

Thermodynamic Correlation between Liquid–Liquid Phase Separation and Crystalline Solubility of Drug-Like Molecules

Abstract
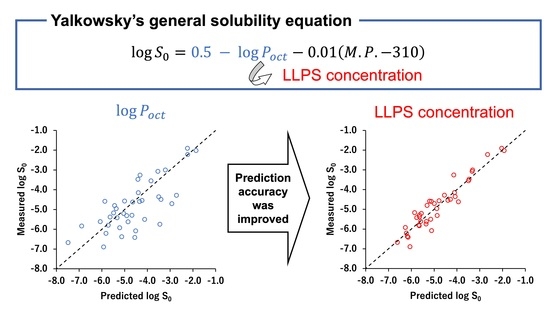
1. Introduction
2. Theory
3. Materials and Methods
3.1. Materials
3.2. Methods
3.2.1. Crystallization Time Measurement
3.2.2. Measurement by the Precipitation Tests Coupled with Laser-Assisted Visual Turbidity Detection (LAVTD)
3.2.3. Measurement by Turbidity Detection Using a UV/VIS Spectrophotometer
3.2.4. Intrinsic Solubility Measurement
3.2.5. Differential Scanning Calorimetry Measurement
3.2.6. Thermodynamic Correlation between the Intrinsic Liquid–Liquid Phase Separation Concentration and Crystalline Solubility
4. Results
4.1. Crystallization Time
4.2. Validation of the Precipitation Tests Coupled with Laser-Assisted Visual Turbidity Detection
4.3. Thermodynamic Correlation between the Intrinsic Liquid–Liquid Phase Separation Concentration and Crystalline Solubility

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drugs | MW | Ref. | |||||
|---|---|---|---|---|---|---|---|
| Atazanavir | 705 | 4.5 (B) | 481 | 5.8 | −4.03 | −5.83 | [23,24] |
| Bifonazole | 310 | 5.7 (B) | 422 2 | 4.8 | −4.70 ± 0.00 2 | −5.42 ± 0.03 2 | [25,26] |
| Carprofen | 274 | 4.2 (A) | 484 2 | 4.3 | −3.66 ± 0.00 2 | −4.80 ± 0.02 2 | [27] |
| Celecoxib | 381 | 11.1 (A) | 437 | 3.4 | −3.95 | −5.50 | [28,29,30] |
| Chlorpromazine 3 | 319 | 9.2 (B) | <298 | 5.4 | −4.70 ± 0.00 2 | - | [27] |
| Cilnidipine | 493 | None | 387 | 5.7 | −5.33 | −6.89 | [31,32,33] |
| Clotrimazole | 345 | 5.9 (B) | 417 | 5.2 | −4.65 | −5.80 | [23,25,34] |
| Clozapine | 327 | 3.8 (B), 7.5 (B) | 458 | 4.1 | −3.38 | −4.57 | [15,25,34] |
| Danazol | 338 | None | 498 | 4.5 | −4.43 | −6.21 | [35,36,37,38] |
| Diclofenac | 296 | 4.0 (A) | 453 | 4.5 | −3.52 ± 0.00 2 | −4.96 | [18,27,39] |
| Diphenhydramine 3 | 255 | 9.1 (B) | <298 | 3.4 | −2.94 ± 0.00 2 | - | [27] |
| Dipyridamole | 505 | 6.4 (B) | 436 | 2.2 | −3.80 ± 0.00 2 | −4.70 | [40,41,42] |
| Efavirenz | 316 | 10.2 (A) | 412 | 5.4 | −4.23 | −4.59 | [15,43,44,45] |
| Enzalutamide | 464 | None between pH 3–11 | 470 | 4.0 | −4.04 | −5.20 | [46,47] |
| Felodipine | 384 | <2 | 415 | 5.6 | −4.59 | −5.61 | [10,15,25,48] |
| Fenofibrate | 361 | None | 354 | 4.6 | −4.70 ± 0.00 2 | −6.08 | [49,50] |
| Flurbiprofen | 244 | 4.0 (A) | 388 2 | 4.2 | −3.37 ± 0.00 2 | −4.15 ± 0.01 2 | [27] |
| Ibuprofen | 206 | 4.4 (A) | 349 | 4.0 | −3.12 ± 0.00 2 | −3.55 | [27,51] |
| Ketoconazole | 531 | 3.3 (B), 6.2 (B) | 423 | 4.3 | −3.80 ± 0.00 2 | −5.31 | [25,52,53,54] |
| Ketoprofen | 254 | 4.2 (A) | 368 | 3.2 | −2.76 ± 0.00 2 | −3.00 | [25,55,56,57] |
| Ketotifen | 309 | 6.7 (B) | 430 | 2.1 | −3.41 ± 0.00 2 | −4.28 | [58,59] |
| Lidocaine | 234 | 8.0 (B) | 342 | 2.4 | −1.74 ± 0.00 2 | −1.90 | [27,60] |
| Loratadine | 383 | 5.3 (B) | 409 | 5.2 | −4.70 | −5.38 | [15,41,61] |
| Losartan | 423 | 3.2 (A) | 457 | 3.5 | −2.07 ± 0.00 2 | −3.47 | [62,63,64,65] |
| Loxoprofen | 246 | 4.2 (A) | 358 2 | 2.3 | −2.21 ± 0.00 2 | −2.22 ± 0.00 2 | [66,67] |
| Meclofenamic-acid | 296 | 4.1 (A) | 530 | 5.9 | −4.52 ± 0.00 2 | −6.68 | [27,68] |
| Miconazole | 416 | 6.1 (B) | 358 | 4.9 | −4.88 | −5.62 | [25,35,69] |
| Orphenadrine 3 | 269 | 9.0 (B) | <298 | 3.8 | −3.26 ± 0.00 2 | - | [27] |
| Paclitaxel | 854 | None | 493 | 3.9 | −4.43 | −6.38 | [35,70,71] |
| Papaverine | 339 | 6.4 (B) | 421 | 3.0 | −3.03 ± 0.00 2 | −4.35 | [27,72] |
| Phenylbutazone | 308 | 4.4 (A) | 379 2 | 3.3 | −3.64 ± 0.00 2 | −4.49 ± 0.01 2 | [27] |
| Posaconazole | 701 | 3.6 (B), 4.6 (B) | 442 | 3.8 | −4.89 | −6.41 | [73,74,75] |
| Pramoxine 3 | 330 | 7.1 (B) | <298 | 3.6 | −3.09 ± 0.00 2 | - | [27] |
| Procaine | 236 | 2.3 (B), 9.0 (B) | 333 2 | 2.1 | −1.71 ± 0.00 2 | −2.02 ± 0.01 2 | [27] |
| Propafenone | 341 | 9.3 (B) | 364 2 | 4.6 | −3.42 ± 0.00 2 | −4.62 ± 0.03 2 | [76,77] |
| Propranolol | 259 | 9.0 (B) | 369 | 3.5 | −2.78 ± 0.00 2 | −3.07 | [27,78] |
| Quinine | 324 | 4.2 (B), 8.6 (B) | 449 2 | 3.5 | −2.82 ± 0.00 2 | −3.26 ± 0.00 2 | [27] |
| Rebamipide | 371 | 3.3 (A) | 579 | 2.6 | −3.09 ± 0.00 2 | −5.29 | [79,80,81] |
| Ritonavir | 721 | 2.4 (B) | 391 | 3.2 | −4.58 | −5.74 | [15,82,83,84] |
| Sulfasalazine | 398 | 2.4 (A), 8.0 (A), 10.9 (A) | 532 2 | 3.6 | −4.15 ± 0.00 2 | −5.92 ± 0.04 2 | [27] |
| Sulindac | 356 | 4.1 (A) | 460 2 | 3.4 | −3.70 ± 0.00 2 | −4.60 ± 0.00 2 | [27] |
| Telaprevir | 680 | 0.3 (B), 11.8 (A) | 519 | 4.0 | −3.87 | −5.17 | [17,85] |
| Terbinafine 3 | 291 | 7.1 (B) | 314 | 6.2 | −5.22 ± 0.00 2 | - | [25,86] |
| Thioridazine 3 | 371 | 8.9 (B) | <298 | 5.3 | −4.30 ± 0.00 2 | - | [87,88,89] |
| Verapamil 3 | 455 | 8.7 (B) | <298 | 4.0 | −4.10 ± 0.00 2 | - | [27] |
| Warfarin | 308 | 4.9 (A) | 436 2 | 3.5 | −3.20 ± 0.00 2 | −4.54 ± 0.01 2 | [27] |


| AAE (log unit) | 0.32 | 0.71 |
| RMSE (log unit) | 0.40 | 0.91 |
| r2 | 0.90 | 0.56 |
5. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Falcón-Cano, G.; Molina, C.; Cabrera-Pérez, M.A. ADME Prediction with KNIME: A Retrospective Contribution to the Second “Solubility Challenge”. ADMET DMPK 2021, 9, 209–218. [Google Scholar] [CrossRef]
- Salahinejad, M.; Le, T.C.; Winkler, D.A. Aqueous Solubility Prediction: Do Crystal Lattice Interactions Help? Mol. Pharm. 2013, 10, 2757–2766. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.; Yalkowsky, S.H. Prediction of Aqueous Solubility from SCRATCH. Int. J. Pharm. 2010, 385, 1–5. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Wang, Y.; Sild, S.; Tamm, T.; Karelson, M. QSPR Studies on Vapor Pressure, Aqueous Solubility, and the Prediction of Water-Air Partition Coefficients. J. Chem. Inf. Comput. Sci. 1998, 38, 720–725. [Google Scholar] [CrossRef]
- Hewitt, M.; Cronin, M.T.D.; Enoch, S.J.; Madden, J.C.; Roberts, D.W.; Dearden, J.C. In Silico Prediction of Aqueous Solubility: The Solubility Challenge. J. Chem. Inf. Model. 2009, 49, 2572–2587. [Google Scholar] [CrossRef] [PubMed]
- Boobier, S.; Hose, D.R.J.; Blacker, A.J.; Nguyen, B.N. Machine Learning with Physicochemical Relationships: Solubility Prediction in Organic Solvents and Water. Nat. Commun. 2020, 11, 5753. [Google Scholar] [CrossRef]
- Llinas, A.; Avdeef, A. Solubility Challenge Revisited after Ten Years, with Multilab Shake-Flask Data, Using Tight (SD ∼0.17 Log) and Loose (SD ∼0.62 Log) Test Sets. J. Chem. Inf. Model. 2019, 59, 3036–3040. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Yalkowsky, S.H. Estimation of the Aqueous Solubility I: Application to Organic Nonelectrolytes. J. Pharm. Sci. 2001, 90, 234–252. [Google Scholar] [CrossRef]
- Ran, Y.; Jain, N.; Yalkowsky, S.H. Prediction of Aqueous Solubility of Organic Compounds by the General Solubility Equation (GSE). J. Chem. Inf. Comput. Sci. 2001, 41, 1208–1217. [Google Scholar] [CrossRef] [PubMed]
- Raina, S.A.; Zhang, G.G.Z.; Alonzo, D.E.; Wu, J.; Zhu, D.; Catron, N.D.; Gao, Y.; Taylor, L.S. Enhancements and Limits in Drug Membrane Transport Using Supersaturated Solutions of Poorly Water Soluble Drugs. J. Pharm. Sci. 2014, 103, 2736–2748. [Google Scholar] [CrossRef]
- Indulkar, A.S.; Gao, Y.; Raina, S.A.; Zhang, G.G.Z.; Taylor, L.S. Exploiting the Phenomenon of Liquid-Liquid Phase Separation for Enhanced and Sustained Membrane Transport of a Poorly Water-Soluble Drug. Mol. Pharm. 2016, 13, 2059–2069. [Google Scholar] [CrossRef]
- Suzuki, K.; Kawakami, K.; Fukiage, M.; Oikawa, M.; Nishida, Y.; Matsuda, M.; Fujita, T. Relevance of Liquid-Liquid Phase Separation of Supersaturated Solution in Oral Absorption of Albendazole from Amorphous Solid Dispersions. Pharmaceutics 2021, 13, 220. [Google Scholar] [CrossRef]
- Van den Mooter, G. The Use of Amorphous Solid Dispersions: A Formulation Strategy to Overcome Poor Solubility and Dissolution Rate. Drug Discov. Today Technol. 2012, 9, e79–e85. [Google Scholar] [CrossRef]
- Ramachandran, G.; Sudheesh, M.S. Role of Permeability on the Biopredictive Dissolution of Amorphous Solid Dispersions. AAPS PharmSciTech 2021, 22, 243. [Google Scholar] [CrossRef]
- Ilevbare, G.A.; Taylor, L.S. Liquid-Liquid Phase Separation in Highly Supersaturated Aqueous Solutions of Poorly Water-Soluble Drugs: Implications for Solubility Enhancing Formulations. Cryst. Growth Des. 2013, 13, 1497–1509. [Google Scholar] [CrossRef]
- Mishra, D.S.; Yalkowsky, S.H. Ideal Solubility of a Solid Solute: Effect of Heat Capacity Assumptions. Pharm. Res. 1992, 9, 958–959. [Google Scholar] [CrossRef] [PubMed]
- Mosquera-Giraldo, L.I.; Taylor, L.S. Glass-Liquid Phase Separation in Highly Supersaturated Aqueous Solutions of Telaprevir. Mol. Pharm. 2015, 12, 496–503. [Google Scholar] [CrossRef]
- Oki, J.; Watanabe, D.; Uekusa, T.; Sugano, K. Mechanism of Supersaturation Suppression in Dissolution Process of Acidic Drug Salt. Mol. Pharm. 2019, 16, 1669–1677. [Google Scholar] [CrossRef] [PubMed]
- Almeida E Sousa, L.; Reutzel-Edens, S.M.; Stephenson, G.A.; Taylor, L.S. Assessment of the Amorphous “Solubility” of a Group of Diverse Drugs Using New Experimental and Theoretical Approaches. Mol. Pharm. 2015, 12, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Ono, A.; Matsumura, N.; Kimoto, T.; Akiyama, Y.; Funaki, S.; Tamura, N.; Hayashi, S.; Kojima, Y.; Fushimi, M.; Sudaki, H.; et al. Harmonizing Solubility Measurement to Lower Inter-Laboratory Variance—Progress of Consortium of Biopharmaceutical Tools (CoBiTo) in Japan. ADMET DMPK 2019, 7, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Völgyi, G.; Csicsák, D.; Takács-Novák, K. Right Filter-Selection for Phase Separation in Equilibrium Solubility Measurement. Eur. J. Pharm. Sci. 2018, 123, 98–105. [Google Scholar] [CrossRef]
- Guerrieri, P.; Jarring, K.; Taylor, L.S. Impact of Counterion on the Chemical Stability of Crystalline Salts of Procaine. J. Pharm. Sci. 2009, 99, 3719–3730. [Google Scholar] [CrossRef]
- Indulkar, A.S.; Box, K.J.; Taylor, R.; Ruiz, R.; Taylor, L.S. PH-Dependent Liquid-Liquid Phase Separation of Highly Supersaturated Solutions of Weakly Basic Drugs. Mol. Pharm. 2015, 12, 2365–2377. [Google Scholar] [CrossRef]
- Duan, J.; Freeling, J.P.; Koehn, J.; Shu, C.; Ho, R.J.Y. Evaluation of Atazanavir and Darunavir Interactions with Lipids for Developing PH-Responsive Anti-HIV Drug Combination Nanoparticles. J. Pharm. Sci. 2014, 103, 2520–2529. [Google Scholar] [CrossRef]
- Avdeef, A. Absorption and Drug Development: Solubility, Permeabiliry, and Charge State, 2nd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012. [Google Scholar]
- Popović, G.; Čakar, M. The Effects of β-Cyclodextrin and PH on Bifonazole Hydrosolubility. J. Serb. Chem. Soc. 2004, 69, 225–231. [Google Scholar] [CrossRef]
- Box, K.J.; Comer, J.E.A. Using Measured PKa, LogP and Solubility to Investigate Supersaturation and Predict BCS Class. Curr. Drug Metab. 2008, 9, 869–878. [Google Scholar] [CrossRef]
- Li, N.; Mosquera-Giraldo, L.I.; Borca, C.H.; Ormes, J.D.; Lowinger, M.; Higgins, J.D.; Slipchenko, L.v.; Taylor, L.S. A Comparison of the Crystallization Inhibition Properties of Bile Salts. Cryst. Growth Des. 2016, 16, 7286–7300. [Google Scholar] [CrossRef]
- Iburahim, M.M.; Abd-Elgawad, A.E.H.; Soliman, O.A.E.; Jablonski, M.M. Nanoparticle-Based Topical Ophthalmic Formulations for Sustained Celecoxib Release. Pharm. Nanotechnol. 2012, 102, 1036–1053. [Google Scholar] [CrossRef]
- Paulson, S.K.; Vaughn, M.B.; Jessen, S.M.; Lawal, Y.; Gresk, C.J.; Yan, B.; Maziasz, T.J.; Cook, C.S.; Karim, A. Pharmacokinetics of Celecoxib after Oral Administration in Dogs and Humans: Effect of Food and Site of Absorption. J. Pharmacol. Exp. Ther. 2001, 297, 638–645. [Google Scholar] [PubMed]
- Liu, Q.; Mai, Y.; Gu, X.; Zhao, Y.; Di, X.; Ma, X.; Yang, J. A Wet-Milling Method for the Preparation of Cilnidipine Nanosuspension with Enhanced Dissolution and Oral Bioavailability. J. Drug Deliv. Sci. Technol. 2020, 55, 101371. [Google Scholar] [CrossRef]
- Indulkar, A.S.; Gao, Y.; Raina, S.A.; Zhang, G.G.Z.; Taylor, L.S. Crystallization from Supersaturated Solutions: Role of Lecithin and Composite Simulated Intestinal Fluid. Pharm. Res. 2018, 35, 158. [Google Scholar] [CrossRef] [PubMed]
- Mochida Pharmaceutical Co., Ltd. Cilnidipine Drug Product Information. Available online: https://med.mochida.co.jp/index/atc-h.html (accessed on 22 November 2022).
- Hsieh, Y.L.; Ilevbare, G.A.; van Eerdenbrugh, B.; Box, K.J.; Sanchez-Felix, M.V.; Taylor, L.S. PH-Induced Precipitation Behavior of Weakly Basic Compounds: Determination of Extent and Duration of Supersaturation Using Potentiometric Titration and Correlation to Solid State Properties. Pharm. Res. 2012, 29, 2738–2753. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Mann, A.K.P.; van Duong, T.; Ormes, J.D.; Okoh, G.A.; Hermans, A.; Taylor, L.S. Drug Release and Nanodroplet Formation from Amorphous Solid Dispersions: Insight into the Roles of Drug Physicochemical Properties and Polymer Selection. Mol. Pharm. 2021, 18, 2066–2081. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.J.; Toth, S.J.; Kestur, U.S.; Huang, J.; Qian, F.; Hussain, M.A.; Simpson, G.J.; Taylor, L.S. Impact of Polymers on the Precipitation Behavior of Highly Supersaturated Aqueous Danazol Solutions. Mol. Pharm. 2014, 11, 3027–3038. [Google Scholar] [CrossRef] [PubMed]
- Mithani, S.D.; Bakatselou, V.; TenHoor, C.N.; Dressman, J.B. Estimation of the Increase in Solubility of Drugs as a Function of Bile Salt Concentration. Pharm. Res. 1996, 13, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, A.; Mano, T.; Sugano, K. Theoretical Dissolution Model of Poly-Disperse Drug Particles in Biorelevant Media. J. Pharm. Sci. 2008, 97, 1843–1852. [Google Scholar] [CrossRef] [PubMed]
- Giordano, F.; Rossi, A.; Pasquali, I.; Bettini, R.; Frigo, E.; Gazzaniga, A.; Sangalli, M.E.; Mileo, V.; Catinella, S. Thermal Degradation and Melting Point Determination of Diclofenac. J. Therm. Anal. Calorim. 2003, 73, 509–518. [Google Scholar] [CrossRef]
- Kostewicz, E.S.; Brauns, U.; Becker, R.; Dressman, J.B. Forecasting the Oral Absorption Behavior of Poorly Soluble Weak Bases Using Solubility and Dissolution Studies in Biorelevant Media. Pharm. Res. 2002, 19, 345–349. [Google Scholar] [CrossRef]
- Sugano, K.; Kato, T.; Suzuki, K.; Keiko, K.; Sujaku, T.; Takashi, M. High Throughput Solubility Measurement with Automated Polarized Light Microscopy Analysis. J. Pharm. Sci. 2006, 95, 2271–2280. [Google Scholar] [CrossRef]
- Xi, Z.; Sharma, N.; Paprikar, A.; Lin, S. Development and Evaluation of Dipyridamole Sustained Release Tablets Containing Micro-Environmental PH Modifiers. J. Drug Deliv. Sci. Technol. 2019, 54, 101231. [Google Scholar] [CrossRef]
- Rabel, S.R.; Michael, M.B.; Susan, R.M.; Hussain, M. Determination of the PKa and PH-Solubility Behavior of an Ionizable Cyclic Carbamate, (S)-6-Chloro-4-(Cyclopropylethynyl)-1,4-Dihydro-4-(Trifluoromethyl)-2H-3,1-Benzoxazin-2-One (DMP 266). Pharm. Dev. Technol. 1996, 1, 91–95. [Google Scholar] [CrossRef]
- Fitriani, L.; Haqi, A.; Zaini, E. Preparation and Characterization of Solid Dispersion Freeze-Dried Efavirenz—Polyvinylpyrrolidone K-30. J. Adv. Pharm. Technol. Res. 2016, 7, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Merck & Co., Inc. Efavirenz Drug Product Information. 2022. Available online: https://www.msdconnect.jp/products/stocrin/ (accessed on 22 November 2022).
- Wilson, V.; Lou, X.; Osterling, D.J.; Stolarik, D.F.; Jenkins, G.; Gao, W.; Zhang, G.G.Z.; Taylor, L.S. Relationship between Amorphous Solid Dispersion In Vivo Absorption and In Vitro Dissolution: Phase Behavior during Dissolution, Speciation, and Membrane Mass Transport. J. Control. Release 2018, 292, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Volkova, T.V.; Drozd, K.V.; Surov, A.O. Effect of Polymers and Cyclodextrins on Solubility, Permeability and Distribution of Enzalutamide and Apalutamide Antiandrogens. J. Mol. Liq. 2021, 322, 114937. [Google Scholar] [CrossRef]
- Kestur, U.S.; Taylor, L.S. Role of Polymer Chemistry in Influencing Crystal Growth Rates from Amorphous Felodipine. CrystEngComm 2010, 12, 2390–2397. [Google Scholar] [CrossRef]
- Vogt, M.; Kunath, K.; Dressman, J.B. Dissolution Enhancement of Fenofibrate by Micronization, Cogrinding and Spray-Drying: Comparison with Commercial Preparations. Eur. J. Pharm. Biopharm. 2008, 68, 283–288. [Google Scholar] [CrossRef]
- Law, D.; Wang, W.; Schmitt, E.A.; Qiu, Y.; Krill, S.L.; Fort, J.J. Properties of Rapidly Dissolving Eutectic Mixtures of Poly(Ethylene Glycol) and Fenofibrate: The Eutectic Microstructure. J. Pharm. Sci. 2003, 92, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Shaw, L.R.; Irwin, W.J.; Grattan, T.J.; Conway, B.R. The Effect of Selected Water-Soluble Excipients on the Dissolution of Paracetamol and Ibuprofen. Drug Dev. Ind. Pharm. 2005, 31, 515–525. [Google Scholar] [CrossRef]
- Viseras, C.; Salem, I.I.; Galan, I.C.R.; Galan, A.C.; Galindo, A.L. The Effect of Recrystallization on the Crystal Growth, Melting Point and Solubility of Ketoconazole. Thermochim. Acta 1995, 268, 143–151. [Google Scholar] [CrossRef]
- Pathak, S.M.; Ruff, A.; Kostewicz, E.S.; Patel, N.; Turner, D.B.; Jamei, M. Model-Based Analysis of Biopharmaceutic Experiments to Improve Mechanistic Oral Absorption Modeling: An Integrated in Vitro in Vivo Extrapolation Perspective Using Ketoconazole as a Model Drug. Mol. Pharm. 2017, 14, 4305–4320. [Google Scholar] [CrossRef]
- Wan, H.; Holmén, A.G.; Wang, Y.; Lindberg, W.; Englund, M.; Någård, M.B.; Thompson, R.A. High-Throughput Screening of PKa Values of Pharmaceuticals by Pressure-Assisted Capillary Electrophoresis and Mass Spectrometry. Rapid Commun. Mass Spectrom. 2003, 17, 2639–2648. [Google Scholar] [CrossRef] [PubMed]
- Umerska, A.; Zotova, J.; Tajber, L. Formation of Low Melting Point Binary Systems Comprising Ketoprofen and an Amide Local Anaesthetic. Int. J. Pharm. 2021, 607, 120969. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.J.; Kasim, N.A.; Chandrasekharan, R.; Amidon, G.L. Solubilization and Dissolution of Insoluble Weak Acid, Ketoprofen: Effects of PH Combined with Surfactant. Eur. J. Pharm. Sci. 2006, 29, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Geiser, L.; Henchoz, Y.; Galland, A.; Carrupt, P.A.; Veuthey, J.L. Determination of PKa Values by Capillary Zone Electrophoresis with a Dynamic Coating Procedure. J. Sep. Sci. 2005, 28, 2374–2380. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Ogawa, K.; Okada, J.; Sugibayashi, K. Enhancement of Skin Permeation of Ketotifen by Supersaturation Generated by Amorphous Form of the Drug. J. Control. Release 2005, 108, 306–318. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-L.; Chiang, C.-H.; Chen, J.-L. In Vitro and in Vivo Percutaneous Absorption Studies of Ketotifen Patches. Drug Dev. Ind. Pharm. 1994, 20, 2965–2976. [Google Scholar] [CrossRef]
- Pedersen, B.T.; Larsen, S.W.; Østergaard, J.; Larsen, C. In Vitro Assessment of Lidocaine Release from Aqueous and Oil Solutions and from Preformed and in Situ Formed Aqueous and Oil Suspensions. Parenteral Depots for Intra-Articular Administration. Drug Deliv. 2008, 15, 23–30. [Google Scholar] [CrossRef]
- Popović, G.; Čakar, M.; Agbaba, D. Acid-Base Equilibria and Solubility of Loratadine and Desloratadine in Water and Micellar Media. J. Pharm. Biomed. Anal. 2009, 49, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Hafez, H.M.; Abdullah, A.E.; Abdelaziz, L.M.; Kamal, M.M. Quantitative Determination of Amlodipine Besylate, Losartan Potassium, Valsartan and Atorvastatin Calcium by HPLC in Their Pharmaceutical Formulations. J. Chromatogr. Sep. Tech. 2014, 5, 1000226. [Google Scholar] [CrossRef]
- Madasu, S.B.; Vekariya, N.A.; Koteswaramma, C.; Islam, A.; Sanasi, P.D.; Korupolu, R.B. An Efficient, Commercially Viable, and Safe Process for Preparation of Losartan Potassium, an Angiotensin II Receptor Antagonist. Org. Process Res. Dev. 2012, 16, 2025–2030. [Google Scholar] [CrossRef]
- Winiwarter, S.; Ax, F.; Lennernäs, H.; Hallberg, A.; Pettersson, C.; Karlén, A. Hydrogen Bonding Descriptors in the Prediction of Human in Vivo Intestinal Permeability. J. Mol. Graph. Model. 2003, 21, 273–287. [Google Scholar] [CrossRef]
- De Souza, J.B.; de Souza, J.; de Castro, L.M.L.; Siqueira, M.F.; Savedra, R.M.L.; Silva-Barcellos, N.M. Evaluation of the Losartan Solubility in the Biowaiver Context by Shake-Flask Method and Intrinsic Dissolution. Pharm. Dev. Technol. 2019, 24, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Narumi, K.; Kobayashi, M.; Kondo, A.; Furugen, A.; Yamada, T.; Takahashi, N.; Iseki, K. Characterization of Loxoprofen Transport in Caco-2 Cells: The Involvement of a Proton-Dependent Transport System in the Intestinal Transport of Loxoprofen. Biopharm. Drug Dispos. 2016, 37, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Daiichi Sankyo Company, Limited. Loxoprofen Drug Product Information. Available online: https://www.medicalcommunity.jp/member/certification?destination=/products/druginfo/loxonin_tablets_60mg& (accessed on 22 November 2022).
- Pobudkowska, A.; DomańSka, U. Study of PH-Dependent Drugs Solubility in Water. Chem. Ind. Chem. Eng. Q. 2014, 20, 115–126. [Google Scholar] [CrossRef]
- Ribeiro, A.; Figueiras, A.; Santos, D.; Veiga, F. Preparation and Solid-State Characterization of Inclusion Complexes Formed between Miconazole and Methyl-β-Cyclodextrin. AAPS PharmSciTech 2008, 9, 1102–1109. [Google Scholar] [CrossRef]
- Martins, K.F.; Messias, A.D.; Leite, F.L.; Duek, E.A.R. Preparation and Characterization of Paclitaxel-Loaded PLDLA Microspheres. Mater. Res. 2014, 17, 650–656. [Google Scholar] [CrossRef]
- Xavier Junior, F.H.; Gueutin, C.; do Vale Morais, A.R.; do Nascimento Alencar, E.; do Egito, E.S.T.; Vauthier, C. HPLC Method for the Dosage of Paclitaxel in Copaiba Oil: Development, Validation, Application to the Determination of the Solubility and Partition Coefficients. Chromatographia 2016, 79, 405–412. [Google Scholar] [CrossRef]
- Serajuddin, A.T.M.; Mufson, D. PH-Solubility Profiles of Organic Bases and Their Hydrochloride Salts. Pharm. Res. Off. J. Am. Assoc. Pharm. Sci. 1985, 2, 65–68. [Google Scholar]
- Courtney, R.; Wexler, D.; Radwanski, E.; Lim, J.; Laughlin, M. Effect of Food on the Relative Bioavailability of Two Oral Formulations of Posaconazole in Healthy Adults. Br. J. Clin. Pharmacol. 2004, 57, 218–222. [Google Scholar] [CrossRef]
- Van Duong, T.; Ni, Z.; Taylor, L.S. Phase Behavior and Crystallization Kinetics of a Poorly Water-Soluble Weakly Basic Drug as a Function of Supersaturation and Media Composition. Mol. Pharm. 2022, 19, 1146–1159. [Google Scholar] [CrossRef]
- Merck & Co., Inc. Posaconzaole Drug Product Information. Available online: https://www.msdconnect.jp/products/noxafil/download/ (accessed on 22 November 2022).
- Völgyi, G.; Baka, E.; Box, K.J.; Comer, J.E.A.; Takács-Novák, K. Study of PH-Dependent Solubility of Organic Bases. Revisit of Henderson-Hasselbalch Relationship. Anal. Chim. Acta 2010, 673, 40–46. [Google Scholar] [CrossRef]
- Moriguchi, I.; Hirono, S.; Nakagome, I.; Hirano, H. Comparision of Reliability of LogP Values for Drugs Calculated by Several Metods. Chem. Pharm. Bull. 1994, 42, 976–978. [Google Scholar] [CrossRef]
- Maitani, Y.; Coutel-Egros, A.; Obata, Y.; Nagai, T. Prediction of Skin Permeabilities of Diclofenac and Propranolol from Theoretical Partition Coefficients Determined from Cohesion Parameters. J. Pharm. Sci. 1993, 82, 416–420. [Google Scholar] [CrossRef] [PubMed]
- Jindal, A.; Singh, R.; Tomar, S.; Dureja, J.; Karan, M.; Chadha, R. Engineering a Remedy to Modulate and Optimize Biopharmaceutical Properties of Rebamipide by Synthesizing New Cocrystal: In Silico and Experimental Studies. Pharm. Res. 2021, 38, 2129–2145. [Google Scholar] [CrossRef]
- Pradhan, R.; Tran, T.H.; Choi, J.Y.; Choi, I.S.; Choi, H.G.; Yong, C.S.; Kim, J.O. Development of a Rebamipide Solid Dispersion System with Improved Dissolution and Oral Bioavailability. Arch. Pharm. Res. 2015, 38, 522–533. [Google Scholar] [CrossRef]
- Otsuka Pharmaceutical, Rebamipide Drug Product Information. Available online: https://www.otsuka-elibrary.jp/check/ (accessed on 22 November 2022).
- Tsinman, O.; Tsinman, K.; Sun, N.; Avdeef, A. Physicochemical Selectivity of the BBB Microenvironment Governing Passive Diffusion—Matching with a Porcine Brain Lipid Extract Artificial Membrane Permeability Model. Pharm. Res. 2011, 28, 337–363. [Google Scholar] [CrossRef] [PubMed]
- Grizić, D.; Lamprecht, A. Predictability of Drug Encapsulation and Release from Propylene Carbonate/PLGA Microparticles. Int. J. Pharm. 2020, 586, 119601. [Google Scholar] [CrossRef] [PubMed]
- Law, D.; Krill, S.L.; Schmitt, E.A.; Fort, J.J.; Qiu, Y.; Wang, W.; Porter, W.R. Physicochemical Considerations in the Preparation of Amorphous Ritonavir-Poly(Ethylene Glycol) 8000 Solid Dispersions. J. Pharm. Sci. 2001, 90, 1015–1025. [Google Scholar] [CrossRef] [PubMed]
- Kwong, A.D.; Kauffman, R.S.; Hurter, P.; Mueller, P. Discovery and Development of Telaprevir: An NS3-4A Protease Inhibitor for Treating Genotype 1 Chronic Hepatitis C Virus. Nat. Biotechnol. 2011, 29, 993–1003. [Google Scholar] [CrossRef] [PubMed]
- Al Hossain, A.S.M.M.; Sil, B.C.; Iliopoulos, F.; Lever, R.; Hadgraft, J.; Lane, M.E. Preparation, Characterisation, and Topical Delivery of Terbinafine. Pharmaceutics 2019, 11, 548. [Google Scholar] [CrossRef] [PubMed]
- Hopkała, H. Preparation and Potentiometric Determination of Quaternary N-Methiodides of Phenothiazine Derivatives. Anal. Lett. 1990, 23, 159–167. [Google Scholar] [CrossRef]
- Franke, U.; Munk, A.; Wiese, M. Ionization Constants and Distribution Coefficients of Phenothiazines and Calcium Channel Antagonists Determined by a PH-Metric Method and Correlation with Calculated Partition Coefficients. J. Pharm. Sci. 1999, 88, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Domańska, U.; Pelczarska, A.; Pobudkowska, A. Solubility and PK a Determination of Six Structurally Related Phenothiazines. Int. J. Pharm. 2011, 421, 135–144. [Google Scholar] [CrossRef]
- Moriguchi, I.; Hirono, S.; Liu, Q.; Nakagome, I.; Matsushita, Y. Simple Method of Calculating Octanol/Water Partition Coefficient. Chem. Pharm. Bull. 1992, 40, 127–130. [Google Scholar] [CrossRef]
- Mannhold, R.; Rekker, R.F. The Hydrophobic Fragmental Constant Approach for Calculating Log P in Octanol/Water and Aliphatic Hydrocarbon/Water Systems. Perspect. Drug Discov. Des. 2000, 18, 1–18. [Google Scholar] [CrossRef]
- Dannenfelser, R.M.; Yalkowsky, S.H. Estimation of Entropy of Melting from Molecular Structure: A Non-Group Contribution Method. Ind. Eng. Chem. Res. 1996, 35, 1483–1486. [Google Scholar] [CrossRef]
- Bogner, R.H.; Murdande, S.B.; Pikal, M.J.; Shanker, R.M. Solubility Advantage of Amorphous Pharmaceuticals: II. Application of Quantitative Thermodynamic Relationships for Prediction of Solubility Enhancement in Structurally Diverse Insoluble Pharmaceuticals. Pharm. Res. 2010, 27, 2704–2714. [Google Scholar] [CrossRef]
- Murdande, S.B.; Pikal, M.J.; Shanker, R.M.; Bogner, R.H. Solubility Advantage of Amorphous Pharmaceuticals: I. A Thermodynamic Analysis. J. Pharm. Sci. 2009, 99, 1254–1264. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, S.; Kushida, I.; Yamashita, T.; Hasebe, T.; Shirai, O.; Kano, K. Evaluation of Drug Supersaturation by Thermodynamic and Kinetic Approaches for the Prediction of Oral Absorbability in Amorphous Pharmaceuticals. J. Pharm. Sci. 2012, 101, 4220–4230. [Google Scholar] [CrossRef]
- Brouwers, J.; Marcus, E.; Brewster, P.A. Supersaturating Drug Delivery Systems: The Answer to Solubility-Limited Oral Bioavailability? J. Pharm. Sci. 2009, 98, 2549–2568. [Google Scholar] [CrossRef]
- Ueda, K.; Higashi, K.; Moribe, K. Unusual Correlation between the Apparent Amorphous Solubility of a Drug and Solubilizer Concentration Revealed by NMR Analysis. Mol. Pharm. 2022, 19, 3336–3349. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Moseson, D.E.; Pathak, V.; Taylor, L.S. Effect of Polymer Species on Maximum Aqueous Phase Supersaturation Revealed by Quantitative Nuclear Magnetic Resonance Spectroscopy. Mol. Pharm. 2021, 18, 1344–1355. [Google Scholar] [CrossRef] [PubMed]



| Drug | Medium | Amount of Drug (mg) | Incubation Time (h) | Wavelength (nm) |
|---|---|---|---|---|
| Bifonazole | pH 9.0 borate buffer 1 | 30 | 72 | 255 |
| Carprofen | 0.1 N HCl | 30 | 48 | 300 |
| Flurbiprofen | 0.1 N HCl | 30 | 48 | 248 |
| Loxoprofen | 0.1 N HCl | 50 | 48 | 220 |
| Phenylbutazone | 0.1 N HCl | 30 | 48 | 264 |
| Procaine | 0.01 N NaOH | 500 | 1 | 280 |
| Propafenone | 0.01 N NaOH | 30 | 48 | 305 |
| Quinine | 0.01 N NaOH | 100 | 48 | 350 |
| Sulfasalazine | 0.1 N HCl | 30 | 48 | 369 |
| Sulindac | 0.1 N HCl | 50 | 48 | 331 |
| Warfarin | 0.1 N HCl | 30 | 48 | 275 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uekusa, T.; Watanabe, T.; Watanabe, D.; Sugano, K. Thermodynamic Correlation between Liquid–Liquid Phase Separation and Crystalline Solubility of Drug-Like Molecules. Pharmaceutics 2022, 14, 2560. https://doi.org/10.3390/pharmaceutics14122560
Uekusa T, Watanabe T, Watanabe D, Sugano K. Thermodynamic Correlation between Liquid–Liquid Phase Separation and Crystalline Solubility of Drug-Like Molecules. Pharmaceutics. 2022; 14(12):2560. https://doi.org/10.3390/pharmaceutics14122560
Chicago/Turabian StyleUekusa, Taiga, Tomohiro Watanabe, Daiju Watanabe, and Kiyohiko Sugano. 2022. "Thermodynamic Correlation between Liquid–Liquid Phase Separation and Crystalline Solubility of Drug-Like Molecules" Pharmaceutics 14, no. 12: 2560. https://doi.org/10.3390/pharmaceutics14122560
APA StyleUekusa, T., Watanabe, T., Watanabe, D., & Sugano, K. (2022). Thermodynamic Correlation between Liquid–Liquid Phase Separation and Crystalline Solubility of Drug-Like Molecules. Pharmaceutics, 14(12), 2560. https://doi.org/10.3390/pharmaceutics14122560