

Time-Based Formulation Strategies for Colon Drug Delivery


 , ,
, ,
Abstract
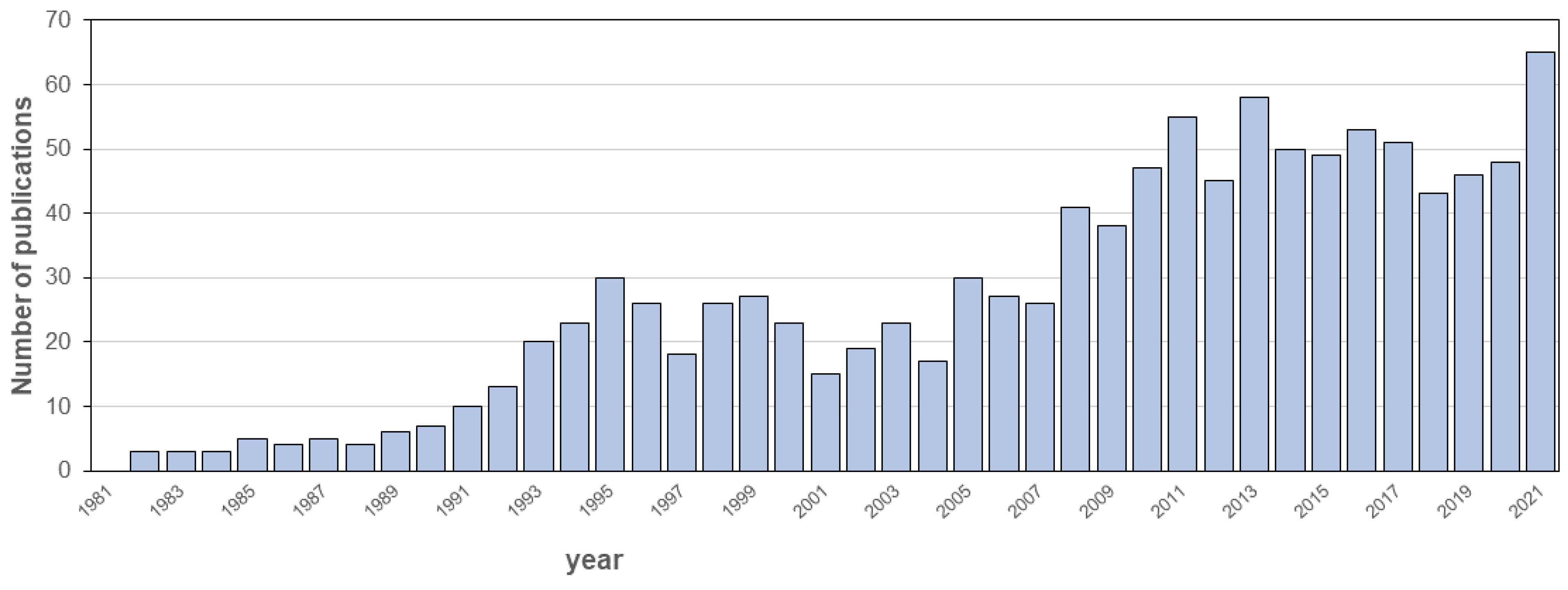
1. Introduction
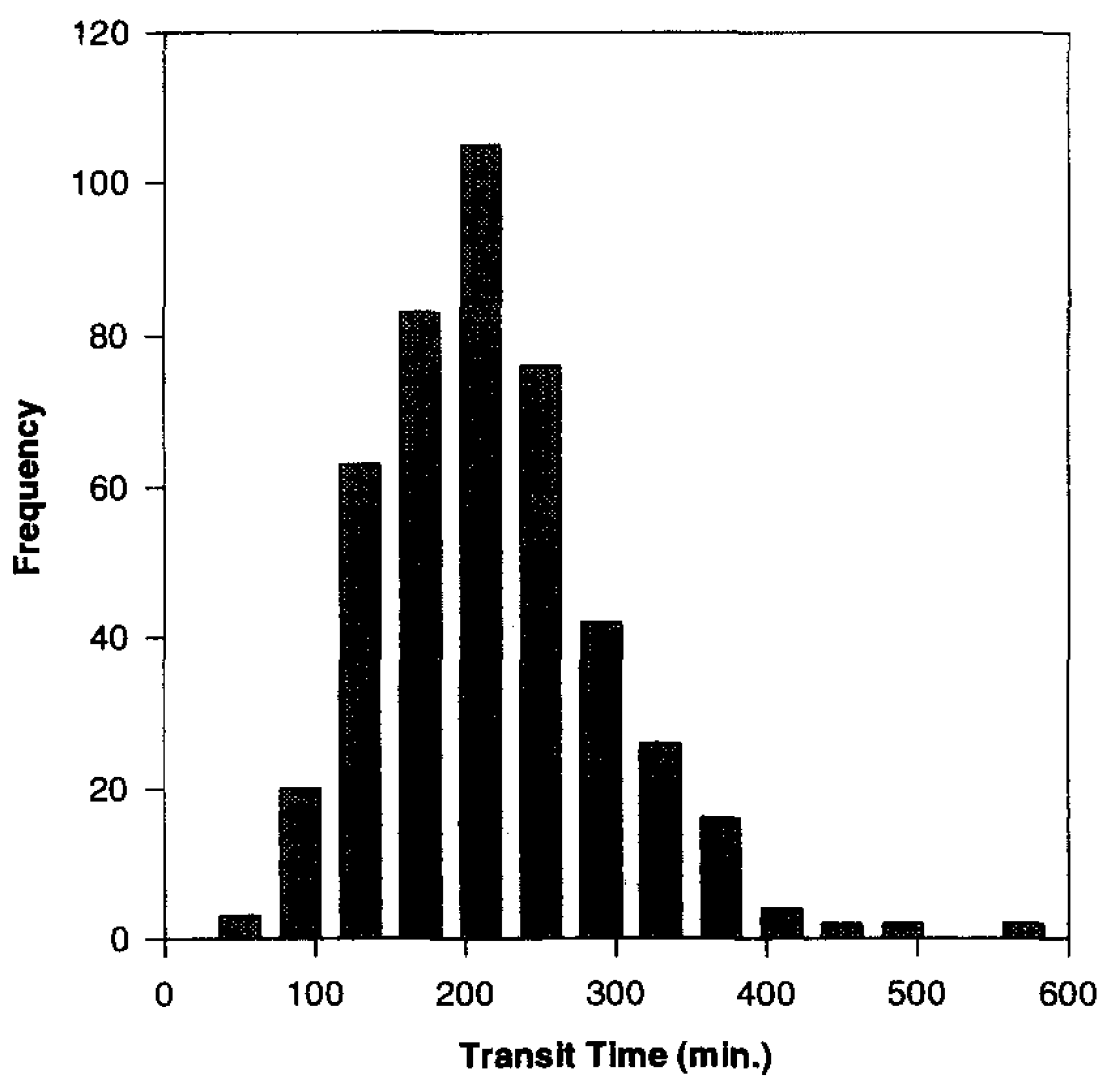
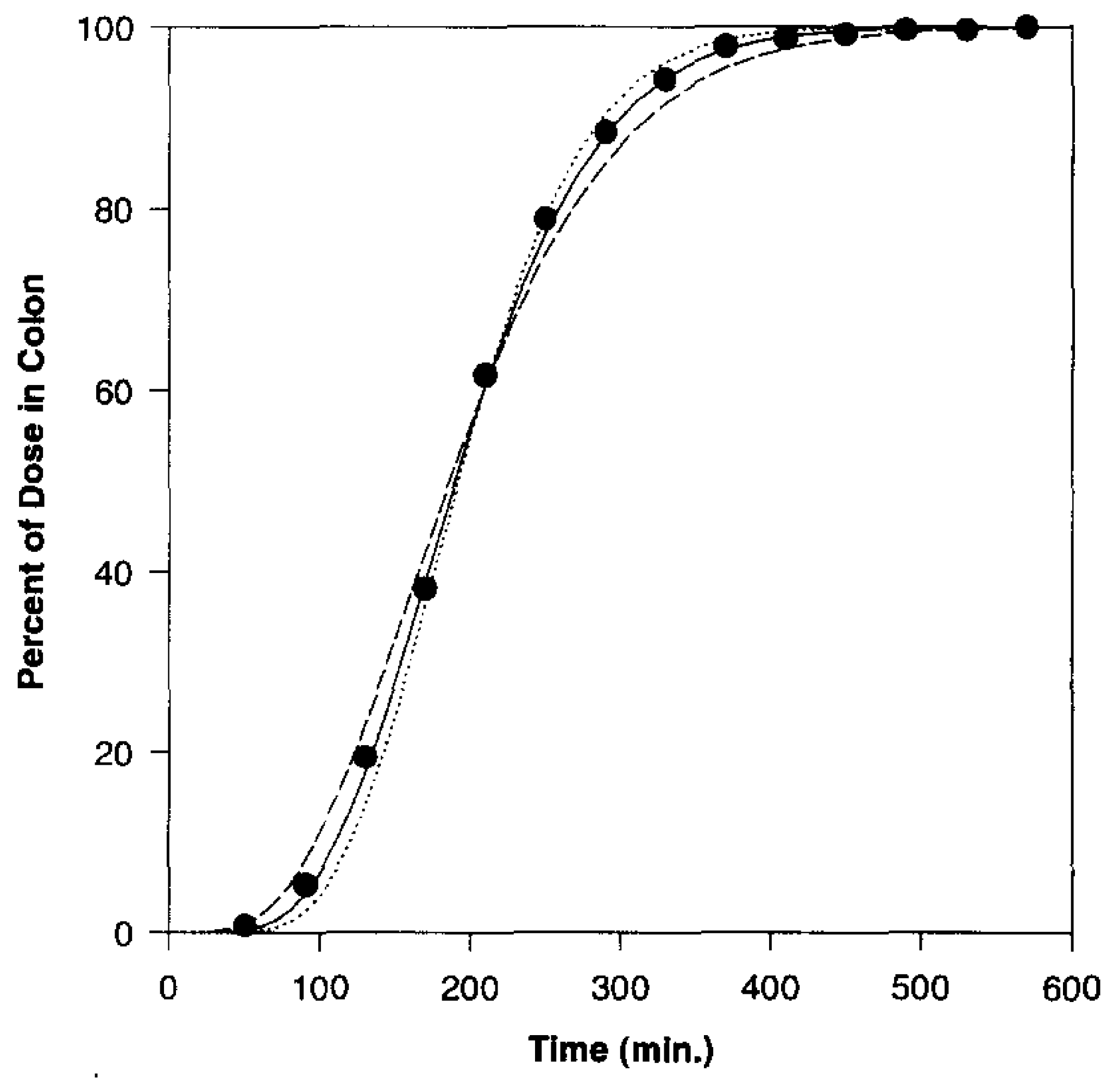
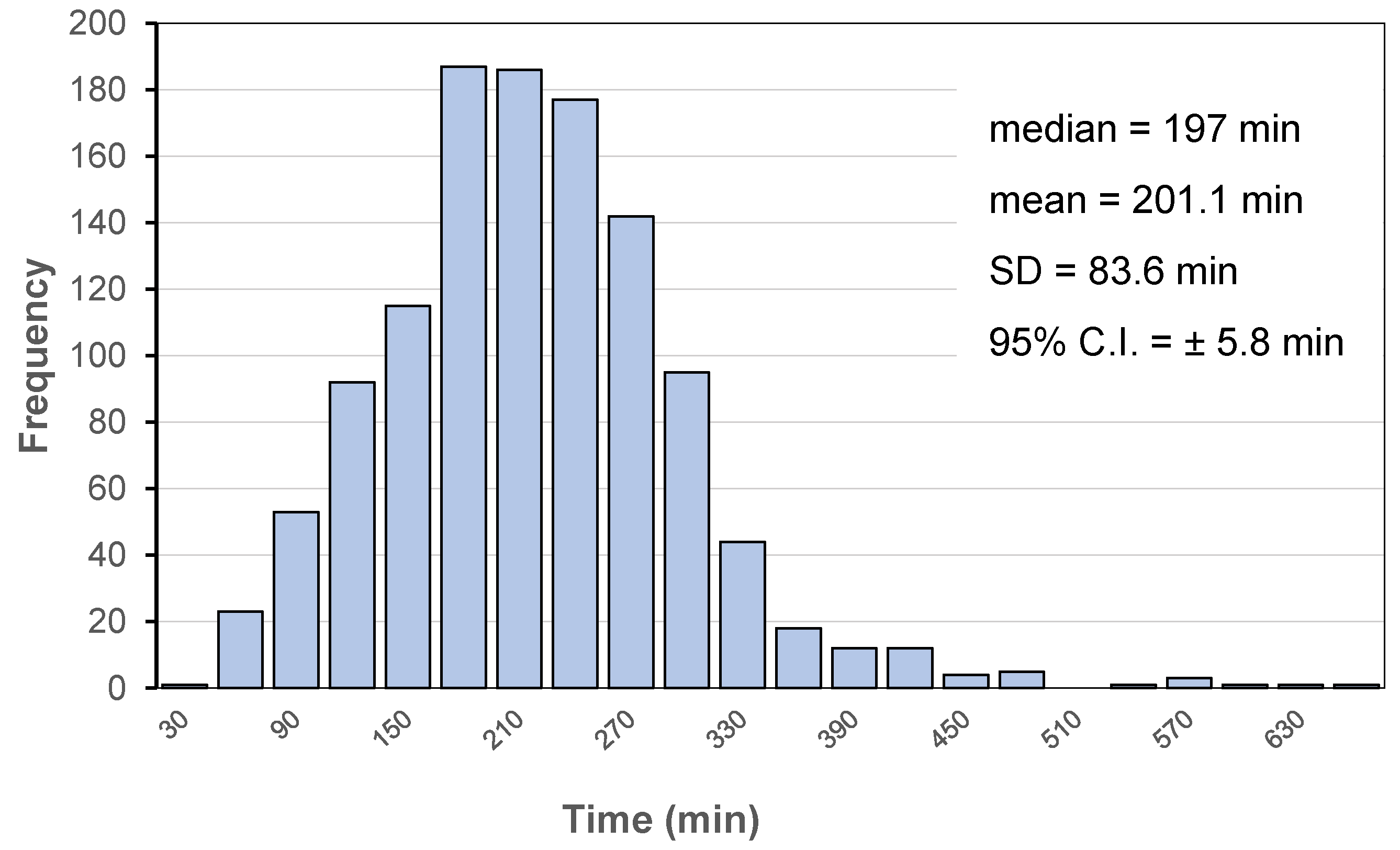
2. Time-Controlled Colon Drug Delivery Systems
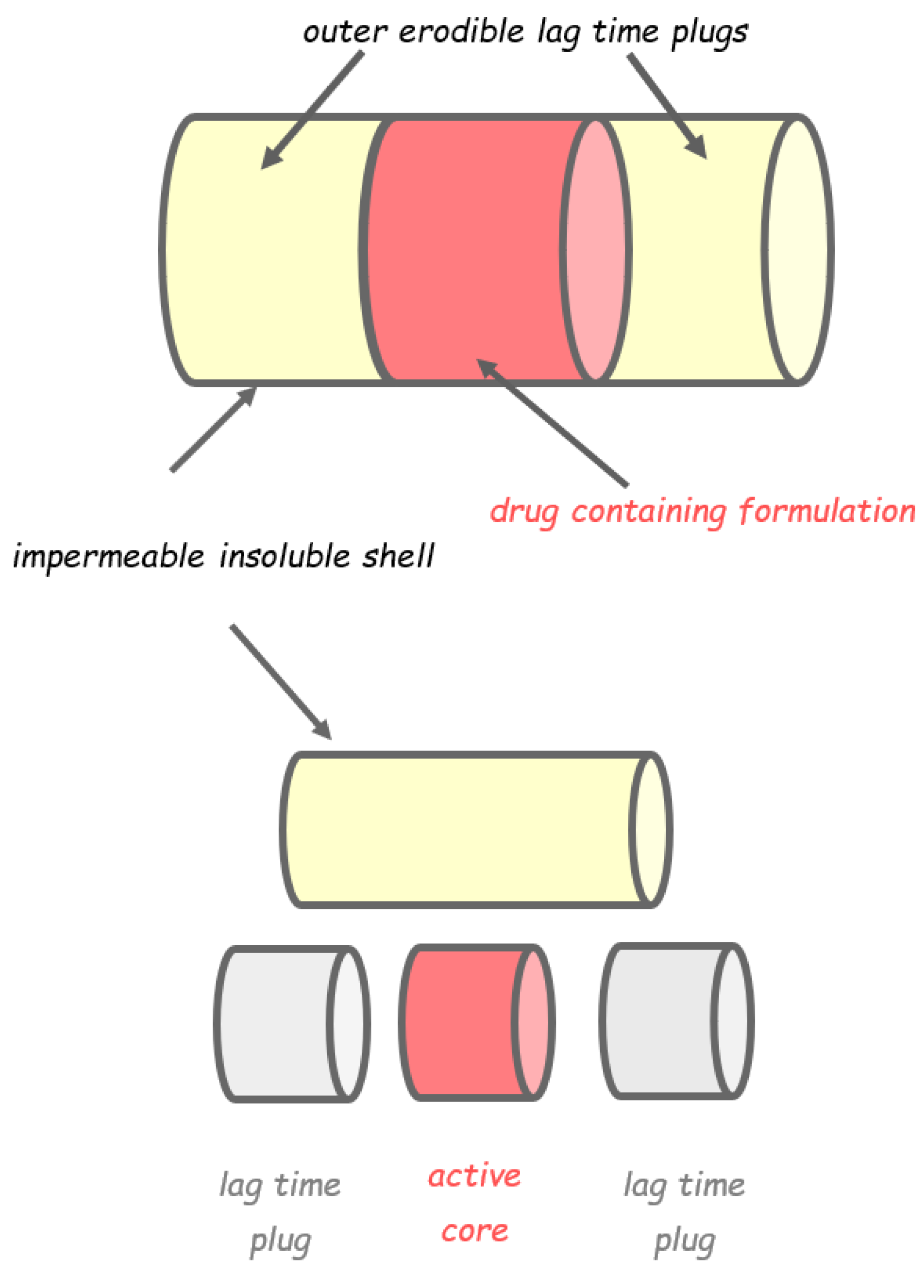
2.1. Capsular Devices with Release-Controlling Plugs
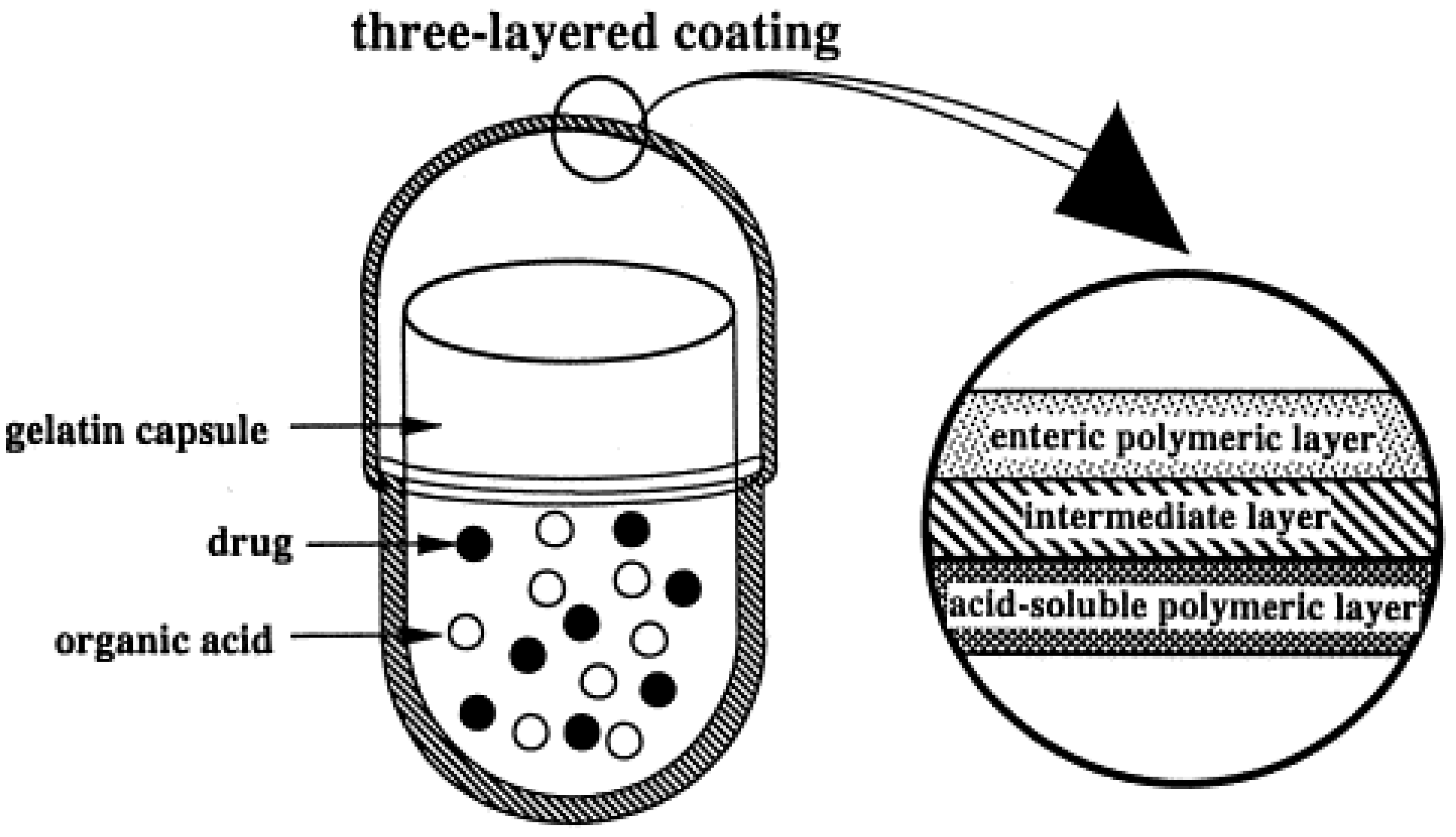
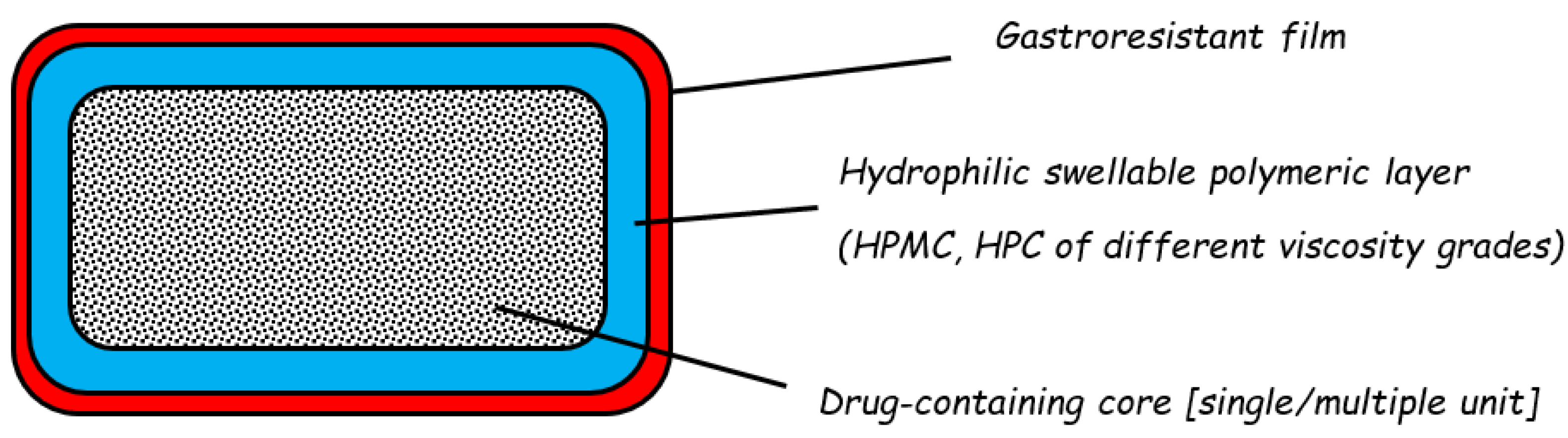
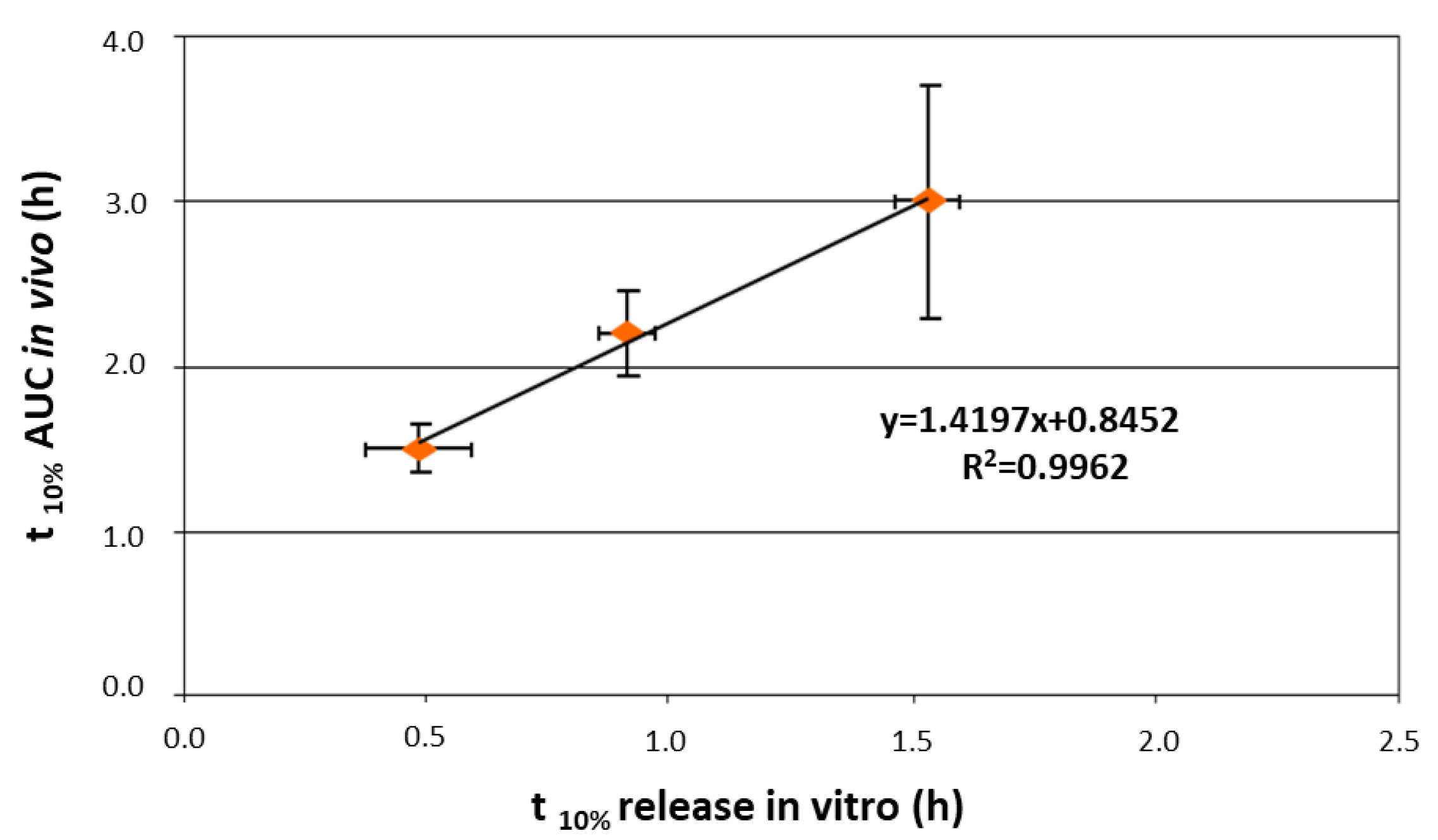
2.2. Reservoir Devices with Release-Controlling Coatings
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gazzaniga, A.; Giordano, F.; Sangalli, M.E.; Zema, L. Oral Colon-Specific Drug Delivery: Design Strategies. S.T.P. Pharma Prat. 1994, 4, 336–343. [Google Scholar]
- Awad, A.; Madla, C.M.; McCoubrey, L.E.; Ferraro, F.; Gavins, F.K.H.; Buanz, A.; Gaisford, S.; Orlu, M.; Siepmann, F.; Siepmann, J.; et al. Clinical Translation of Advanced Colonic Drug Delivery Technologies. Adv. Drug Deliv. Rev. 2022, 181, 114076. [Google Scholar] [CrossRef] [PubMed]
- Friend, D.R. New Oral Delivery Systems for Treatment of Inflammatory Bowel Disease. Adv. Drug Deliv. Rev. 2005, 57, 247–265. [Google Scholar] [CrossRef] [PubMed]
- Klotz, U.; Schwab, M. Topical Delivery of Therapeutic Agents in the Treatment of Inflammatory Bowel Disease. Adv. Drug Deliv. Rev. 2005, 57, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Haupt, S.; Rubinstein, A. The Colon as a Possible Target for Orally Administered Peptide and Protein Drugs. Crit. Rev. Drug Carr. Syst. 2002, 19, 499–551. [Google Scholar] [CrossRef]
- Maroni, A.; Zema, L.; Del Curto, M.D.; Foppoli, A.; Gazzaniga, A. Oral Colon Delivery of Insulin with the Aid of Functional Adjuvants. Adv. Drug Deliv. Rev. 2012, 64, 540–556. [Google Scholar] [CrossRef]
- Bourgeois, S.; Laham, A.; Besnard, M.; Andremont, A.; Fattal, E. In Vitro and in Vivo Evaluation of Pectin Beads for the Colon Delivery of β-Lactamases. J. Drug Target. 2005, 13, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Bak, A.; Ashford, M.; Brayden, D.J. Local Delivery of Macromolecules to Treat Diseases Associated with the Colon. Adv. Drug Deliv. Rev. 2018, 136–137, 2–27. [Google Scholar] [CrossRef]
- Tozer, T.N. Colonic Drug Delivery. In Proceedings of the 17th International Symposium on Controlled Release of Bioactive Materials, Reno, NV, USA, 22–25 July 1990; p. 126. [Google Scholar]
- Rubinstein, A. Natural Polysaccharides as Targeting Tools of Drugs to the Human Colon. Drug Dev. Res. 2000, 50, 435–439. [Google Scholar] [CrossRef]
- Van den Mooter, G.; Samyn, C.; Kinget, R. Azo Polymers for Colon-Specific Drug Delivery. Int. J. Pharm. 1992, 87, 37–46. [Google Scholar] [CrossRef]
- Van den Mooter, G.; Maris, B.; Samyn, C.; Augustus, P.; Kinget, R. Use of Azo Polymers for Colon-Specific Drug Delivery. J. Pharm. Sci. 1997, 86, 1321–1327. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.H.; Milojevic, S.; Harding, M.; Coward, W.A.; Gibson, G.R.; Botham, R.L.; Ring, S.G.; Wraight, E.P.; Stockham, M.A.; Allwood, M.C.; et al. In Vivo Studies of Amylose-and Ethylcellulose-Coated [13C]Glucose Microspheres as a Model for Drug Delivery to the Colon. J. Control. Release 1996, 40, 123–131. [Google Scholar] [CrossRef]
- Milojevic, S.; Newton, J.M.; Cummings, J.H.; Gibson, G.R.; Louise Botham, R.; Ring, S.G.; Stockham, M.; Allwood, M.C. Amylose as a Coating for Drug Delivery to the Colon: Preparation and in vitro Evaluation Using 5-Aminosalicylic Acid Pellets. J. Control. Release 1996, 38, 75–84. [Google Scholar] [CrossRef]
- Rasmussen, S.N.; Bondesen, S.; Hvidberg, E.F.; Hansen, S.H.; Binder, V.; Halskov, S.; Flachs, H. 5-Aminosalicylic Acid in a Slow-Release Preparation: Bioavailability, Plasma Level, and Excretion in Humans. Gastroenterology 1982, 83, 1062–1070. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.J.; Hanauer, S.B.; Buchà, A. Comparative Pharmacokinetics of Equimolar Doses of 5-Aminosalicylate Administered as Oral Mesalamine (Asacol) and Balsalazide: A Randomized, Single-Dose, Crossover Study in Healthy Volunteers. Aliment Pharm. 2004, 19, 1089–1098. [Google Scholar] [CrossRef]
- Dew, M.; Ryder, R.; Evans, N.; Evans, B.; Rhodes, J. Colonic Release of 5-amino Salicylic Acid from an Oral Preparation in Active Ulcerative Colitis. Br. J. Clin. Pharm. 1983, 16, 185–187. [Google Scholar] [CrossRef]
- Dew, M.; Hughes, P.; Lee, M.; Evans, B.; Rhodes, J. An Oral Preparation to Release Drugs in the Human Colon. Br. J. Clin. Pharm. 1982, 14, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Dew, M.J.; Hughes, P.; Harries, A.D.; Williams, G.; Evans, B.K.; Rhodes, J. Maintenance of Remission in Ulcerative Colitis with Oral Preparation of 5-Aminosalicylic Acid. Br. Med. J. 1982, 285, 1012. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.F.; Pye, G.; Bramley, R.; Clark, A.G.; Dyson, J.; Hardcastle, J.D. Measurement of Gastrointestinal pH Profiles in Normal Ambulant Human Subjects. Gut 1988, 29, 1035–1041. [Google Scholar] [CrossRef] [PubMed]
- Maroni, A.; Moutaharrik, S.; Zema, L.; Gazzaniga, A. Enteric Coatings for Colonic Drug Delivery: State of the Art. Expert Opin. Drug Deliv. 2017, 14, 1027–1029. [Google Scholar] [CrossRef]
- Ibekwe, V.C.; Khela, M.K.; Evans, D.F.; Basit, A.W. A New Concept in Colonic Drug Targeting: A Combined pH-Responsive and Bacterially-Triggered Drug Delivery Technology. Aliment. Pharmacol. Ther. 2008, 28, 911–916. [Google Scholar] [CrossRef]
- Varum, F.; Freire, A.C.; Fadda, H.M.; Bravo, R.; Basit, A.W. A Dual pH and Microbiota-Triggered Coating (PhloralTM) for Fail-Safe Colonic Drug Release. Int. J. Pharm. 2020, 583, 119379. [Google Scholar] [CrossRef] [PubMed]
- Varum, F.; Freire, A.C.; Bravo, R.; Basit, A.W. OPTICORETM, an Innovative and Accurate Colonic Targeting Technology. Int. J. Pharm. 2020, 583, 119372. [Google Scholar] [CrossRef] [PubMed]
- Moutaharrik, S.; Maroni, A.; Melocchi, A.; Zema, L.; Foppoli, A.; Cerea, M.; Palugan, L.; Neut, C.; Siepmann, F.; Siepmann, J.; et al. Oral Colon Delivery Platform Based on a Novel Combination Approach: Design Concept and Preliminary Evaluation. J. Drug Deliv. Sci. Technol. 2021, 66, 102919. [Google Scholar] [CrossRef]
- Varum, F.; Bravo, R.; Basit, A. OPTICORETM: A First-in-Class Colonic Targeting Technology. ONdrugDelivery 2020, 2020, 40–44. [Google Scholar]
- Davis, S.S. The Design and Evaluation of Controlled Release Systems for the Gastrointestinal Tract. J. Control. Release 1985, 2, 27–38. [Google Scholar] [CrossRef]
- Davis, S.S.; Hardy, J.G.; Fara, J.W. Transit of Pharmaceutical Dosage Forms through the Small Intestine. Gut 1986, 27, 886–892. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.S.; Hardy, J.G.; Taylor, M.J.; Whalley, D.R.; Wilson, C.G. The Effect of Food on the Gastrointestinal Transit of Pellets and an Osmotic Device (Osmet). Int. J. Pharm. 1984, 21, 331–340. [Google Scholar] [CrossRef]
- Davis, S.S.; Hardy, J.G.; Wilson, C.G.; Feely, L.C.; Palin, K.J. Gastrointestinal Transit of a Controlled Release Naproxen Tablet Formulation. Int. J. Pharm. 1986, 32, 85–90. [Google Scholar] [CrossRef]
- Gazzaniga, A.; Busetti, C.; Moro, L.; Sangalli, M.E.; Giordano, F. Time-Dependent Oral Delivery Systems for Colon Targeting. S.T.P. Pharma Sci. 1995, 5, 83–88. [Google Scholar]
- Iamartino, P.; Maffione, G.; Pontello, L. Orally-Pharmaceutical Preparations with Colon Selective Delivery. U.S. Patent 5,171,580, 15 December 1992. [Google Scholar]
- McNeil, M.E.; Rashid, A.; Stevens, H.N.E. Dispensing Device. WO1990009168A1, 23 August 1990. [Google Scholar]
- Pozzi, F.; Furlani, P. Programmed Release Oral Solid Pharmaceutical Dosage Form. GB 2,245,492, 8 January 1992. [Google Scholar]
- Hatano, H.A.; Ito, T.Y.; Ishibashi, T.S.; Yoshino, H.S.; Mizobe, M.T. Pharmaceutical Preparation in Form of Coated Capsule Release able at Lower Part of Digestive Tract. U.S. Patent 6,309,666, 30 October 2001. [Google Scholar]
- Bar-Shalom, D. Controlled Release Composition. CA 2,327,685A1, 14 October 1999. [Google Scholar]
- Weitschies, W.; Blume, H.; Mönnikes, H. Magnetic Marker Monitoring: High Resolution Real-Time Tracking of Oral Solid Dosage Forms in the Gastrointestinal Tract. Eur. J. Pharm. Biopharm. 2010, 74, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.X.; Crison, J.R.; Amidon, G.L. Compartmental Transit and Dispersion Model Analysis of Small Intestinal Transit Flow in Humans. Int. J. Pharm. 1996, 140, 111–118. [Google Scholar] [CrossRef]
- Yuen, K.H. The Transit of Dosage Forms through the Small Intestine. Int. J. Pharm. 2010, 395, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Fadda, H.M.; McConnell, E.L.; Short, M.D.; Basit, A.W. Meal-Induced Acceleration of Tablet Transit through the Human Small Intestine. Pharm. Res. 2009, 26, 356–360. [Google Scholar] [CrossRef]
- Koch-Weser, J.; Schechter, P.J. Slow-Release Preparations in Clinical Perspective. In Drug Absorption; Prescott, L.F., Nimmo, W.S., Eds.; MTP Press: Lancaster, UK, 1981; pp. 217–227. [Google Scholar]
- Abuhelwa, A.Y.; Foster, D.J.R.; Upton, R.N. A Quantitative Review and Meta-Models of the Variability and Factors Affecting Oral Drug Absorption-Part I: Gastrointestinal pH. AAPS J. 2016, 18, 1309–1321. [Google Scholar] [CrossRef]
- Abuhelwa, A.Y.; Foster, D.J.R.; Upton, R.N. A Quantitative Review and Meta-Models of the Variability and Factors Affecting Oral Drug Absorption—Part II: Gastrointestinal Transit Time. AAPS J. 2016, 18, 1322–1333. [Google Scholar] [CrossRef]
- Liu, F.; Basit, A.W. A Paradigm Shift in Enteric Coating: Achieving Rapid Release in the Proximal Small Intestine of Man. J. Control. Release 2010, 147, 242–245. [Google Scholar] [CrossRef]
- Hénin, E.; Bergstrand, M.; Weitschies, W.; Karlsson, M.O. Meta-Analysis of Magnetic Marker Monitoring Data to Characterize the Movement of Single Unit Dosage Forms Though the Gastrointestinal Tract under Fed and Fasting Conditions. Pharm. Res. 2016, 33, 751–762. [Google Scholar] [CrossRef]
- Wilding, I.R.; Davis, S.S.; Bakhshaee, M.; Stevens, H.N.E.; Sparrow, R.A.; Brennan, J. Gastrointestinal transit and systemic absorption of captopril from a pulsed-release formulation. Pharm. Res. 1992, 9, 654–657. [Google Scholar] [CrossRef]
- Wilson, C.G.; Bakhshaee, M.; Stevens, H.N.E.; Perkins, A.C.; Frier, M.; Blackshaw, E.P.; Binns, J.S. Evaluation of a Gastro-Resistant Pulsed Release Delivery System (Pulsincap) in Humans. Drug Deliv. 1997, 4, 201–206. [Google Scholar] [CrossRef]
- Bar-Shalom, D.; Slot, L.; Lee, W.W.; Wilson, C.G. Development of the Egalet® Technology. In Modified-Release Drug Delivery Technology; Rathbone, M.J., Hadgraft, J., Roberts, M.S., Eds.; Marcel Dekker: New York, NY, USA, 2003; pp. 263–271. ISBN 978-0-8247-0869-6. [Google Scholar]
- Lee, W.W.; Mahony, B.O.; Bar-Shalom, D.; Slot, L.; Wilson, C.G.; Blackshaw, P.E.; Perkins, A.C. Scintigraphic Characterisation of a Novel Injection-Moulded Dosage Form. In Proceedings of the 27th International Symposium on Controlled Release of Bioactive Materials, Paris, France, 7–13 July 2000. [Google Scholar]
- Pozzi, F.; Furlani, P.; Gazzaniga, A.; Davis, S.; Wilding, I. The Time-Clock System: A New Oral Dosage Form for Fast and Complete Release of Drug after a Predetermined Lag Time. J. Control. Release 1994, 31, 99–108. [Google Scholar] [CrossRef]
- Wilding, I.; Davis, S.; Pozzi, F.; Furlani, P.; Gazzaniga, A. Enteric Coated Timed Release Systems for Colonic Targeting. Int. J. Pharm. 1994, 111, 99–102. [Google Scholar] [CrossRef]
- Steed, K.P.; Hooper, G.; Monti, N.; Strolin Benedetti, M.; Fornasini, G.; Wilding, I.R. The Use of Pharmacoscintigraphy to Focus the Development Strategy for a Novel 5-ASA Colon Targeting System (“Time Clock®” System). J. Control. Release 1997, 49, 115–122. [Google Scholar] [CrossRef]
- Ishibashi, T.; Hatano, H.; Kobayashi, M.; Mizobe, M.; Yoshino, H. Design and Evaluation of a New Capsule-Type Dosage Form for Colon-Targeted Delivery of Drugs. Int. J. Pharm. 1998, 168, 31–40. [Google Scholar] [CrossRef]
- Ishibashi, T.; Ikegami, K.; Kubo, H.; Kobayashi, M.; Mizobe, M.; Yoshino, H. Evaluation of Colonic Absorbability of Drugs in Dogs Using a Novel Colon-Targeted Delivery Capsule (CTDC). J. Control. Release 1999, 59, 361–376. [Google Scholar] [CrossRef]
- Ishibashi, T.; Pitcairn, G.R.; Yoshino, H.; Mizobe, M.; Wilding, I.R. Scintigraphic Evaluation of a New Capsule-Type Colon Specific Drug Delivery System in Healthy Volunteers. J. Pharm. Sci. 1998, 87, 531–535. [Google Scholar] [CrossRef]
- Gazzaniga, A.; Sangalli, M.E.; Giordano, F. Oral Chronotopic Drug-Delivery Systems—Achievement of Time and or Site-Specificity. Eur. Pharm. Biopharm. 1994, 40, 246–250. [Google Scholar]
- Gazzaniga, A.; Iamartino, P.; Maffione, G.; Sangalli, M.E. Oral Delayed-Release System for Colonic Specific Delivery. Int. J. Pharm. 1994, 108, 77–83. [Google Scholar] [CrossRef]
- Sangalli, M.E.E.; Maroni, A.; Zema, L.; Cerea, M.; Gazzaniga, A. ChronotopicTM Technology. In Chronopharmaceutics: Science and Technology for Biological Rhythm-Guided Therapy and Prevention of Diseases; John Wiley & Sons: New York, NY, USA, 2009; ISBN 978-0-47174-343-9. [Google Scholar]
- Maffione, G.; Iamartino, P.; Guglielmini, G.; Gazzaniga, A. High-Viscosity HPMC as a Film-Coating Agent. Drug Dev. Ind. Pharm. 1993, 19, 2043–2053. [Google Scholar] [CrossRef]
- Sangalli, M.E.; Maroni, A.; Foppoli, A.; Zema, L.; Giordano, F.; Gazzaniga, A. Different HPMC Viscosity Grades as Coating Agents for an Oral Time and/or Site-Controlled Delivery System: A Study on Process Parameters and in Vitro Performances. Eur. J. Pharm. Sci. 2004, 22, 469–476. [Google Scholar] [CrossRef]
- Sangalli, M.E.; Maroni, A.; Zema, L.; Busetti, C.; Giordano, F.; Gazzaniga, A. In Vitro and in Vivo Evaluation of an Oral System for Time and/or Site-Specific Drug Delivery. J. Control. Release 2001, 73, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Foppoli, A.; Maroni, A.; Palugan, L.; Zema, L.; Moutaharrik, S.; Melocchi, A.; Cerea, M.; Gazzaniga, A. Erodible Coatings Based on HPMC and Cellulase for Oral Time-Controlled Release of Drugs. Int. J. Pharm. 2020, 585, 119425. [Google Scholar] [CrossRef] [PubMed]
- Foppoli, A.; Cerea, M.; Palugan, L.; Zema, L.; Melocchi, A.; Maroni, A.; Gazzaniga, A. Evaluation of Powder-Layering vs. Spray-Coating Techniques in the Manufacturing of a Swellable/Erodible Pulsatile Delivery System. Drug Dev. Ind. Pharm. 2020, 46, 1230–1237. [Google Scholar] [CrossRef] [PubMed]
- Cerea, M.; Maroni, A.; Palugan, L.; Moutaharrik, S.; Melocchi, A.; Zema, L.; Foppoli, A.; Gazzaniga, A. Oral Hydrophilic Matrices Having Non Uniform Drug Distribution for Zero-Order Release: A Literature Review. J. Control. Release 2020, 325, 72–83. [Google Scholar] [CrossRef]
- Gazzaniga, A.; Cerea, M.; Cozzi, A.; Foppoli, A.; Maroni, A.; Zema, L. A Novel Injection-Molded Capsular Device for Oral Pulsatile Delivery Based on Swellable/Erodible Polymers. AAPS PharmSciTech 2011, 12, 295–303. [Google Scholar] [CrossRef]
- Macchi, E.; Zema, L.; Maroni, A.; Gazzaniga, A.; Felton, L.A. Enteric-Coating of Pulsatile-Release HPC Capsules Prepared by Injection Molding. Eur. J. Pharm. Sci. 2015, 70, 1–11. [Google Scholar] [CrossRef]
- Cozzi, A. Applicazioni Farmaceutiche delle Tecniche di Estrusione. Ph.D. Thesis, Università degli Studi di Milano, Milan, Italy, 2008. [Google Scholar]
- Melocchi, A.; Parietti, F.; Loreti, G.; Maroni, A.; Gazzaniga, A.; Zema, L. 3D Printing by Fused Deposition Modeling (FDM) of a Swellable/Erodible Capsular Device for Oral Pulsatile Release of Drugs. J. Drug Deliv. Sci. Technol. 2015, 30, 360–367. [Google Scholar] [CrossRef]
- Melocchi, A.; Parietti, F.; Maroni, A.; Foppoli, A.; Gazzaniga, A.; Zema, L. Hot-Melt Extruded Filaments Based on Pharmaceutical Grade Polymers for 3D Printing by Fused Deposition Modeling. Int. J. Pharm. 2016, 509, 255–263. [Google Scholar] [CrossRef]
- Maroni, A.; Melocchi, A.; Parietti, F.; Foppoli, A.; Zema, L.; Gazzaniga, A. 3D Printed Multi-Compartment Capsular Devices for Two-Pulse Oral Drug Delivery. J. Control. Release 2017, 268, 10–18. [Google Scholar] [CrossRef]
- Foppoli, A.; Maroni, A.; Moutaharrik, S.; Melocchi, A.; Zema, L.; Palugan, L.; Cerea, M.; Gazzaniga, A. In Vitro and Human Pharmacoscintigraphic Evaluation of an Oral 5-ASA Delivery System for Colonic Release. Int. J. Pharm. 2019, 572, 118723. [Google Scholar] [CrossRef]
- Maroni, A.; Del Curto, M.D.; Cerea, M.; Zema, L.; Foppoli, A.; Gazzaniga, A. Polymeric Coatings for a Multiple-Unit Pulsatile Delivery System: Preliminary Study on Free and Applied Films. Int. J. Pharm. 2013, 440, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Del Curto, M.D.; Palugan, L.; Foppoli, A.; Zema, L.; Gazzaniga, A.; Maroni, A. Erodible Time-Dependent Colon Delivery Systems with Improved Efficiency in Delaying the Onset of Drug Release. J. Pharm. Sci. 2014, 103, 3585–3593. [Google Scholar] [CrossRef] [PubMed]
- Maroni, A.; Del Curto, M.D.; Salmaso, S.; Zema, L.; Melocchi, A.; Caliceti, P.; Gazzaniga, A. In Vitro and in Vivo Evaluation of an Oral Multiple-Unit Formulation for Colonic Delivery of Insulin. Eur. J. Pharm. Biopharm. 2016, 108, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Del Curto, M.D.; Maroni, A.; Palugan, L.; Zema, L.; Gazzaniga, A.; Sangalli, M.E. Oral Delivery System for Two-Pulse Colonic Release of Protein Drugs and Protease Inhibitor/Absorption Enhancer Compounds. J. Pharm. Sci. 2011, 100, 3251–3259. [Google Scholar] [CrossRef]
- Davis, S.S.; Wilding, I.R. Oral Drug Absorption Studies: The Best Model for Man Is Man! Drug Discov. Today 2001, 6, 127–130. [Google Scholar] [CrossRef]
- Sjögren, E.; Abrahamsson, B.; Augustijns, P.; Becker, D.; Bolger, M.B.; Brewster, M.; Brouwers, J.; Flanagan, T.; Harwood, M.; Heinen, C.; et al. In Vivo Methods for Drug Absorption–comparative Physiologies, Model Selection, Correlations with in Vitro Methods (IVIVC), and Applications for Formulation/API/Excipient Characterization Including Food Effects. Eur. J. Pharm. Sci. 2014, 57, 99–151. [Google Scholar] [CrossRef]


































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
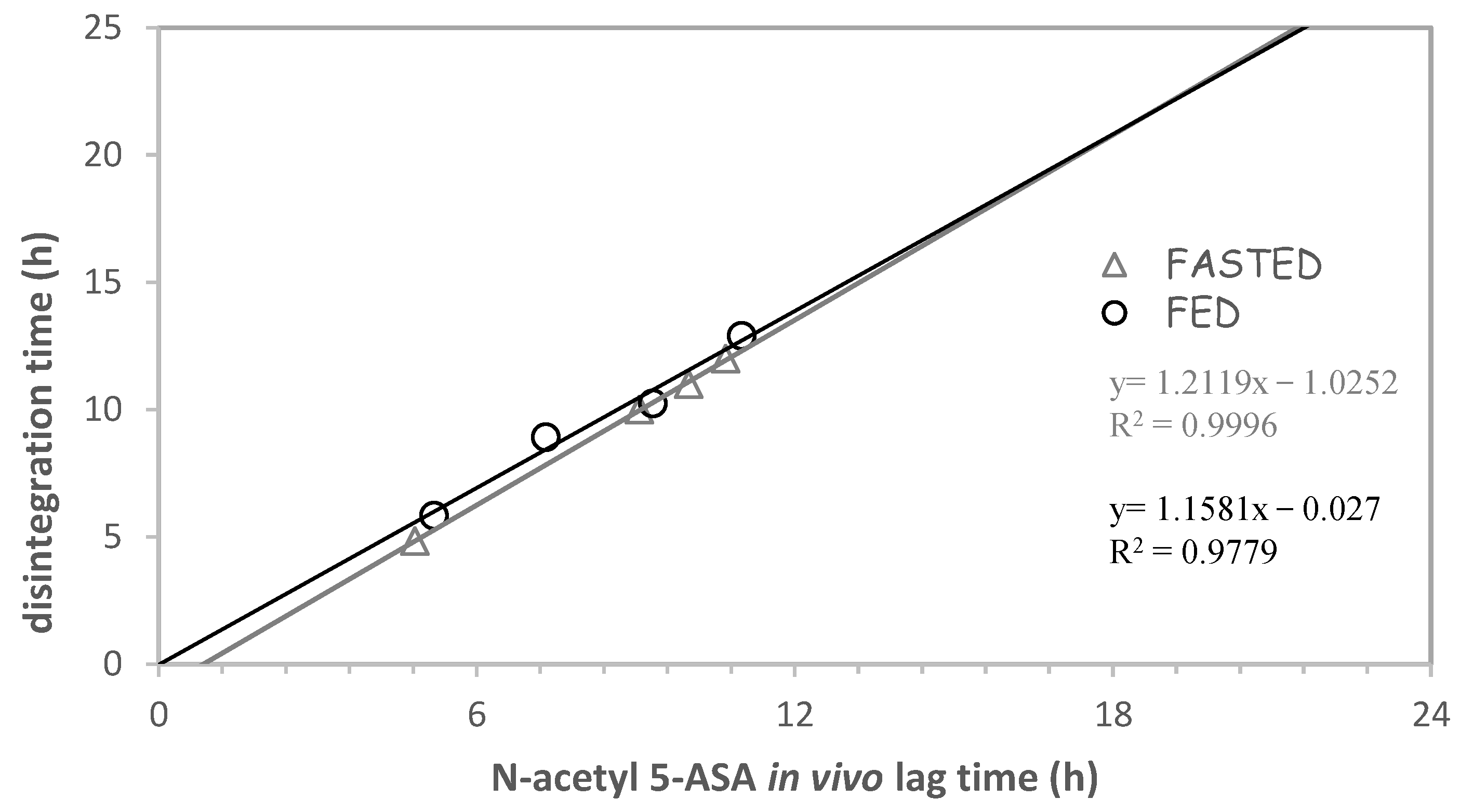{kind=link}
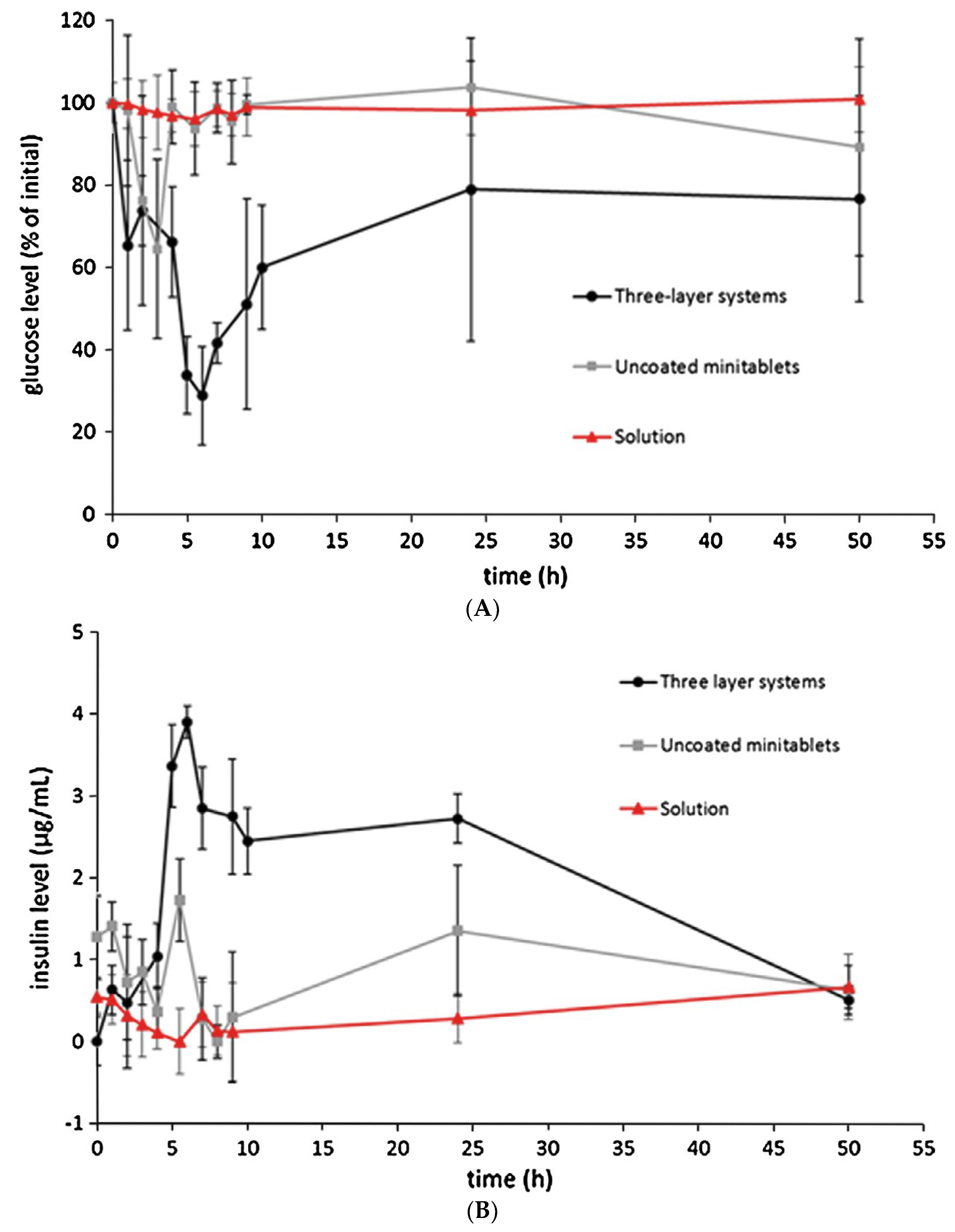{kind=link}
| Delivery Platform | Type | Patent Priority Date | Patent n. |
|---|---|---|---|
| Chronotopic® | Tablet device | 20 October 1988 | US 5,171,580 [32] |
| Pulsincap™ | Capsule device | 16 February 1989 | WO 90/09168 [33] |
| Time Clock® | Tablet device | 4 July 1990 | GB 2,245,492 [34] |
| CTDC | Capsule device | 20 July 1995 | US 6,309,666B1 [35] |
| Egalet® | Cylindrical container device | 3 April 1997 | CA 2,327,685 [36] |
| Subject | Gastric Residence | Small Intestine Transit | Colon Arrival | Plug Separation Post-Dose | Plug Separation Post-GE | Ascending Colon Residence |
|---|---|---|---|---|---|---|
| 1 | 0.19 | 3.31 | 3.50 | 3.68 | 3.49 | 6.33 |
| 2 | 0.56 | 3.38 | 3.94 | 4.52 | 3.96 | 5.14 |
| 3 | 0.86 | 2.77 | 3.63 | 5.07 | 4.21 | 7.73 |
| 4 | 0.27 | 3.33 | 3.60 | 4.13 | 3.86 | 2.63 |
| 5 | 0.81 | 3.07 | 3.88 | 4.50 | 3.69 | 6.03 |
| 6 | 0.26 | 3.32 | 3.58 | 10.48 1 | 10.22 1 | 8.20 |
| Mean | 0.49 | 3.20 | 3.69 | 5.40 | 4.91 | 6.01 |
| SD | 0.30 | 0.24 | 0.18 | 2.53 | 2.62 | 2.00 |
| Subject | Gastric Residence | Release Time | Release Site |
|---|---|---|---|
| 1 | 1.00 | 5.67 | Ascending colon |
| 2 | 0.67 | 6.33 | Ascending colon |
| 3 | 0.33 | 7.00 | Transverse colon |
| 4 | 1.67 | 8.00 | Transverse colon |
| 5 | 0.67 | 5.67 | Ascending colon |
| 6 | 0.67 | 8.00 | Ascending colon |
| Mean | 0.84 | 6.78 | |
| SD | 0.46 | 1.07 |
| Subject | Gastric Residence | Small Intestine Transit | Colon Arrival | Tablet Dispersion | Position of Dispersion |
|---|---|---|---|---|---|
| 1 | 103 | 248 | 351 | 655 | Cecum |
| 2 | 251 | 168 | 419 | 656 | Proximal colon |
| 3 | 154 | 267 | 421 | 655 | Cecum |
| 4 | 123 | 186 | 319 | 593 | Proximal colon |
| 5 | 87 | 163 | 250 | 523 | Descending colon |
| 6 | 201 | 251 | 452 | 575 | Proximal colon |
| Mean | 153 | 261 | 369 | 610 | |
| SE | 27 | 19 | 31 | 23 |
| Subject | Initial Disintegration | Complete Disintegration | |||
|---|---|---|---|---|---|
| Minutes Post-Dose | Minutes Post-GE | Anatomical Position | Minutes Post-Dose | Anatomical Position | |
| 1 | 371 | 324 | Ileo-cecal junction | 422 | Ascending colon |
| 2 | 310 | 282 | Ascending colon | 421 | Ascending colon |
| 3 | 304 | 241 | Ileo-cecal junction | 514 | Ascending colon |
| 4 | 298 | 272 | Descending colon | 469 | Descending colon |
| 5 | 385 | 349 | Ascending colon | 495 | Ascending colon |
| 6 | 663 | 590 | Ascending colon | 685 | Ascending colon |
| 7 | 240 | 201 | Ascending colon | 301 | Ascending colon |
| 8 | 283 | 270 | Ascending colon | 502 | Transverse colon |
| Mean | 357 | 316 | 476 | ||
| SD | 132 | 120 | 109 | ||
| Subject | Gastric Residence | Small Intestine Transit | Colon Arrival | Breakup Time after Gastric Emptying | Breakup Site |
|---|---|---|---|---|---|
| 1 | 1.0 | 7.0 | 8.0 | 7.0 | Cecum/ascending colon |
| 2 | 2.0 | 5.0 | 7.0 | 6.0 | Ascending colon |
| 3 | 0.5 | 3.5 | 4.0 | 4.5 | Cecum/ascending colon |
| 4 | 0.5 | 4.5 | 5.0 | 5.5 | Ascending colon |
| 5 | 1.0 | 4.5 | 5.5 | 5.0 | Cecum/ascending colon |
| 6 | 0.5 | 4.5 | 6.0 | 6.0 | Ascending colon |
| Mean | 0.9 | 5.0 | 5.9 | 5.7 | |
| SD | 0.5 | 1.1 | 1.3 | 0.8 |
| Fasted | |||||
| Subject | Gastric Residence | Small Intestine Transit | Colon Arrival | Breakup Time after Gastric Emptying | Breakup Site |
| 1 | 1.75 | 3.00 | 4.75 | 10.25 | Transverse colon |
| 2 | 0.33 | 4.50 | 4.84 | 4.50 | Cecum |
| 3 | 0.42 | 1.50 | 1.92 | 10.58 | Ascending colon |
| 4 | 0.42 | 4.58 | 5.00 | 9.58 | Ascending colon |
| 5 | 0.58 | 4.50 | 5.08 | 10.52 1 | Transverse colon |
| 6 | 0.58 | 1.59 | 2.17 | 12.59 | Transverse colon |
| Mean | 0.68 | 3.28 | 3.96 | 8.73 | |
| SD | 0.53 | 1.47 | 1.49 | 2.85 | |
| Fed | |||||
| Subject | Gastric Residence | Small Intestine Transit | Colon Arrival | Breakup Time after Gastric Emptying | Breakup Site |
| 1 | 1.83 | 1.92 | 3.75 | - | - |
| 2 | 2.33 | 2.50 | 4.83 | 3.50 | Ascending colon |
| 3 | 1.41 | 3.51 | 4.92 | 7.51 | Ascending colon |
| 4 | 2.50 | 2.50 | 5.00 | 10.40 | Ascending colon |
| 5 | 1.92 | 3.00 | 4.92 | 11.95 1 | Ascending colon |
| 6 | 0.58 | 3.59 | 4.17 | 9.67 | Transverse colon |
| Mean | 1.76 | 2.84 | 4.60 | 7.77 | |
| SD | 0.70 | 0.65 | 0.51 | 3.10 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gazzaniga, A.; Moutaharrik, S.; Filippin, I.; Foppoli, A.; Palugan, L.; Maroni, A.; Cerea, M. Time-Based Formulation Strategies for Colon Drug Delivery. Pharmaceutics 2022, 14, 2762. https://doi.org/10.3390/pharmaceutics14122762
Gazzaniga A, Moutaharrik S, Filippin I, Foppoli A, Palugan L, Maroni A, Cerea M. Time-Based Formulation Strategies for Colon Drug Delivery. Pharmaceutics. 2022; 14(12):2762. https://doi.org/10.3390/pharmaceutics14122762
Chicago/Turabian StyleGazzaniga, Andrea, Saliha Moutaharrik, Ilaria Filippin, Anastasia Foppoli, Luca Palugan, Alessandra Maroni, and Matteo Cerea. 2022. "Time-Based Formulation Strategies for Colon Drug Delivery" Pharmaceutics 14, no. 12: 2762. https://doi.org/10.3390/pharmaceutics14122762
APA StyleGazzaniga, A., Moutaharrik, S., Filippin, I., Foppoli, A., Palugan, L., Maroni, A., & Cerea, M. (2022). Time-Based Formulation Strategies for Colon Drug Delivery. Pharmaceutics, 14(12), 2762. https://doi.org/10.3390/pharmaceutics14122762