
Lipid/Clay-Based Solid Dispersion Formulation for Improving the Oral Bioavailability of Curcumin


Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Lipid/Clay-Based Solid Dispersion
2.3. Structural and Morphological Characterization
2.4. Solubility Study
2.5. In Vitro Drug Release Study
2.6. Dissolution Studies in Simulated Intestinal Fluids
2.7. Storage Stability
2.8. Pharmacokinetic Studies
2.9. Analytical Assay
2.10. Pharmacokinetic and Statistical Analysis
3. Results and Discussion
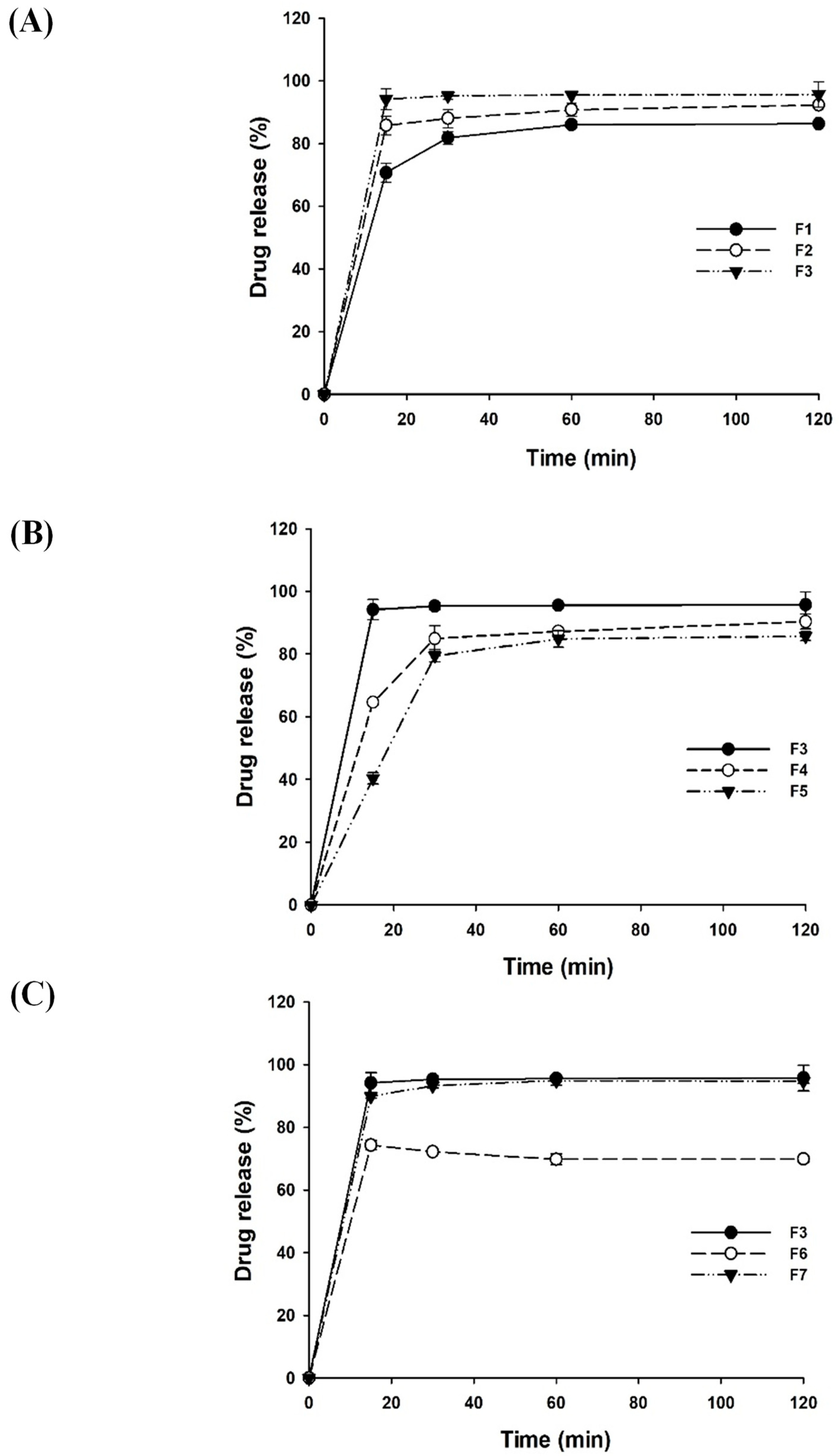
3.1. Optimization of LSD Formulations
3.2. Characterization of the Optimized LSD Formulation
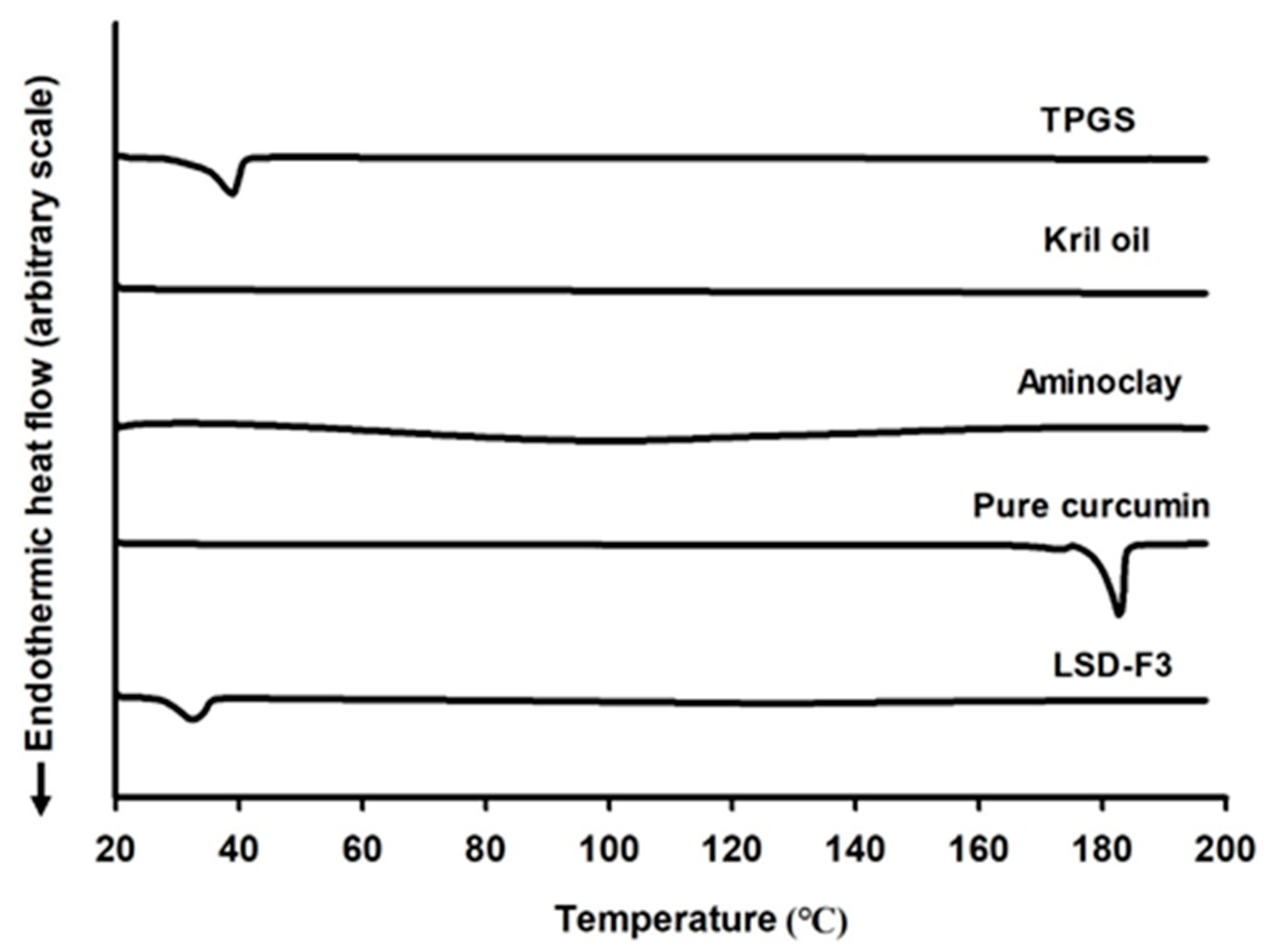
3.2.1. Structural and Morphological Characteristics
3.2.2. Solubility
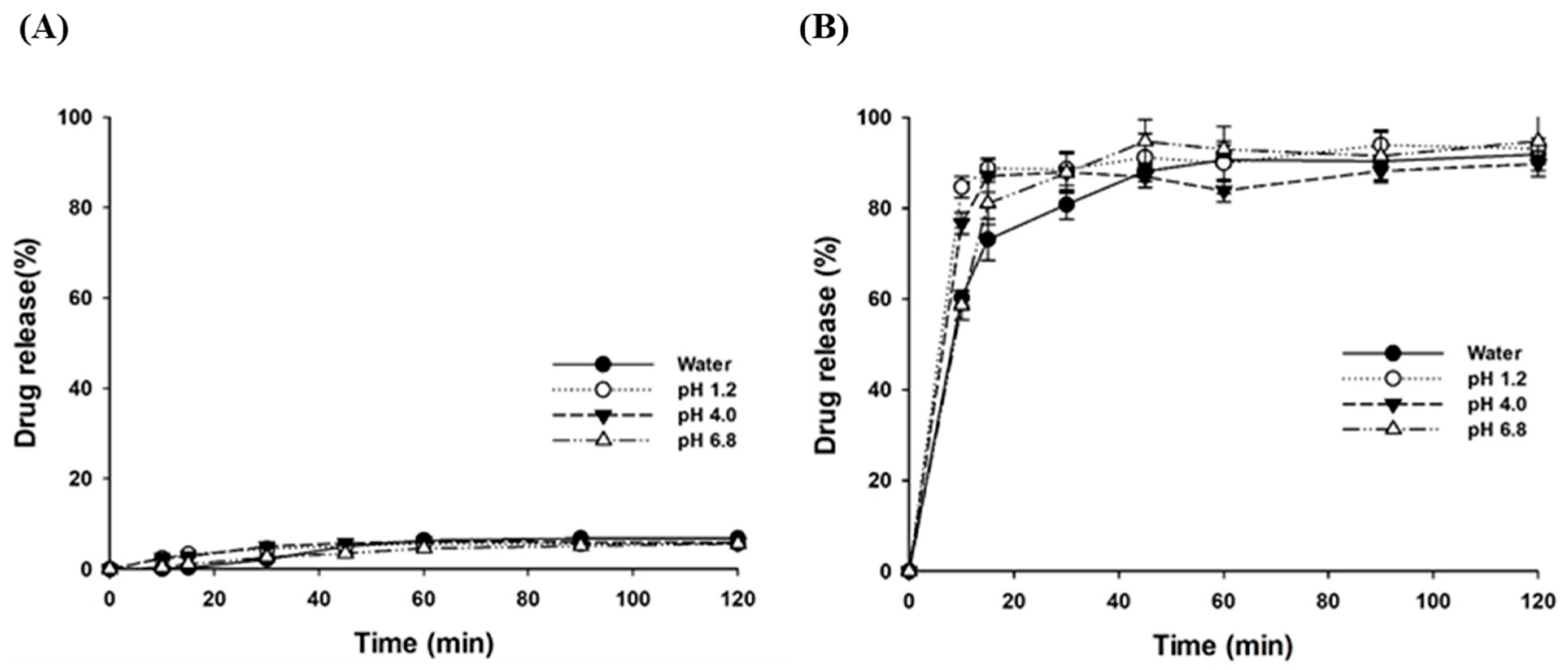
3.2.3. Dissolution Characteristics
3.2.4. Storage Stability
3.2.5. Pharmacokinetics
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Artiga-Artigas, M.; Lanjari-Pérez, Y.; Martín-Belloso, O. Curcumin-loaded nanoemulsions stability as affected by the nature and concentration of surfactant. Food Chem. 2018, 266, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Kunnumakkara, A.B.; Newman, R.A.; Aggarwal, B.B. Bioavailability of curcumin: Problems and promises. Mol. Pharm. 2007, 4, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Shin, G.H.; Li, J.; Cho, J.H.; Kim, J.T.; Park, H.J. Enhancement of curcumin solubility by phase change from crystalline to amorphous in Cur-TPGS nanosuspension. J. Food Sci. 2016, 81, N494–N501. [Google Scholar] [CrossRef] [PubMed]
- Song, I.-S.; Cha, J.-S.; Choi, M.-K. Characterization, in vivo and in vitro evaluation of solid dispersion of curcumin containing d-α-Tocopheryl polyethylene glycol 1000 succinate and mannitol. Molecules 2016, 21, 1386. [Google Scholar] [CrossRef]
- Seo, S.-W.; Han, H.-K.; Chun, M.-K.; Choi, H.-K. Preparation and pharmacokinetic evaluation of curcumin solid dispersion using Solutol® HS15 as a carrier. Int. J. Pharm. 2012, 424, 18–25. [Google Scholar] [CrossRef]
- Homayouni, A.; Amini, M.; Sohrabi, M.; Varshosaz, J.; Nokhodchi, A. Curcumin nanoparticles containing poloxamer or soluplus tailored by high pressure homogenization using antisolvent crystallization. Int. J. Pharm. 2019, 562, 124–134. [Google Scholar] [CrossRef]
- Li, Z.-L.; Peng, S.-F.; Chen, X.; Zhu, Y.-Q.; Zou, L.-Q.; Liu, W.; Liu, C.-M. Pluronics modified liposomes for curcumin encapsulation: Sustained release, stability and bioaccessibility. Food Res. Int. 2018, 108, 246–253. [Google Scholar] [CrossRef]
- Takahashi, M.; Uechi, S.; Takara, K.; Asikin, Y.; Wada, K. Evaluation of an oral carrier system in rats: Bioavailability and antioxidant properties of liposome-encapsulated curcumin. J. Agric. Food Chem. 2009, 57, 9141–9146. [Google Scholar] [CrossRef]
- Lertpairod, J.; Tiyaboonchai, W. pH-sensitive beads containing curcumin loaded nanostructured lipid carriers for a colon targeted oral delivery system. J. Pharm. Investig. 2022, 52, 387–396. [Google Scholar] [CrossRef]
- Bapat, P.; Ghadi, R.; Chaudhari, D.; Katiyar, S.S.; Jain, S. Tocophersolan stabilized lipid nanocapsules with high drug loading to improve the permeability and oral bioavailability of curcumin. Int. J. Pharm. 2019, 560, 219–227. [Google Scholar] [CrossRef]
- Porter, C.J.; Trevaskis, N.L.; Charman, W.N. Lipids and lipid-based formulations: Optimizing the oral delivery of lipophilic drugs. Nat. Rev. Drug Discov. 2007, 6, 231–248. [Google Scholar] [CrossRef] [PubMed]
- Tran, P.; Park, J.-S. Recent trends of self-emulsifying drug delivery system for enhancing the oral bioavailability of poorly water-soluble drugs. J. Pharm. Investig. 2021, 51, 439–463. [Google Scholar] [CrossRef]
- Ryu, S.; Jin, M.; Lee, H.-K.; Wang, M.-H.; Baek, J.-S.; Cho, C.-W. Effects of lipid nanoparticles on physicochemical properties, cellular uptake, and lymphatic uptake of 6-methoxflavone. J. Pharm. Investig. 2022, 52, 233–241. [Google Scholar] [CrossRef]
- Chhitij, T.; Seo, J.-E.; Keum, T.; Noh, G.; Bashyal, S.; Lamichhane, S.; Kim, J.H.; Lee, J.H.; Park, J.H.; Choi, J. Optimized self-microemulsifying drug delivery system improves the oral bioavailability and brain delivery of coenzyme Q10. Drug Deliv. 2022, 29, 2330–2342. [Google Scholar] [CrossRef]
- Maki, K.C.; Reeves, M.S.; Farmer, M.; Griinari, M.; Berge, K.; Vik, H.; Hubacher, R.; Rains, T.M. Krill oil supplementation increases plasma concentrations of eicosapentaenoic and docosahexaenoic acids in overweight and obese men and women. Nutr. Res. 2009, 29, 609–615. [Google Scholar] [CrossRef]
- Jacobsen, A.-C.; Ejskjær, L.; Brandl, M.; Holm, R.; Bauer-Brandl, A. Do phospholipids boost or attenuate drug absorption? In vitro and in vivo evaluation of mono-and diacyl phospholipid-based solid dispersions of celecoxib. J. Pharm. Sci. 2021, 110, 198–207. [Google Scholar] [CrossRef]
- Seto, Y.; Morizane, C.; Ueno, K.; Sato, H.; Onoue, S. Supersaturable self-emulsifying drug delivery system of Krill oil with improved oral absorption and hypotriglyceridemic function. J. Agric. Food Chem. 2018, 66, 5352–5358. [Google Scholar] [CrossRef]
- Bunea, R.; El Farrah, K.; Deutsch, L. Evaluation of the effects of Neptune Krill Oil on the clinical course of hyperlipidemia. Altern. Med. Rev. 2004, 9, 420–428. [Google Scholar]
- Kesisoglou, F.; Panmai, S.; Wu, Y. Nanosizing—Oral formulation development and biopharmaceutical evaluation. Adv. Drug Deliv. Rev. 2007, 59, 631–644. [Google Scholar] [CrossRef]
- Song, J.G.; Lee, S.H.; Han, H.-K. Biophysical evaluation of aminoclay as an effective protectant for protein stabilization during freeze-drying and storage. Int. J. Nanomed. 2016, 11, 6609. [Google Scholar] [CrossRef]
- Yang, L.; Shao, Y.; Han, H.-K. Aminoclay–lipid hybrid composite as a novel drug carrier of fenofibrate for the enhancement of drug release and oral absorption. Int. J. Nanomed. 2016, 11, 1067. [Google Scholar]
- Chaudhari, S.P.; Dugar, R.P. Application of surfactants in solid dispersion technology for improving solubility of poorly water soluble drugs. J. Drug Deliv. Sci. Technol. 2017, 41, 68–77. [Google Scholar] [CrossRef]
- Rathod, S.; Bahadur, P.; Tiwari, S. Nanocarriers based on vitamin E-TPGS: Design principle and molecular insights into improving the efficacy of anticancer drugs. Int. J. Pharm. 2021, 592, 120045. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Choi, S.-K.; Shin, H.-J.; Han, H.-K. 3-aminopropyl functionalized magnesium phyllosilicate as an organoclay based drug carrier for improving the bioavailability of flurbiprofen. Int. J. Nanomed. 2013, 8, 4147. [Google Scholar]
- Bou-Chacra, N.; Melo, K.J.C.; Morales, I.A.C.; Stippler, E.S.; Kesisoglou, F.; Yazdanian, M.; Löbenberg, R. Evolution of choice of solubility and dissolution media after two decades of biopharmaceutical classification system. AAPS J. 2017, 19, 989–1001. [Google Scholar] [CrossRef]
- Jantratid, E.; Janssen, N.; Reppas, C.; Dressman, J.B. Dissolution media simulating conditions in the proximal human gastrointestinal tract: An update. Pharm. Res. 2008, 25, 1663–1676. [Google Scholar] [CrossRef]
- Marques, M.R.; Loebenberg, R.; Almukainzi, M. Simulated biological fluids with possible application in dissolution testing. Dissolution Technol. 2011, 18, 15–28. [Google Scholar] [CrossRef]
- Yang, L.; Shao, Y.; Han, H.-K. Development of omega-3 phospholipid-based solid dispersion of fenofibrate for the enhancement of oral bioavailability. Eur. J. Pharm. Sci. 2015, 78, 103–110. [Google Scholar] [CrossRef]
- Zu, Y.; Li, N.; Zhao, X.; Li, Y.; Ge, Y.; Wang, W.; Wang, K.; Liu, Y. In vitro dissolution enhancement of micronized l-nimodipine by antisolvent re-crystallization from its crystal form H. Int. J. Pharm. 2014, 464, 1–9. [Google Scholar] [CrossRef]
- Zu, Y.; Wu, W.; Zhao, X.; Li, Y.; Wang, W.; Zhong, C.; Zhang, Y.; Zhao, X. Enhancement of solubility, antioxidant ability and bioavailability of taxifolin nanoparticles by liquid antisolvent precipitation technique. Int. J. Pharm. 2014, 471, 366–376. [Google Scholar] [CrossRef]
- Strauss, G.; Schurtenberger, P.; Hauser, H. The interaction of saccharides with lipid bilayer vesicles: Stabilization during freeze-thawing and freeze-drying. Biochim. Biophys. Acta Biomembr. BBA-Biomembr. 1986, 858, 169–180. [Google Scholar] [CrossRef]
- Miyajima, K. Role of saccharides for the freeze-thawing and freeze drying of liposome. Adv. Drug Deliv. Rev. 1997, 24, 151–159. [Google Scholar] [CrossRef]
- Collnot, E.-M.; Baldes, C.; Schaefer, U.F.; Edgar, K.J.; Wempe, M.F.; Lehr, C.-M. Vitamin E TPGS P-glycoprotein inhibition mechanism: Influence on conformational flexibility, intracellular ATP levels, and role of time and site of access. Mol. Pharm. 2010, 7, 642–651. [Google Scholar] [CrossRef]
- Zhang, Z.; Tan, S.; Feng, S.-S. Vitamin E TPGS as a molecular biomaterial for drug delivery. Biomaterials 2012, 33, 4889–4906. [Google Scholar] [CrossRef] [PubMed]
- Donsi, F.; Wang, Y.; Li, J.; Huang, Q. Preparation of curcumin sub-micrometer dispersions by high-pressure homogenization. J. Agric. Food Chem. 2010, 58, 2848–2853. [Google Scholar] [CrossRef]
- Vasconcelos, T.; Sarmento, B.; Costa, P. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug Discov. 2007, 12, 1068–1075. [Google Scholar] [CrossRef] [PubMed]
- Hallouard, F.; Mehenni, L.; Lahiani-Skiba, M.; Anouar, Y.; Skiba, M. Solid dispersions for oral administration: An overview of the methods for their preparation. Curr. Pharm. Des. 2016, 22, 4942–4958. [Google Scholar] [CrossRef]
- Kim, N.A.; Oh, H.K.; Lee, J.C.; Choi, Y.H.; Jeong, S.H. Comparison of solubility enhancement by solid dispersion and micronized butein and its correlation with in vivo study. J. Pharm. Investig. 2021, 51, 53–60. [Google Scholar] [CrossRef]
- Burgess, D.J.; Duffy, E.; Etzler, F.; Hickey, A.J. Particle size analysis: AAPS workshop report, cosponsored by the Food and Drug Administration and the United States Pharmacopeia. AAPS J. 2004, 6, 23–34. [Google Scholar] [CrossRef]
- Baral, K.C.; Song, J.-G.; Lee, S.H.; Bajracharya, R.; Sreenivasulu, G.; Kim, M.; Lee, K.; Han, H.-K. Enhanced Bioavailability of AC1497, a Novel Anticancer Drug Candidate, via a Self-Nanoemulsifying Drug Delivery System. Pharmaceutics 2021, 13, 1142. [Google Scholar] [CrossRef]
- Klein, S. The use of biorelevant dissolution media to forecast the in vivo performance of a drug. AAPS J. 2010, 12, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Dahlgren, D.; Venczel, M.; Ridoux, J.-P.; Skjöld, C.; Müllertz, A.; Holm, R.; Augustijns, P.; Hellström, P.M.; Lennernäs, H. Fasted and fed state human duodenal fluids: Characterization, drug solubility, and comparison to simulated fluids and with human bioavailability. Eur. J. Pharm. Biopharm. 2021, 163, 240–251. [Google Scholar] [CrossRef] [PubMed]
- Pichot, R.; Watson, R.L.; Norton, I.T. Phospholipids at the interface: Current trends and challenges. Int. J. Mol. Sci. 2013, 14, 11767–11794. [Google Scholar] [CrossRef] [PubMed]
- Saadati, R.; Dadashzadeh, S. Marked effects of combined TPGS and PVA emulsifiers in the fabrication of etoposide-loaded PLGA-PEG nanoparticles: In vitro and in vivo evaluation. Int. J. Pharm. 2014, 464, 135–144. [Google Scholar] [CrossRef]
- Wu, X.; Xu, J.; Huang, X.; Wen, C. Self-microemulsifying drug delivery system improves curcumin dissolution and bioavailability. Drug Dev. Ind. Pharm 2011, 37, 15–23. [Google Scholar] [CrossRef]
- Kalepu, S.; Manthina, M.; Padavala, V. Oral lipid-based drug delivery systems–an overview. Acta Pharm. Sin. B 2013, 3, 361–372. [Google Scholar] [CrossRef]
- Hassanzadeh, K.; Buccarello, L.; Dragotto, J.; Mohammadi, A.; Corbo, M.; Feligioni, M. Obstacles against the Marketing of Curcumin as a Drug. Int. J. Mol. Sci. 2020, 21, 6619. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Ratio (w/w/w) | |||
|---|---|---|---|---|
| Curcumin | Krill Oil | Aminoclay | TPGS (%) | |
| F1 | 1 | 1 | 5 | 0.5 |
| F2 | 1 | 3 | 5 | 0.5 |
| F3 | 1 | 5 | 5 | 0.5 |
| F4 | 1 | 5 | 3 | 0.5 |
| F5 | 1 | 5 | 1 | 0.5 |
| F6 | 1 | 5 | 5 | 0.1 |
| F7 | 1 | 5 | 5 | 1 |
| Temperature (°C) | Dissolution (%) | ||
|---|---|---|---|
| Day 0 | 1 Month | 2 Month | |
| 4 | 91.9 ± 2.14 | 93.6 ± 5.62 | 92.8 ± 3.77 |
| 25 | 91.9 ± 2.14 | 92.0 ± 5.35 | 90.1 ± 1.98 |
| Parameter | Pure Curcumin | LSD-F3 |
|---|---|---|
| Cmax (ng/mL) | 11.8 ± 7.19 | 147 ± 49.5 * |
| Tmax (h) | 0.38 ± 0.26 | 0.42 ± 0.20 |
| AUC0–8h (ng·h/mL) | 7.82 ± 7.02 | 185 ± 31.7 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, J.G.; Noh, H.-M.; Lee, S.H.; Han, H.-K. Lipid/Clay-Based Solid Dispersion Formulation for Improving the Oral Bioavailability of Curcumin. Pharmaceutics 2022, 14, 2269. https://doi.org/10.3390/pharmaceutics14112269
Song JG, Noh H-M, Lee SH, Han H-K. Lipid/Clay-Based Solid Dispersion Formulation for Improving the Oral Bioavailability of Curcumin. Pharmaceutics. 2022; 14(11):2269. https://doi.org/10.3390/pharmaceutics14112269
Chicago/Turabian StyleSong, Jae Geun, Hye-Mi Noh, Sang Hoon Lee, and Hyo-Kyung Han. 2022. "Lipid/Clay-Based Solid Dispersion Formulation for Improving the Oral Bioavailability of Curcumin" Pharmaceutics 14, no. 11: 2269. https://doi.org/10.3390/pharmaceutics14112269
APA StyleSong, J. G., Noh, H.-M., Lee, S. H., & Han, H.-K. (2022). Lipid/Clay-Based Solid Dispersion Formulation for Improving the Oral Bioavailability of Curcumin. Pharmaceutics, 14(11), 2269. https://doi.org/10.3390/pharmaceutics14112269