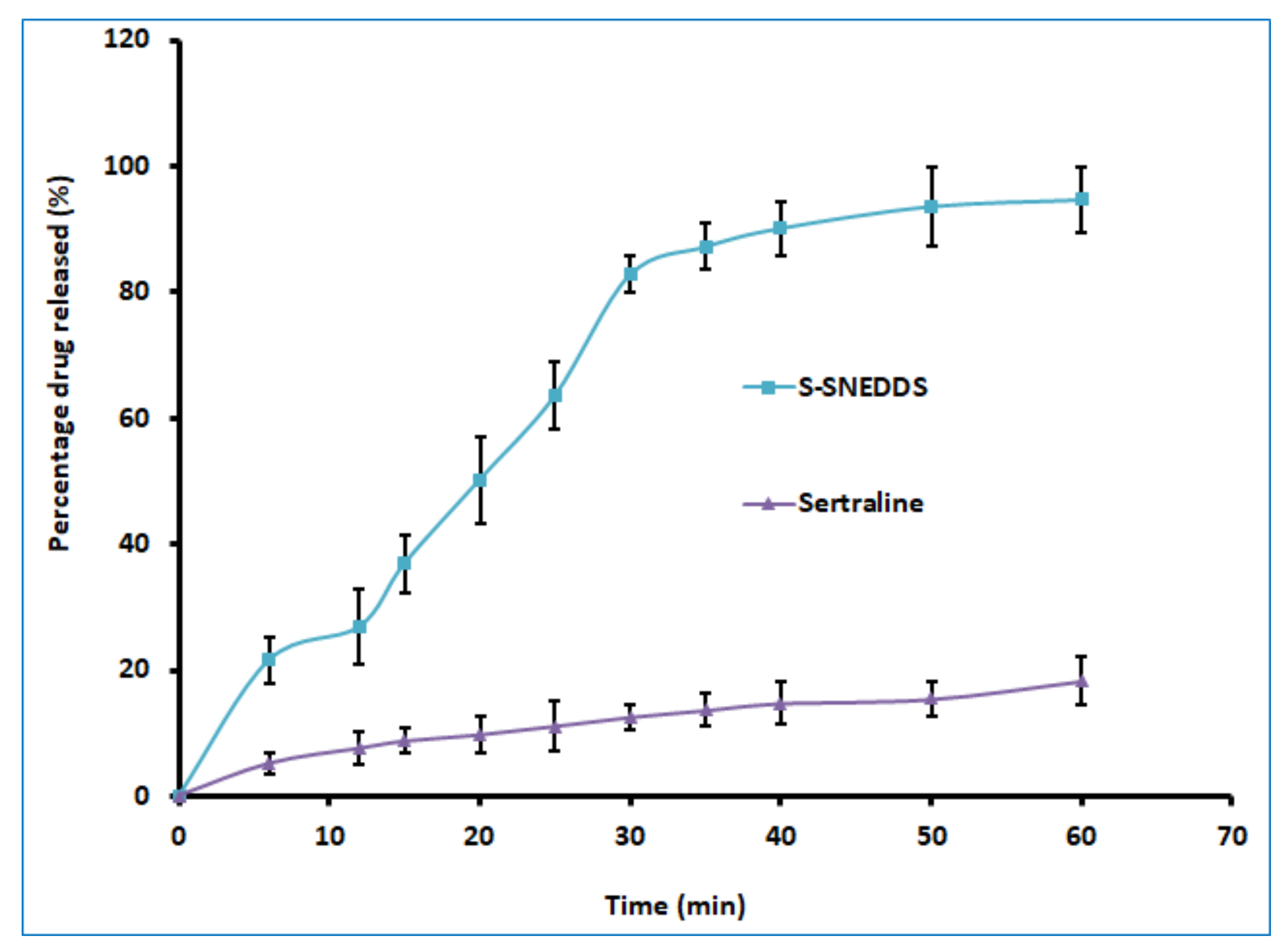
3.6.5. Drug Release
3.6.6. DSC
3.6.7. XRD
3.6.8. FTIR
3.6.9. SEM
3.7. Oral Bioavailability Studies
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sutton, S.C. The use of gastrointestinal intubation studies for controlled release development. Br. J. Clin. Pharmacol. 2009, 68, 342–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alhadab, A.A.; Brundage, R.C. Population Pharmacokinetics of Sertraline in Healthy Subjects: A Model-Based Meta-analysis. AAPS J. 2020, 22, 73. [Google Scholar] [CrossRef] [PubMed]
- Homayun, B.; Lin, X.; Choi, H.J. Challenges and Recent Progress in Oral Drug Delivery Systems for Biopharmaceuticals. Pharmaceutics 2019, 11, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McRae, A.L.; Brady, K.T. Review of sertraline and its clinical applications in psychiatric disorders. Expert Opin. Pharmacother. 2001, 2, 883–892. [Google Scholar] [CrossRef]
- Kim, S.J.; Lee, H.K.; Na, Y.G.; Bang, K.H.; Lee, H.J.; Wang, M.; Huh, H.W.; Cho, C.W. A novel composition of ticagrelor by solid dispersion technique for increasing solubility and intestinal permeability. Int. J. Pharm. 2019, 555, 11–18. [Google Scholar] [CrossRef]
- Rasenack, N.; Müller, B.W. Dissolution rate enhancement by in situ micronization of poorly water-soluble drugs. Pharm. Res. 2002, 19, 1894–1900. [Google Scholar] [CrossRef]
- Taniguchi, C.; Kawabata, Y.; Wada, K.; Yamada, S.; Onoue, S. Microenvironmental pH-modification to improve dissolution behavior and oral absorption for drugs with pH-dependent solubility. Expert Opin. Drug Deliv. 2014, 11, 505–516. [Google Scholar] [CrossRef]
- Ishikawa, M.; Hashimoto, Y. Improvement in aqueous solubility in small molecule drug discovery programs by disruption of molecular planarity and symmetry. J. Med. Chem. 2011, 54, 1539–1554. [Google Scholar] [CrossRef]
- Van Staden, D.; Du Plessis, J.; Viljoen, J. Development of topical/transdermal self-emulsifying drug delivery systems, not as simple as expected. Sci. Pharm. 2020, 88, 17. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.A.; Harwansh, R.K.; Iqbal, Z. Systematic Development of Sertraline Loaded Solid Lipid Nanoparticle (SLN) by Emulsification-Ultrasonication Method and Pharmacokinetic Study in Sprague-Dawley Rats. Pharm. Nanotechnol. 2019, 7, 162–176. [Google Scholar] [CrossRef]
- Nunes, C.D.; Vaz, P.D.; Fernandes, A.C.; Ferreira, P.; Romão, C.C.; Calhorda, M.J. Loading and delivery of sertraline using inorganic micro and mesoporous materials. Eur. J. Pharm. Biopharm. 2007, 66, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Vijaya, R.; Ruckmani, K. In vitro and In vivo characterization of the transdermal delivery of sertraline hydrochloride Films. DARU J. Pharm. Sci. 2011, 19, 424–432. [Google Scholar]
- Gupta, A.; Aggarwal, G.; Singla, S.; Arora, R. Transfersomes: A novel vesicular carrier for enhanced transdermal delivery of sertraline: Development, characterization, and performance evaluation. Sci. Pharm. 2012, 80, 1061–1080. [Google Scholar] [CrossRef] [Green Version]
- Kubackova, J.; Holas, O.; Zbytovska, J.; Vranikova, B.; Zeng, G.; Pavek, P.; Mullertz, A. Oligonucleotide Delivery across the Caco-2 Monolayer: The Design and Evaluation of Self-Emulsifying Drug Delivery Systems (SEDDS). Pharmaceutics 2021, 13, 459. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Liu, J.; Yang, Y.; Adu-Frimpong, M.; Ji, H.; Toreniyazov, E.; Wang, Q.; Yu, J.; Xu, X. SMEDDS for improved oral bioavailability and anti-hyperuricemic activity of licochalcone A. J. Microencapsul. 2021, 38, 459–471. [Google Scholar] [CrossRef]
- Singh, D. Self-nanoemulsifying Drug Delivery System: A Versatile Carrier for Lipophilic Drugs. Pharm. Nanotechnol. 2021, 9, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Rasoanirina, B.N.V.; Lassoued, M.A.; Miladi, K.; Razafindrakoto, Z.; Chaâbane-Banaoues, R.; Ramanitrahasimbola, D.; Cornet, M.; Sfar, S. Self-nanoemulsifying drug delivery system to improve transcorneal permeability of voriconazole: In-vivo studies. J. Pharm. Pharmacol. 2020, 72, 889–896. [Google Scholar] [CrossRef]
- Buya, A.B.; Beloqui, A.; Memvanga, P.B.; Préat, V. Self-Nano-Emulsifying Drug-Delivery Systems: From the Development to the Current Applications and Challenges in Oral Drug Delivery. Pharmaceutics 2020, 12, 1194. [Google Scholar] [CrossRef]
- Porter, C.J.; Kaukonen, A.M.; Taillardat-Bertschinger, A.; Boyd, B.J.; O’Connor, J.M.; Edwards, G.A.; Charman, W.N. Use of in vitro lipid digestion data to explain the in vivo performance of triglyceride-based oral lipid formulations of poorly water-soluble drugs: Studies with halofantrine. J. Pharm. Sci. 2004, 93, 1110–1121. [Google Scholar] [CrossRef]
- Rahman, M.A.; Harwansh, R.; Mirza, M.A.; Hussain, S.; Hussain, A. Oral lipid based drug delivery system (LBDDS): Formulation, characterization and application: A review. Curr. Drug Deliv. 2011, 8, 330–345. [Google Scholar] [CrossRef]
- Abbaspour, M.; Jalayer, N.; Sharif Makhmalzadeh, B. Development and evaluation of a solid self-nanoemulsifying drug delivery system for loratadin by extrusion-spheronization. Adv. Pharm. Bull. 2014, 4, 113–119. [Google Scholar] [CrossRef] [PubMed]
- McClements, D.J. Nanoemulsions versus microemulsions: Terminology, differences, and similarities. Soft Matter 2012, 8, 1719–1729. [Google Scholar] [CrossRef]
- Azmi, N.A.N.; Elgharbawy, A.A.; Motlagh, S.R.; Samsudin, N.; Salleh, H.M. Nanoemulsions: Factory for food, pharmaceutical and cosmetics. Processes 2019, 7, 617. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Eral, H.B.; Hatton, T.A.; Doyle, P.S. Nanoemulsions: Formation, properties and applications. Soft Matter 2016, 12, 2826–2841. [Google Scholar] [CrossRef] [Green Version]
- Prajapat, M.D.; Patel, N.J.; Bariya, A.; Patel, S.S.; Butani, S.B. Formulation and evaluation of self-emulsifying drug delivery system for nimodipine, a BCS class II drug. J. Drug Deliv. Sci. Technol. 2017, 39, 59–68. [Google Scholar] [CrossRef]
- Sanka, K.; Suda, D.; Bakshi, V. Optimization of solid-self nanoemulsifying drug delivery system for solubility and release profile of clonazepam using simplex lattice design. J. Drug Deliv. Sci. Technol. 2016, 33, 114–124. [Google Scholar] [CrossRef]
- Kohli, K.; Chopra, S.; Dhar, D.; Arora, S.; Khar, R.K. Self-emulsifying drug delivery systems: An approach to enhance oral bioavailability. Drug Discov. Today 2010, 15, 958–965. [Google Scholar] [CrossRef]
- Khadka, P.; Ro, J.; Kim, H.; Kim, I.; Kim, J.T.; Kim, H.; Cho, J.M.; Yun, G.; Lee, J. Pharmaceutical particle technologies: An approach to improve drug solubility, dissolution and bioavailability. Asian J. Pharm. Sci. 2014, 9, 304–316. [Google Scholar] [CrossRef] [Green Version]
- Melis, V.; Usach, I.; Peris, J.E. Determination of sertraline in rat plasma by HPLC and fluorescence detection and its application to in vivo pharmacokinetic studies. J. Sep. Sci. 2012, 35, 3302–3307. [Google Scholar] [CrossRef]
- Guideline, I.H.T. Validation of analytical procedures: Text and methodology. Q2 (R1) 2005, 1, 5. [Google Scholar]
- Shah, J.; Nair, A.B.; Jacob, S.; Patel, R.K.; Shah, H.; Shehata, T.M.; Morsy, M.A. Nanoemulsion based vehicle for effective ocular delivery of moxifloxacin using experimental design and pharmacokinetic study in rabbits. Pharmaceutics 2019, 11, 230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akrawi, S.H.; Gorain, B.; Nair, A.B.; Choudhury, H.; Pandey, M.; Shah, J.N.; Venugopala, K.N. Development and optimization of naringenin-loaded chitosan-coated nanoemulsion for topical therapy in wound healing. Pharmaceutics 2020, 12, 893. [Google Scholar] [CrossRef] [PubMed]
- Nasr, A.; Gardouh, A.; Ghorab, M. Novel Solid Self-Nanoemulsifying Drug Delivery System (S-SNEDDS) for Oral Delivery of Olmesartan Medoxomil: Design, Formulation, Pharmacokinetic and Bioavailability Evaluation. Pharmaceutics 2016, 8, 20. [Google Scholar] [CrossRef] [PubMed]
- Shafiq, S.; Shakeel, F.; Talegaonkar, S.; Ahmad, F.J.; Khar, R.K.; Ali, M. Development and bioavailability assessment of ramipril nanoemulsion formulation. Eur. J. Pharm. Biopharm. 2007, 66, 227–243. [Google Scholar] [CrossRef]
- Shah, J.; Vasanti, S.; Anroop, B.; Vyas, H. Enhancement of dissolution rate of valdecoxib by solid dispersions technique with PVP K 30 & PEG 4000: Preparation and in vitro evaluation. J. Incl. Phenom. Macrocycl. Chem. 2009, 63, 69–75. [Google Scholar]
- Jakki, R.; Afzal Syed, M.; Kandadi, P.; Veerabrahma, K. Development of a self-microemulsifying drug delivery system of domperidone: In vitro and in vivo characterization. Acta Pharm. 2013, 63, 241–251. [Google Scholar] [CrossRef] [Green Version]
- Jhaveri, M.; Nair, A.B.; Shah, J.; Jacob, S.; Patel, V.; Mehta, T. Improvement of oral bioavailability of carvedilol by liquisolid compact: Optimization and pharmacokinetic study. Drug Deliv. Transl. Res. 2020, 10, 975–985. [Google Scholar] [CrossRef]
- Jacob, S.; Shirwaikar, A.; Nair, A. Preparation and evaluation of fast-disintegrating effervescent tablets of glibenclamide. Drug Dev. Ind. Pharm. 2009, 35, 321–328. [Google Scholar] [CrossRef]
- Nair, A.; Gupta, R.; Vasanti, S. In vitro controlled release of alfuzosin hydrochloride using HPMC-based matrix tablets and its comparison with marketed product. Pharm. Dev. Technol. 2007, 12, 621–625. [Google Scholar] [CrossRef]
- Eleftheriadis, G.K.; Mantelou, P.; Karavasili, C.; Chatzopoulou, P.; Katsantonis, D.; Irakli, M.; Mygdalia, A.; Vizirianakis, I.S.; Fatouros, D.G. Development and Characterization of a Self-Nanoemulsifying Drug Delivery System Comprised of Rice Bran Oil for Poorly Soluble Drugs. AAPS PharmSciTech 2019, 20, 78. [Google Scholar] [CrossRef]
- Dalal, R.; Shah, J.; Gorain, B.; Choudhury, H.; Jacob, S.; Mehta, T.A.; Shah, H.; Nair, A.B. Development and Optimization of Asenapine Sublingual Film Using QbD Approach. AAPS PharmSciTech 2021, 22, 244. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, S.; Nair, A.B.; Shah, J.; Gorain, B.; Jacob, S.; Shah, H.; Patel, V. Enhanced Solubility and Bioavailability of Dolutegravir by Solid Dispersion Method: In Vitro and In Vivo Evaluation—A Potential Approach for HIV Therapy. AAPS PharmSciTech 2021, 22, 127. [Google Scholar] [CrossRef] [PubMed]
- Sree Harsha, N.; Hiremath, J.G.; Sarudkar, S.; Attimarad, M.; Al-Dhubiab, B.; Nair, A.B.; Venugopala, K.N.; Asif, A.H. Spray dried amorphous form of simvastatin: Preparation and evaluation of the buccal tablet. Indian J. Pharm. Educ. Res. 2020, 54, 46–54. [Google Scholar] [CrossRef] [Green Version]
- Nair, A.; Morsy, M.A.; Jacob, S. Dose translation between laboratory animals and human in preclinical and clinical phases of drug development. Drug Dev. Res. 2018, 79, 373–382. [Google Scholar] [CrossRef]
- Satyavert; Gupta, S.; Choudhury, H.; Jacob, S.; Nair, A.B.; Dhanawat, M.; Munjal, K. Pharmacokinetics and tissue distribution of hydrazinocurcumin in rats. Pharmacol. Rep. 2021, 73, 1734–1743. [Google Scholar] [CrossRef]
- Kumar, S.; Yadav Ravulapalli, S.; Kumar Tiwari, S.; Gupta, S.; Nair, A.B.; Jacob, S. Effect of sex and food on the pharmacokinetics of different classes of BCS drugs in rats after cassette administration. Int. J. Pharm. 2021, 610, 121221. [Google Scholar] [CrossRef]
- Shen, J.; Bi, J.; Tian, H.; Jin, Y.; Wang, Y.; Yang, X.; Yang, Z.; Kou, J.; Li, F. Preparation and evaluation of a self-nanoemulsifying drug delivery system loaded with Akebia saponin D-phospholipid complex. Int. J. Nanomed. 2016, 11, 4919–4929. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, K.; Yoshikawa, T.; Moroto, Y.; Kanaoka, E.; Takahashi, K.; Nishihara, Y.; Masuda, K. Microemulsion formulation for enhanced absorption of poorly soluble drugs. I. Prescription design. J. Control. Release 2002, 81, 65–74. [Google Scholar] [CrossRef]
- Kreilgaard, M.; Pedersen, E.J.; Jaroszewski, J.W. NMR characterisation and transdermal drug delivery potential of microemulsion systems. J. Control. Release 2000, 69, 421–433. [Google Scholar] [CrossRef]
- Azeem, A.; Rizwan, M.; Ahmad, F.J.; Iqbal, Z.; Khar, R.K.; Aqil, M.; Talegaonkar, S. Nanoemulsion components screening and selection: A technical note. AAPS PharmSciTech 2009, 10, 69–76. [Google Scholar] [CrossRef]
- Attwood, D.; Mallon, C.; Ktistis, G.; Taylor, C. A study on factors influencing the droplet size in nonionic oil-in-water microemulsions. Int. J. Pharm. 1992, 88, 417–422. [Google Scholar] [CrossRef]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug solubility: Importance and enhancement techniques. ISRN Pharm. 2012, 2012, 195727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joung, H.J.; Choi, M.J.; Kim, J.T.; Park, S.H.; Park, H.J.; Shin, G.H. Development of Food-Grade Curcumin Nanoemulsion and its Potential Application to Food Beverage System: Antioxidant Property and In Vitro Digestion. J. Food Sci. 2016, 81, N745–N753. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.; Carvajal, M.; Patel, C.; Infeld, M.; Malick, A. Self-emulsifying drug delivery systems (SEDDS) with polyglycolyzed glycerides for improving in vitro dissolution and oral absorption of lipophilic drugs. Int. J. Pharm. 1994, 106, 15–23. [Google Scholar] [CrossRef]
- Vraníková, B.; Gajdziok, J. Liquisolid systems and aspects influencing their research and development. Acta Pharm. 2013, 63, 447–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Trinkle, D.; Derbin, G.; Martin, K.; Sharif, S.; Timmins, P.; Desa, D. Moisture adsorption and desorption properties of colloidal silicon dioxide and its impact on layer adhesion of a bilayer tablet formulation. J. Excip. Food Chem. 2016, 5, 1004. [Google Scholar]
- Shah, R.B.; Tawakkul, M.A.; Khan, M.A. Comparative evaluation of flow for pharmaceutical powders and granules. AAPS PharmSciTech 2008, 9, 250–258. [Google Scholar] [CrossRef] [Green Version]
- Butler, J.; Hens, B.; Vertzoni, M.; Brouwers, J.; Berben, P.; Dressman, J.; Andreas, C.J.; Schaefer, K.J.; Mann, J.; McAllister, M.; et al. In vitro models for the prediction of in vivo performance of oral dosage forms: Recent progress from partnership through the IMI OrBiTo collaboration. Eur. J. Pharm. Biopharm. 2019, 136, 70–83. [Google Scholar] [CrossRef]
- Jacob, S.; Nair, A.B. An updated overview with simple and practical approach for developing in vitro-in vivo correlation. Drug Dev. Res. 2018, 79, 97–110. [Google Scholar] [CrossRef]
- Granero, G.E.; Longhi, M.R.; Mora, M.J.; Junginger, H.E.; Midha, K.K.; Shah, V.P.; Stavchansky, S.; Dressman, J.B.; Barends, D.M. Biowaiver monographs for immediate release solid oral dosage forms: Furosemide. J. Pharm. Sci. 2010, 99, 2544–2556. [Google Scholar] [CrossRef]
- Mendes, C.; Buttchevitz, A.; Kruger, J.H.; Caon, T.; de Oliveira Benedet, P.; Lemos-Senna, E.; Silva, M.A.S. Self-Nanoemulsified Drug Delivery System of Hydrochlorothiazide for Increasing Dissolution Rate and Diuretic Activity. AAPS PharmSciTech 2017, 18, 2494–2504. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Siddiqui, A.; Ali, H.; Nazzal, S. Dissolution and powder flow characterization of solid self-emulsified drug delivery system (SEDDS). Int. J. Pharm. 2009, 366, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.T.D.; Tran, P.H.L. Molecular Interactions in Solid Dispersions of Poorly Water-Soluble Drugs. Pharmaceutics 2020, 12, 745. [Google Scholar] [CrossRef] [PubMed]
- Hancock, B.C.; Shamblin, S.L.; Zografi, G. Molecular mobility of amorphous pharmaceutical solids below their glass transition temperatures. Pharm. Res. 1995, 12, 799–806. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Batch No. | Amount of Oil (mg) (X1) | Amount of Surfactant (mg) (X2) | Amount of Co-Surfactant (mg) (X3) | |||
|---|---|---|---|---|---|---|
| Coded Values | Actual Values | Coded Values | Actual Values | Coded Values | Actual Values | |
| F1 | −1 | 100 | −1 | 66.5 | −1 | 33.5 |
| F2 | −1 | 100 | −1 | 66.5 | +1 | 66.5 |
| F3 | −1 | 100 | +1 | 133.5 | −1 | 33.5 |
| F4 | −1 | 100 | +1 | 133.5 | +1 | 66.5 |
| F5 | +1 | 200 | −1 | 66.5 | −1 | 33.5 |
| F6 | +1 | 200 | −1 | 66.5 | +1 | 66.5 |
| F7 | +1 | 200 | +1 | 133.5 | −1 | 33.5 |
| F8 | +1 | 200 | +1 | 133.5 | +1 | 66.5 |
| Ingredients | Category | Quantity (% w/w) | Amount in (mg) |
|---|---|---|---|
| The powder contains 50 mg of the drug | Drug loaded solid self-nanoemulsifying drug delivery system | 83.33 | 500 |
| l-hydroxypropyl cellulose | Binder | 8.67 | 50 |
| Croscarmellose sodium | Disintegrant | 6 | 36 |
| Magnesium stearate | Lubricant | 1 | 6 |
| Talc | Glidant | 1 | 6 |
| Smix Ratio | Oil: Water Ratio | % w/w | Thermodynamic Stability | Dispersibility Test | |||||
|---|---|---|---|---|---|---|---|---|---|
| Oil | Water | Smix | Heating Cooling Cycles | Centrifugation | Freeze-Thaw Cycles | Water | 0.1 N HCl | ||
| 2:1 | 9:1 | 58.05 | 6.45 | 35.5 | P | P | P | A | A |
| 56.25 | 6.25 | 37.5 | P | P | P | A | A | ||
| 8:2 | 48.48 | 12.12 | 39.4 | P | P | P | A | A | |
| 47.04 | 11.76 | 41.2 | P | P | P | A | A | ||
| 7:3 | 41.16 | 17.65 | 41.19 | P | P | P | A | A | |
| 39.99 | 17.14 | 42.87 | P | P | P | A | A | ||
| 6:4 | 35.29 | 23.53 | 41.18 | P | P | P | A | A | |
| 34.29 | 22.86 | 42.85 | P | P | P | A | A | ||
| 5:5 | 22.73 | 22.73 | 54.54 | P | P | P | A | A | |
| 20.83 | 20.83 | 58.34 | P | P | P | A | A | ||
| 4:6 | 16.67 | 25.00 | 58.33 | P | P | P | A | A | |
| 15.38 | 23.07 | 61.55 | P | P | P | A | A | ||
| 3:7 | 13.04 | 30.43 | 56.53 | P | P | P | A | A | |
| 11.54 | 26.92 | 61.54 | P | P | P | A | A | ||
| 2:8 | 8.33 | 33.32 | 58.35 | P | P | P | A | A | |
| 7.69 | 30.76 | 61.55 | P | P | P | A | A | ||
| 1:9 | 4.35 | 39.15 | 56.5 | P | P | P | A | A | |
| 3.85 | 34.65 | 61.5 | P | P | P | A | A | ||
| Batch No. | Y1 (break)Dissolution Efficiency (%) | Y2 (break)Globule Size (nm) | Y3Self-Emulsification Time (s) |
|---|---|---|---|
| F1 | 55.67 ± 2.02 | 241.51 ± 40.92 | 38.52 ± 3.11 |
| F2 | 65.55 ± 1.82 | 153.8 ± 42. 55 | 45.05 ± 4.35 |
| F3 | 75.20 ± 2.18 | 112.3 ± 35.58 | 22.78 ± 3.42 |
| F4 | 92.98 ± 2.92 | 75.66 ± 24.47 | 31.14 ± 2.67 |
| F5 | 81.02 ±2.07 | 94.15 ± 29.51 | 58.37 ± 5.14 |
| F6 | 67.65 ± 2.15 | 149.2 ± 38.18 | 68.33 ± 5.48 |
| F7 | 80.19 ± 3.11 | 96.65 ± 24.72 | 59.18 ± 5.22 |
| F8 | 58.95 ± 2.19 | 195.7 ± 39.77 | 18.28 ± 2.46 |
| Dependent Variables | Experimental Value | Predicted Value |
|---|---|---|
| Y1 Dissolution efficiency (%) | 92.12 | 90.08 |
| Y2 Globule size (nm) | 76.03 | 77.92 |
| Y3 Self-emulsification time (s) | 29 | 25.04 |
| Parameter | SNEDDS | Control |
|---|---|---|
| Tmax a (h) | 2 | 6 |
| Cmax b (ng/mL) | 110.26 ± 13.93 * | 29.19 ± 5.63 |
| AUC0-α c (ng.h/mL) | 3115.73 ± 482.58 * | 685.73 ± 150.64 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nair, A.B.; Singh, B.; Shah, J.; Jacob, S.; Aldhubiab, B.; Sreeharsha, N.; Morsy, M.A.; Venugopala, K.N.; Attimarad, M.; Shinu, P. Formulation and Evaluation of Self-Nanoemulsifying Drug Delivery System Derived Tablet Containing Sertraline. Pharmaceutics 2022, 14, 336. https://doi.org/10.3390/pharmaceutics14020336
Nair AB, Singh B, Shah J, Jacob S, Aldhubiab B, Sreeharsha N, Morsy MA, Venugopala KN, Attimarad M, Shinu P. Formulation and Evaluation of Self-Nanoemulsifying Drug Delivery System Derived Tablet Containing Sertraline. Pharmaceutics. 2022; 14(2):336. https://doi.org/10.3390/pharmaceutics14020336
Chicago/Turabian StyleNair, Anroop B., Bhavna Singh, Jigar Shah, Shery Jacob, Bandar Aldhubiab, Nagaraja Sreeharsha, Mohamed A. Morsy, Katharigatta N. Venugopala, Mahesh Attimarad, and Pottathil Shinu. 2022. "Formulation and Evaluation of Self-Nanoemulsifying Drug Delivery System Derived Tablet Containing Sertraline" Pharmaceutics 14, no. 2: 336. https://doi.org/10.3390/pharmaceutics14020336
APA StyleNair, A. B., Singh, B., Shah, J., Jacob, S., Aldhubiab, B., Sreeharsha, N., Morsy, M. A., Venugopala, K. N., Attimarad, M., & Shinu, P. (2022). Formulation and Evaluation of Self-Nanoemulsifying Drug Delivery System Derived Tablet Containing Sertraline. Pharmaceutics, 14(2), 336. https://doi.org/10.3390/pharmaceutics14020336