
Heterocyclic Compounds as Hsp90 Inhibitors: A Perspective on Anticancer Applications

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
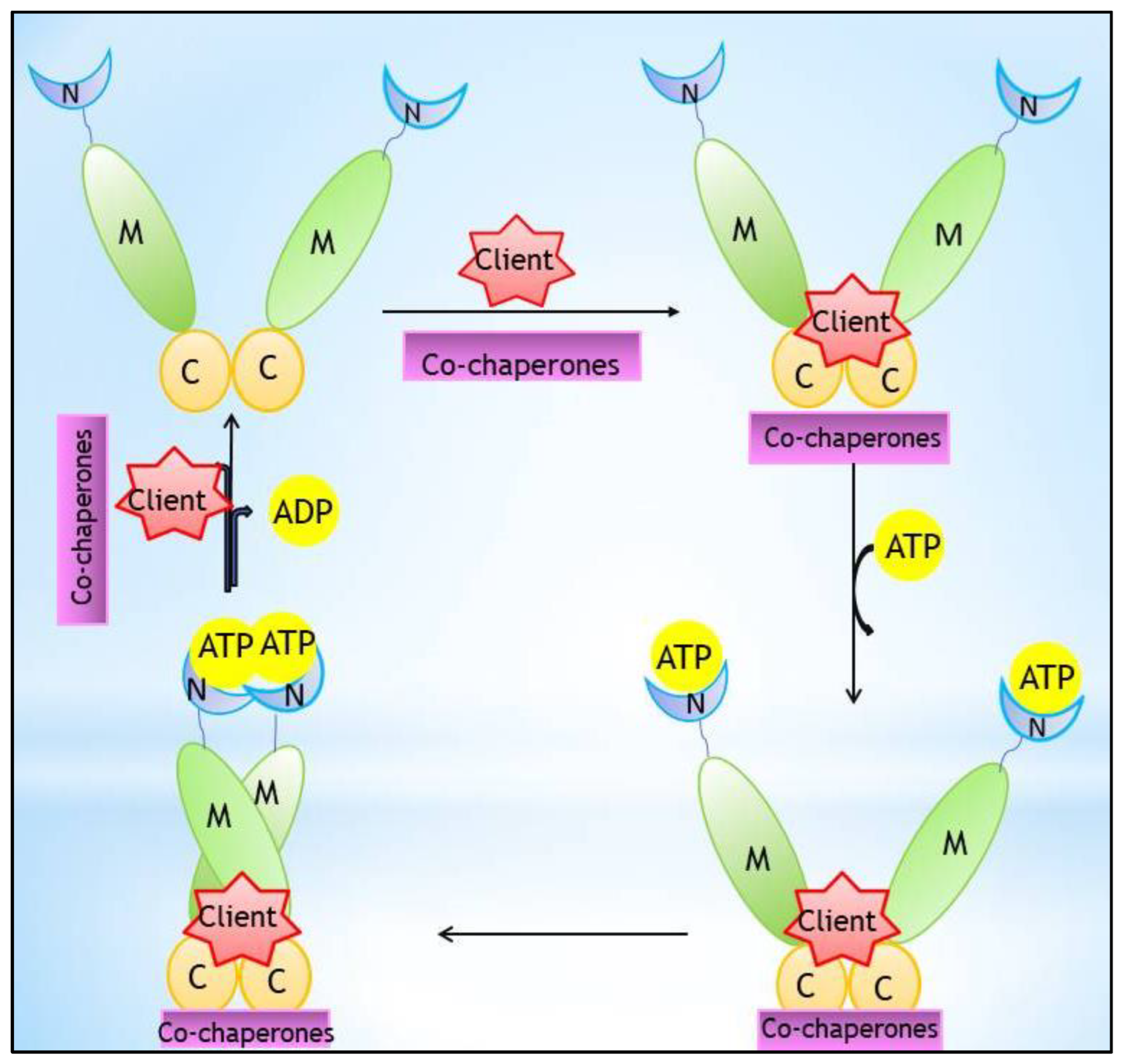
1. Introduction
2. Hsp90 Inhibitors
2.1. Natural Inhibitors
2.2. Synthetic Hsp90 Inhibitors
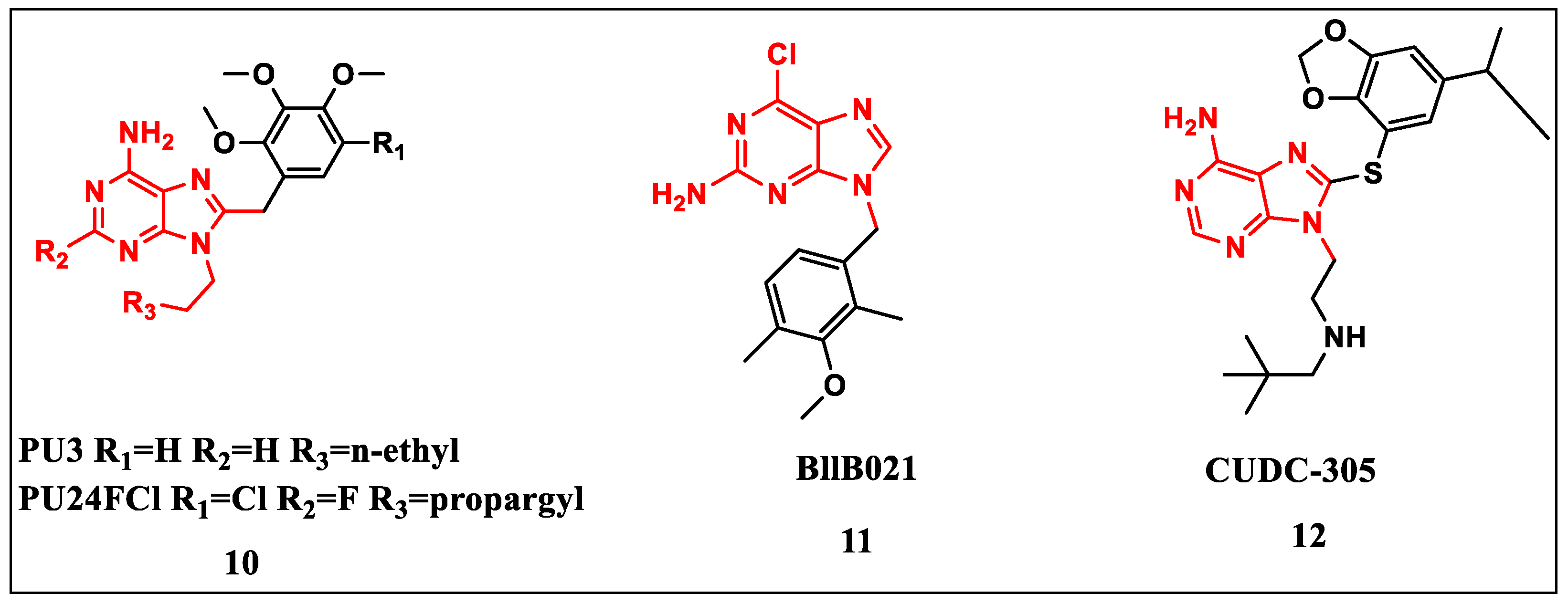
2.2.1. Purine-Based Structures
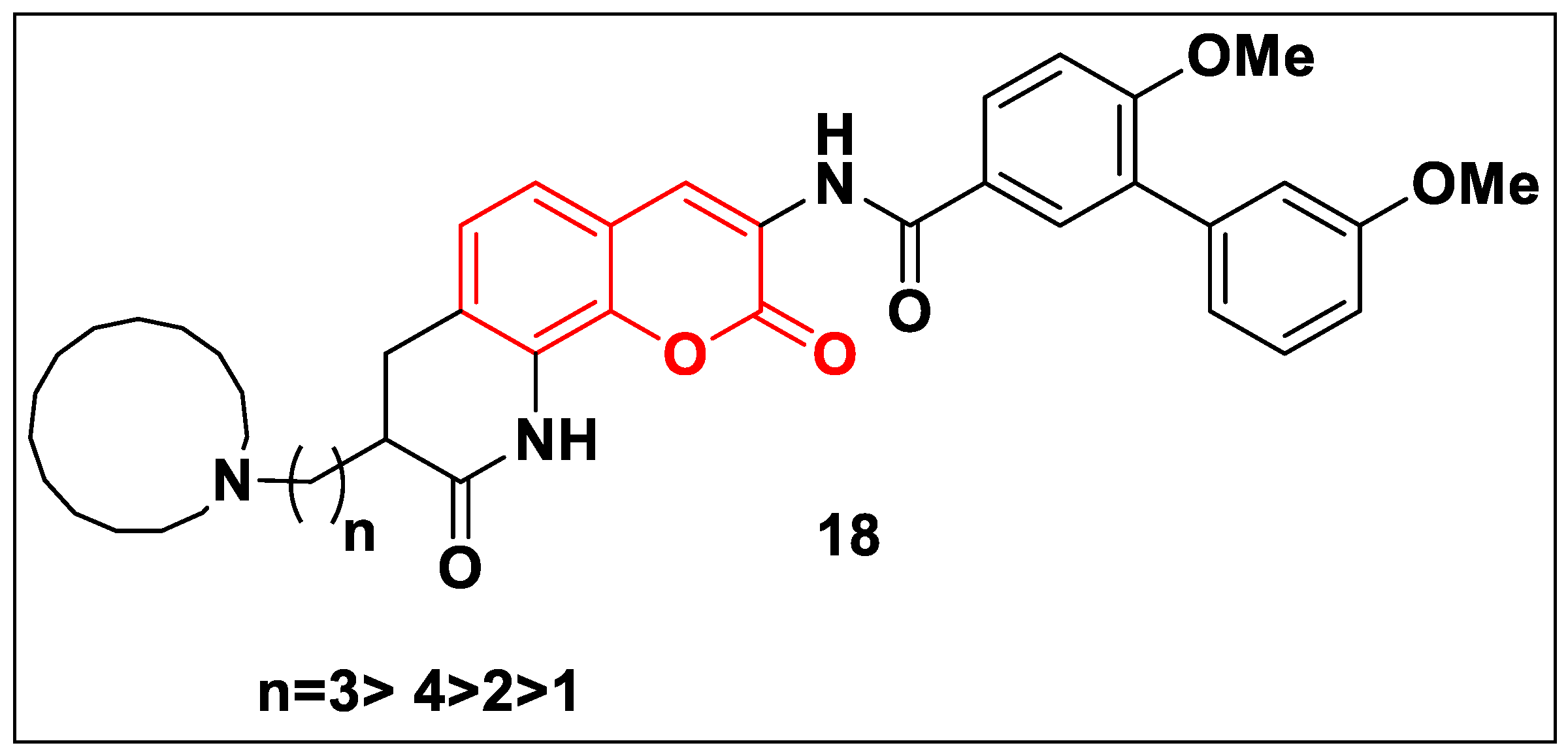
2.2.2. Coumarin-Based Structures
2.2.3. Quinolone-Based Structures
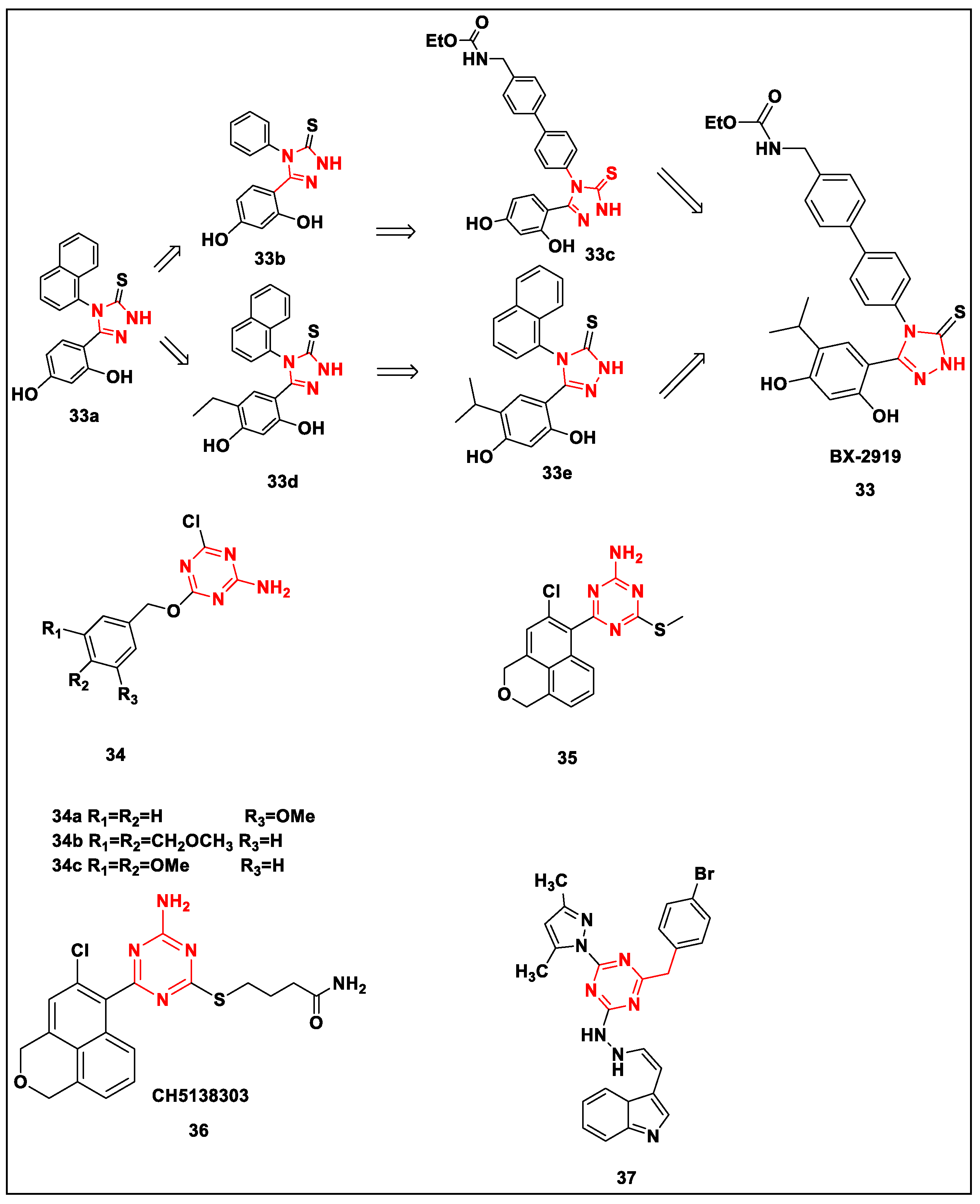
2.2.4. Triazine and Triazolothione-Based Structures
2.2.5. Isoxazole-Based Structures
2.2.6. Pyrazole-Based Structure
3. Conclusions and Future Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mahboubi-Rabbani, M.; Zarghi, A. Lipoxygenase Inhibitors as Cancer Chemopreventives: Discovery, Recent Developments and Future Perspectives. Curr. Med. Chem. 2021, 28, 1143–1175. [Google Scholar] [CrossRef] [PubMed]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The Hsp90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, D.; Swords, R.; Carew, J.S.; Nawrocki, S.T.; Bhalla, K.; Giles, F.J. Targeting HSP90 for cancer therapy. Br. J. Cancer 2009, 100, 1523–1529. [Google Scholar] [CrossRef] [PubMed]
- Ischia, J.; So, A.I. The role of heat shock proteins in bladder cancer. Nat. Rev. Urol. 2013, 10, 386–395. [Google Scholar] [CrossRef]
- Davenport, J.; Manjarrez, J.R.; Peterson, L.; Krumm, B.; Blagg, B.S.J.; Matts, R.L. Gambogic Acid, a Natural Product Inhibitor of Hsp90. J. Nat. Prod. 2011, 74, 1085–1092. [Google Scholar] [CrossRef] [PubMed]
- Bhat, R.; Tummalapalli, S.R.; Rotella, D.P.J. Progress in the Discovery and Development of Heat Shock Protein 90 (Hsp90) Inhibitors: Miniperspective. Med. Chem. 2014, 57, 8718–8728. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, A.M.; Whitesell, L. Hsp90: Enabler of Cancer Adaptation. Annu. Rev. Cancer Biol. 2019, 3, 275–297. [Google Scholar] [CrossRef]
- Gupta, S.D.; Bommaka, M.K.; Banerjee, A. Inhibiting protein-protein interactions of Hsp90 as a novel approach for targeting cancer. Eur. J. Med. Chem. 2019, 178, 48–63. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, Q.; You, Q. Targeting the HSP90–CDC37–kinase chaperone cycle: A promising therapeutic strategy for cancer. Med. Res. Rev. 2022, 42, 156–182. [Google Scholar] [CrossRef]
- Li, L.; Chen, N.-N.; You, Q.-D.; Xu, X.-L. An updated patent review of anticancer Hsp90 inhibitors (2013–present). Expert Opin. Ther. Patents 2021, 31, 67–80. [Google Scholar] [CrossRef]
- Niu, M.; Song, S.; Su, Z.; Wei, L.; Li, L.; Pu, W.; Zhao, C.; Ding, Y.; Wang, J.; Cao, W.; et al. Inhibition of heat shock protein (HSP) 90 reverses signal transducer and activator of transcription (STAT) 3-mediated muscle wasting in cancer cachexia mice. Br. J. Pharmacol. 2021, 178, 4485–4500. [Google Scholar] [CrossRef] [PubMed]
- Jafari, A.; Rezaei-Tavirani, M.; Farhadihosseinabadi, B.; Taranejoo, S.; Zali, H. HSP90 and Co-chaperones: Impact on Tumor Progression and Prospects for Molecular-Targeted Cancer Therapy. Cancer Investig. 2020, 38, 310–328. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, J.; Carter, T.R.; Cohen, M.S.; Blagg, B.S.J. Old and New Approaches to Target the Hsp90 Chaperone. Curr. Cancer Drug Targets 2020, 20, 253–270. [Google Scholar] [CrossRef] [PubMed]
- Trepel, J.; Mollapour, M.; Giaccone, G.; Neckers, L. Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 2010, 10, 537–549. [Google Scholar] [CrossRef]
- Nazaripour, E.; Mousazadeh, F.; Moghadam, M.D.; Najafi, K.; Borhani, F.; Sarani, M.; Ghasemi, M.; Rahdar, A.; Iravani, S.; Khatami, M. Biosynthesis of lead oxide and cerium oxide nanoparticles and their cytotoxic activities against colon cancer cell line. Inorg. Chem. Commun. 2021, 131, 108800. [Google Scholar] [CrossRef]
- Iravani, S.; Varma, R.S. MXenes for Cancer Therapy and Diagnosis: Recent Advances and Current Challenges. ACS Biomater. Sci. Eng. 2021, 7, 1900–1913. [Google Scholar] [CrossRef]
- Delfi, M.; Sartorius, R.; Ashrafizadeh, M.; Sharifi, E.; Zhang, Y.; De Berardinis, P.; Zarrabi, A.; Varma, R.S.; Tay, F.R.; Smith, B.R.; et al. Self-assembled peptide and protein nanostructures for anti-cancer therapy: Targeted delivery, stimuli-responsive devices and immunotherapy. Nano Today 2021, 38, 101119. [Google Scholar] [CrossRef]
- Alavi, M.; Varma, R.S. Overview of Novel Strategies for the Delivery of Anthracyclines to Cancer Cells by Liposomal and Polymeric Nano Formulations. Int. J. Biol. Macromol. 2020, 164, 2197–2203. [Google Scholar] [CrossRef]
- Makvandi, P.; Baghbantaraghdari, Z.; Zhou, W.; Zhang, Y.; Manchanda, R.; Agarwal, T.; Wu, A.; Maiti, T.K.; Varma, R.S.; Smith, B.R. Gum polysaccharide/nanometal hybrid biocomposites in cancer diagnosis and therapy. Biotechnol. Adv. 2021, 48, 107711. [Google Scholar] [CrossRef]
- Hajipour, A.R.; Khorsandi, Z.; Mortazavi, M.; Farrokhpour, H. Green, efficient and large-scale synthesis of benzimidazoles, benzoxazoles and benzothiazoles derivatives using ligand-free cobalt-nanoparticles: As potential anti-estrogen breast cancer agents, and study of their interactions with estrogen receptor by molecular docking. RSC Adv. 2015, 5, 107822–107828. [Google Scholar] [CrossRef]
- Khorsandi, Z.; Hajipour, A.R.; Sarfjoo, M.R.; Varma, R.S. A Pd/Cu-Free magnetic cobalt catalyst for C–N cross coupling reactions: Synthesis of abemaciclib and fedratinib. Green Chem. 2021, 23, 5222–5229. [Google Scholar] [CrossRef]
- Khorsandi, Z.; Keshavarzipour, F.; Varma, R.S.; Hajipour, A.R.; Sadeghi-Aliabadi, H. Sustainable synthesis of potential antitumor new derivatives of Abemaciclib and Fedratinib via C-N cross coupling reactions using Pd/Cu-free Co-catalyst. Mol. Catal. 2021, 517, 112011. [Google Scholar] [CrossRef]
- Whitesell, L.; Lindquist, S.L. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer. 2005, 5, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Jego, G.; Hazoumé, A.; Seigneuric, R.; Garrido, C. Targeting heat shock proteins in cancer. Cancer Lett. 2013, 332, 275–285. [Google Scholar] [CrossRef]
- Calderwood, S.K.; Khaleque, M.A.; Sawyer, D.B.; Ciocca, D.R. Heat Shock Proteins in Cancer: Chaperones of Tum Origenesis. J. Pet. Sci. Eng. 2006, 31, 164–172. [Google Scholar]
- Patel, H.J.; Patel, P.D.; Ochiana, S.O.; Yan, P.; Sun, W.; Patel, M.R.; Shah, S.K.; Tramentozzi, E.; Brooks, J.; Bolaender, A.J. Structure–Activity Relationship in a Purine-Scaffold Compound Series with Selectivity for the Endoplasmic Reticulum Hsp90 ParalogGrp94. Med. Chem. 2015, 58, 3922–3943. [Google Scholar] [CrossRef]
- Johnson, J.L. Evolution and function of diverse Hsp90 homologs and cochaperone proteins. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 607–613. [Google Scholar] [CrossRef]
- Csermely, P.; Miyata, Y.; Schnaider, T.; Yahara, I. Autophosphorylation of grp94 (Endoplasmin). J. Biol. Chem. 1995, 270, 6381–6388. [Google Scholar] [CrossRef]
- Mishra, P.; Bolon, D.N. Designed Hsp90 Heterodimers Reveal an Asymmetric ATPase-Driven Mechanism In Vivo. Mol. Cell 2014, 53, 344–350. [Google Scholar] [CrossRef]
- Meyer, P.; Prodromou, C.; Hu, B.; Vaughan, C.; Roe, S.M.; Panaretou, B.; Piper, P.W.; Pearl, L.H. Structural and Functional Analysis of the Middle Segment of Hsp90: Implications for ATP Hydrolysis and Client Protein and Cochaperone Interactions. Mol. Cell 2003, 11, 647–658. [Google Scholar] [CrossRef]
- Gupta, S.D. Hsp90 Flexibility and Development of its Inhibitors for the Treatment of Cancer. Curr. Chem. Biol. 2018, 12, 53–64. [Google Scholar] [CrossRef]
- Gupta, S.D. Novel Anti-Cancer Drugs Based on Hsp90 Inhibitory Mechanisms: A Recent Report. In Medicinal Chemistry with Pharma-Ceutical Product Development; Apple Academic Press: New York, NY, USA, 2019; pp. 57–104. [Google Scholar]
- Gupta, S.D.; Pan, C.H. Recent update on discovery and development of Hsp90 inhibitors as senolytic agents. Int. J. Biol. Macromol. 2020, 161, 1086–1098. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Vartholomaiou, E.; Echeverría, P.C.; Picard, D. Unusual Suspects in the Twilight Zone Between the Hsp90 Interactome and Carcinogenesis. Adv. Cancer Res. 2016, 129, 1–30. [Google Scholar] [CrossRef]
- Hayat, U.; Elliott, G.T.; Olszanski, A.J.; Altieri, D.C. Feasibility and safety of targeting mitochondria for cancer therapy–preclinical characterization of gamitrinib, a first-in-class, mitochondriaL-targeted small molecule Hsp90 inhibitor. Cancer Biol. Ther. 2022, 23, 117–126. [Google Scholar] [CrossRef]
- Biondini, M.; Kiepas, A.; El-Houjeiri, L.; Annis, M.G.; Hsu, B.E.; Fortier, A.-M.; Morin, G.; Martina, J.A.; Sirois, I.; Aguilar-Mahecha, A.; et al. HSP90 inhibitors induce GPNMB cell-surface expression by modulating lysosomal positioning and sensitize breast cancer cells to glembatumumab vedotin. Oncogene 2022, 41, 1701–1717. [Google Scholar] [CrossRef]
- Epp-Ducharme, B.; Dunne, M.; Fan, L.; Evans, J.C.; Ahmed, L.; Bannigan, P.; Allen, C. Heat-activated nanomedicine formulation improves the anticancer potential of the HSP90 inhibitor luminespib in vitro. Sci. Rep. 2021, 11, 11103. [Google Scholar] [CrossRef]
- Wu, Y.-W.; Chao, M.-W.; Tu, H.-J.; Chen, L.-C.; Hsu, K.-C.; Liou, J.-P.; Yang, C.-R.; Yen, S.-C.; HuangFu, W.-C.; Pan, S.-L. A novel dual HDAC and HSP90 inhibitor, MPT0G449, downregulates oncogenic pathways in human acute leukemia in vitro and in vivo. Oncogenesis 2021, 10, 39. [Google Scholar] [CrossRef]
- Mshaik, R.; Simonet, J.; Georgievski, A.; Jamal, L.; Bechoua, S.; Ballerini, P.; Bellaye, P.-S.; Mlamla, Z.; de Barros, J.-P.P.; Geissler, A.; et al. HSP90 inhibitor NVP-BEP800 affects stability of SRC kinases and growth of T-cell and B-cell acute lymphoblastic leukemias. Blood Cancer J. 2021, 11, 61. [Google Scholar] [CrossRef]
- Abdelmoaty, A.A.A.; Zhang, P.; Lin, W.; Fan, Y.-J.; Ye, S.-N.; Xu, J.-H. C0818, a novel curcumin derivative, induces ROS-dependent cytotoxicity in human hepatocellular carcinoma cells in vitro via disruption of Hsp90 function. Acta Pharmacol. Sin. 2022, 43, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Zhou, L.; Dicker, D.T.; Lev, A.; Zhang, S.; Ross, E.; El-Deiry, W.S. Anti-Cancer Efficacy Including Rb-Deficient Tumors and VHL-Independent HIF1α Proteasomal Destabilization by Dual Targeting of CDK1 or CDK4/6 and Hsp90. Sci. Rep. 2021, 11, 20871. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-L.; Liu, P.; Zhu, W.-L.; Lou, L.-G. DCZ5248, a novel dual inhibitor of Hsp90 and autophagy, exerts antitumor activity against colon cancer. Acta Pharmacol. Sin. 2021, 42, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.-J.; Liu, K.-X.; Zhang, M.; Shen, F.-K.; Ye, L.-L.; Wu, W.-B.; Hou, X.-T.; Hao, E.-W.; Hou, Y.-Y.; Bai, G. Okicamelliaside targets the N-terminal chaperone pocket of HSP90 disrupts the chaperone protein interaction of HSP90-CDC37 and exerts antitumor activity. Acta Pharmacol. Sin. 2021, 43, 1046–1058. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Cheng, S.-C.; Supko, J.G.; Polak, M.; Wahner-Hendrickson, A.E.; Ivy, S.P.; Bowes, B.; Sawyer, H.; Basada, P.; Hayes, M.; et al. Combined PARP and HSP90 inhibition: Preclinical and Phase 1 evaluation in patients with advanced solid tumours. Br. J. Cancer 2021, 126, 1027–1036. [Google Scholar] [CrossRef]
- Magwenyane, A.M.; Lawal, M.M.; Amoako, D.G.; Somboro, A.M.; Agoni, C.; Khan, R.B.; Mhlongo, N.N.; Kumalo, H.M. Exploring the inhibitory mechanism of resorcinylic isoxazole amine NVP-AUY922 towards the discovery of potential heat shock protein 90 (Hsp90) inhibitors. Sci. Afr. 2022, 15, e01107. [Google Scholar] [CrossRef]
- Serwetnyk, M.A.; Blagg, S.J. The Disruption of Protein−Protein Interactions with Co-chaperones and Client Substrates as a Strategy Towards Hsp90 Inhibition. Acta. Pharm. Sin. B 2021, 11, 1446–1468. [Google Scholar] [CrossRef]
- Roe, S.M.; Prodromou, C.; O’Brien, R.; Ladbury, J.E.; Piper, P.W.; Pearl, L.H. Structural Basis for Inhibition of the Hsp90 Molecular Chaperone by the Antitumor Antibiotics Radicicol and Geldanamycin. J. Med. Chem. 1999, 42, 260–266. [Google Scholar] [CrossRef]
- Supko, J.G.; Hickman, R.L.; Grever, M.R.; Malspeis, L. Preclinical Pharmacologic Evaluation of Geldanamycin as an Antitumor Agent. Cancer Chemother. Pharmacol. 1995, 36, 305–315. [Google Scholar] [CrossRef]
- Banerji, U.; O’Donnell, A.; Scurr, M.; Benson, C.; Hanwell, J.; Clark, S.; Raynaud, F.; Turner, A.; Walton, M.; Workman, P.; et al. Phase I Trial of the Heat Shock Protein 90 (Hsp90) Inhibitor 17-Allylamino 17-Demethoxygeldanamycin (17AAG). Pharmacokinetic (PK) Profile and Pharmacodynamic (PD) Endpoints. Proc. Am. Soc. Clin. Oncol. 2001, 20, 326. [Google Scholar]
- Egorin, M.J.; Zuhowski, E.G.; Rosen, D.M.; Sentz, D.L.; Covey, J.M.; Eiseman, J.L. Plasma pharmacokinetics and tissue distribution of 17-(allylamino)-17-demethoxygeldanamycin (NSC 330507) in CD2F1 mice1. Cancer Chemother. Pharmacol. 2001, 47, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Egorin, M.J.; Lagattuta, T.F.; Hamburger, D.R.; Covey, J.M.; White, K.D.; Musser, S.M.; Eiseman, J.L. Pharmacokinetics, tissue distribution, and metabolism of 17-(dimethylaminoethylamino)-17-demethoxygeldanamycin (NSC 707545) in CD2F1 mice and Fischer 344 rats. Cancer Chemother. Pharmacol. 2002, 49, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Munster, P.N.; Tong, L.; Schwartz, L.; Larson, S.; Kenneson, K.; De La Cruz, A.; Rosen, N.; Scher, H. Phase I Trial of 17(Allylamino)-17-demethoxygeldanamycin (17AAG) in Patients (Pts) with Advanced Solid Malignancies. Proc. Am. Soc. Clin. Oncol. 2001, 20, 83–91. [Google Scholar]
- Soga, S.; Sharma, S.V.; Shiotsu, Y.; Shimizu, M.; Tahara, H.; Yamaguchi, K.; Ikuina, Y.; Murakata, C.; Tamaoki, T.; Kurebayashi, J.; et al. Stereospecific antitumor activity of radicicol oxime derivatives. Cancer Chemother. Pharmacol. 2001, 48, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Johnson, V.A.; Singh, E.K.; Nazarova, L.A.; Alexander, L.D.; McAlpine, S.R. Macrocyclic inhibitors of hsp90. Curr. Top. Med. Chem. 2010, 10, 1380–1402. [Google Scholar] [CrossRef] [PubMed]
- Porter, J.R.; Ge, J.; Lee, J.; Normant, E.; West, K. Ansamycin Inhibitors of Hsp90: Nature’s Prototype for Anti-chaperone Therapy. Curr. Top. Med. 2009, 9, 1386–1418. [Google Scholar] [CrossRef]
- Tanida, S.; Hasegawa, T.; Higashide, E.; Macbecins, I.; Macbecins, I.I. New Antitumor Antibiotics. I. Producing Organism, Fermentation and Antimicrobial Activities. J. Antibiot. 1980, 33, 199–204. [Google Scholar] [CrossRef]
- Tian, Z.-Q.; Liu, Y.; Zhang, D.; Wang, Z.; Dong, S.D.; Carreras, C.W.; Zhou, Y.; Rastelli, G.; Santi, D.V.; Myles, D.C. Synthesis and biological activities of novel 17-aminogeldanamycin derivatives. Bioorg. Med. Chem. 2004, 12, 5317–5329. [Google Scholar] [CrossRef]
- Wagner, A.J.; Chugh, R.; Rosen, L.S.; Morgan, J.A.; George, S.; Gordon, M.; Dunbar, J.; Normant, E.; Grayzel, D.; Demetri, G.D. A Phase I Study of the HSP90 Inhibitor Retaspimycin Hydrochloride (IPI-504) in Patients with Gastrointestinal Stromal Tumors or Soft-Tissue Sarcomas. Clin. Cancer Res. 2013, 19, 6020–6029. [Google Scholar] [CrossRef]
- Floris, G.; Debiec-Rychter, M.; Wozniak, A.; Stefan, C.; Normant, E.; Faa, G.; Machiels, K.; Vanleeuw, U.; Sciot, R.; Schöffski, P. The Heat Shock Protein 90 Inhibitor IPI-504 Induces KIT Degradation, Tumor Shrinkage, and Cell Proliferation Arrest in Xenograft Models of Gastrointestinal Stromal Tumors. Mol. Cancer Ther. 2011, 10, 1897–1908. [Google Scholar] [CrossRef]
- Floris, G.; Sciot, R.; Wozniak, A.; Van Looy, T.; Wellens, J.; Faa, G.; Normant, E.; Debiec-Rychter, M.; Schöffski, P. The Novel Hsp90 inhibitor, IPI-493, is Highly Effective in Human Gastrostrointestinal Stromal Tumor Xenografts Carrying Heterogeneous KIT Mutations. Clin. Cancer Res. 2011, 17, 5604–5614. [Google Scholar] [CrossRef] [PubMed]
- Whitesell, L.; Mimnaugh, E.G.; De Costa, B.; Myers, C.E.; Neckers, L.M. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: Essential role for stress proteins in oncogenic transformation. Proc. Natl. Acad. Sci. USA 1994, 91, 8324–8328. [Google Scholar] [CrossRef] [PubMed]
- Stebbins, C.E.; Russo, A.A.; Schneider, C.; Rosen, N.; Hartl, F.; Pavletich, N.P. Crystal Structure of an Hsp90–Geldanamycin Complex: Targeting of a Protein Chaperone by an Antitumor Agent. Cell 1997, 89, 239–250. [Google Scholar] [CrossRef]
- Soga, S.; Shiotsu, Y.; Akinaga, S.; Sharma, S. Development of Radicicol Analogues. Curr. Cancer Drug Targets 2003, 3, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Miyata, Y. Hsp90 Inhibitor Geldanamycin and Its Derivatives as Novel Cancer Chemotherapeutic Agents. Curr. Pharm. Des. 2005, 11, 1131–1138. [Google Scholar] [CrossRef]
- Chiosis, G.; Lucas, B.; Huezo, H.; Solit, D.; Basso, A.; Rosen, N. Development of Purine-Scaffold Small Molecule Inhibitors of Hsp90. Curr. Cancer Drug Targets 2003, 3, 371–376. [Google Scholar] [CrossRef]
- Conejo-García, A.; García-Rubiño, M.E.; Marchal, J.A.; Núñez, M.C.; Ramírez, A.; Cimino, S.; García, M.; Aránega, A.; Gallo, M.A.; Campos, J.M. Synthesis and anticancer activity of (RS)-9-(2,3-dihydro-1,4-benzoxaheteroin-2-ylmethyl)-9H-purines. Eur. J. Med. Chem. 2011, 46, 3795–3801. [Google Scholar] [CrossRef]
- Pelliccia, S.; Amato, J.; Capasso, D.; Di Gaetano, S.; Massarotti, A.; Piccolo, M.; Irace, C.; Tron, G.C.; Pagano, B.; Randazzo, A.J. Bio-Inspired Dual-Selective BCL-2/c-MYC G-Quadruplex Binders: Design, Synthesis, and Anticancer Activity of Drug-like Imidazo [2,1-i] purine Derivatives. Med. Chem. 2019, 63, 2035–2050. [Google Scholar] [CrossRef]
- Ashour, F.A.; Rida, S.M.; El-Hawash, S.A.; ElSemary, M.M.; Badr, M.H. Synthesis, Anticancer, Anti-HIV-1, and Antimicrobial Activity of Some Tricyclic Triazino and Triazolo [4,3-e] purine Derivatives. Cancer Causes Control 2012, 21, 1107–1119. [Google Scholar] [CrossRef]
- Kinali-Demirci, S.; İdil, Ö.; Dişli, A. Synthesis of Some Novel Purine Derivatives Incorporating Tetrazole Ring and Investigation of their Antimicrobial Activity and DNA Interactions. Cancer Cell Int. J. Mol. Histol. 2015, 24, 1218–1225. [Google Scholar] [CrossRef]
- Abdallah, A.E.; Elgemeie, G.H. Development; Therapy, Design, Synthesis, Docking, and Antimicrobial Evaluation of Some Novel Pyrazolo [1,5-a] Pyrimidines and their Corresponding Cycloalkane Ring-fused Derivatives as Purine Analogs. Drug Des. Devel. Ther. 2018, 12, 1785–1798. [Google Scholar] [CrossRef] [PubMed]
- Rosemeyer, H. The Chemodiversity of Purine as a Constituent of Natural Products. Chem. Biodivers. 2004, 1, 361–401. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, K.; Aoyagi, Y.; Fukuuchi, T.; Inazawa, K.; Yamaoka, N. Total Purine and Purine Base Content of Common Foodstuffs for Facilitating Nutritional Therapy for Gout and Hyperuricemia. Biol. Pharm. Bull. 2014, 37, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Ren, W.; Huang, X.; Deng, J.; Li, T.; Yin, Y. Potential Mechanisms Connecting Purine Metabolism and Cancer Therapy. Front. Immunol. 2018, 9, 1697. [Google Scholar] [CrossRef] [PubMed]
- Chiosis, G.; Timaul, M.N.; Lucas, B.; Munster, P.N.; Zheng, F.F.; Sepp-Lorenzino, L.; Rosen, N. A small molecule designed to bind to the adenine nucleotide pocket of Hsp90 causes Her2 degradation and the growth arrest and differentiation of breast cancer cells. Chem. Biol. 2001, 8, 289–299. [Google Scholar] [CrossRef]
- Bao, R.; Lai, C.-J.; Qu, H.; Wang, D.; Yin, L.; Zifcak, B.; Atoyan, R.; Wang, J.; Samson, M.; Forrester, J.; et al. CUDC-305, a Novel Synthetic HSP90 Inhibitor with Unique Pharmacologic Properties for Cancer Therapy. Clin. Cancer Res. 2009, 15, 4046–4057. [Google Scholar] [CrossRef]
- Zhang, H.; Neely, L.; Lundgren, K.; Yang, Y.-C.; Lough, R.; Timple, N.; Burrows, F. BIIB021, a synthetic HSP90 inhibitor, has broad application against tumors with acquired multidrug resistance. Int. J. Cancer 2010, 126, 1226–1234. [Google Scholar] [CrossRef]
- Yin, X.; Zhang, H.; Lundgren, K.; Wilson, L.; Burrows, F.; Shores, C.G. BIIB021, a novel HSP90 inhibitor, sensitizes head and neck squamous cell carcinoma to radiotherapy. Int. J. Cancer 2010, 126, 1216–1225. [Google Scholar] [CrossRef]
- Chaurasiya, A.; Wahan, S.K.; Sahu, C.; Chawla, P.A. An insight into the rational design of recent purine-based scaffolds in targeting various cancer pathways. J. Mol. Struct. 2022, 134308. [Google Scholar] [CrossRef]
- Wang, X.-T.; Bao, C.-H.; Jia, Y.-B.; Wang, N.; Ma, W.; Liu, F.; Wang, C.; Wang, J.-B.; Song, Q.-X.; Cheng, Y.-F. BIIB021, a novel Hsp90 inhibitor, sensitizes esophageal squamous cell carcinoma to radiation. Biochem. Biophys. Res. Commun. 2014, 452, 945–950. [Google Scholar] [CrossRef]
- ElFiky, A.; Saif, M.W.; Beeram, M.; Brien, S.O.; Lammanna, N.; Castro, J.E.; Woodworth, J.; Perea, R.; Storgard, C.; Von Hoff, D.D. BIIB021, an oral, synthetic non-ansamycin Hsp90 inhibitor: Phase I experience. J. Clin. Oncol. 2008, 26, 2503. [Google Scholar] [CrossRef]
- Dickson, M.A.; Okuno, S.H.; Keohan, M.L.; Maki, R.G.; D’Adamo, D.R.; Akhurst, T.J.; Antonescu, C.R.; Schwartz, G.K. Phase II study of the HSP90-inhibitor BIIB021 in gastrointestinal stromal tumors. Ann. Oncol. 2013, 24, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Emami, S.; Dadashpour, S. Current developments of coumarin-based anti-cancer agents in medicinal chemistry. Eur. J. Med. Chem. 2015, 102, 611–630. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Xu, Z. Coumarin-containing hybrids and their anticancer activities. Eur. J. Med. Chem. 2019, 181, 111587. [Google Scholar] [CrossRef] [PubMed]
- Al-Warhi, T.; Sabt, A.; Elkaeed, E.B.; Eldehna, W.M. Recent advancements of coumarin-based anticancer agents: An up-to-date review. Bioorg. Chem. 2020, 103, 104163. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Fan, J.; Liu, L.; Liu, X.; Gao, F. Coumarin derivatives with anticancer activities: An update. Arch. Pharm. 2020, 353, 2000025. [Google Scholar] [CrossRef]
- Qin, H.-L.; Zhang, Z.-W.; Ravindar, L.; Rakesh, K. Antibacterial activities with the structure-activity relationship of coumarin derivatives. Eur. J. Med. Chem. 2020, 207, 112832. [Google Scholar] [CrossRef]
- Keshavarzipour, F.; Tavakol, H. The synthesis of coumarin derivatives using choline chloride/zinc chloride as a deep eutectic solvent. J. Iran. Chem. Soc. 2016, 13, 149–153. [Google Scholar] [CrossRef]
- Ahmed, E.Y.; Elserwy, W.S.; El-Mansy, M.F.; Serry, A.M.; Salem, A.M.; Abdou, A.M.; Abdelrahman, B.A.; Elsayed, K.H.; Elaziz, M.R.A. Angiokinase inhibition of VEGFR-2, PDGFR and FGFR and cell growth inhibition in lung cancer: Design, synthesis, biological evaluation and molecular docking of novel azaheterocyclic coumarin derivatives. Bioorg. Med. Chem. Lett. 2021, 48, 128258. [Google Scholar] [CrossRef]
- Burlison, J.A.; Neckers, L.; Smith, A.B.; Maxwell, A.A.; Blagg, B.S.J. Novobiocin: Redesigning a DNA Gyrase Inhibitor for Selective Inhibition of Hsp90. J. Am. Chem. Soc. 2006, 128, 15529–15536. [Google Scholar] [CrossRef]
- Shelton, S.N.; Shawgo, M.E.; Matthews, S.B.; Lu, Y.; Donnelly, A.C.; Szabla, K.; Tanol, M.; Vielhauer, G.A.; Rajewski, R.A.; Matts, R.L.; et al. KU135, a Novel Novobiocin-Derived C-Terminal Inhibitor of the 90-kDa Heat Shock Protein, Exerts Potent Antiproliferative Effects in Human Leukemic Cells. Mol. Pharmacol. 2009, 76, 1314–1322. [Google Scholar] [CrossRef] [PubMed]
- Garg, G.; Zhao, H.; Blagg, B.S.J. Design, Synthesis, and Biological Evaluation of Ring-Constrained Novobiocin Analogues as Hsp90 C-Terminal Inhibitors. ACS Med. Chem. Lett. 2015, 6, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Ning, J.-Y.; Dai, X.; Gao, Y.-D.; Su, L.; Zhao, B.-X.; Miao, J.-Y. Discovery of novel HSP90 inhibitors that induced apoptosis and impaired autophagic flux in A549 lung cancer cells. Eur. J. Med. Chem. 2018, 145, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Kouznetsov, V.V.; Méndez, L.Y.V.; Galvis, C.E.P.; Villamizar, M.C.O. The direct C–H alkenylation of quinoline N-oxides as a suitable strategy for the synthesis of promising antiparasitic drugs. New J. Chem. 2020, 44, 12–19. [Google Scholar] [CrossRef]
- Keshavarzipour, F.; Tavakol, H. Zinc Cation Supported on Carrageenan Magnetic Nanoparticles: A Novel, Green and Efficient Catalytic System for One-pot Three-Component Synthesis of Quinoline Derivatives. Appl. Organomet. Chem. 2017, 31, 3682–3692. [Google Scholar] [CrossRef]
- Insuasty, D.; García, S.; Abonia, R.; Insuasty, B.; Quiroga, J.; Nogueras, M.; Laali, K.K. Design, Synthesis, and Molecular Docking Study of Novel Quinoline-Based Bis-Chalcones as Potential Antitumor Agents. Arch. Pharm. 2021, 354, 2100094–2100103. [Google Scholar] [CrossRef]
- Jin, G.; Li, Z.; Xiao, F.; Qi, X.; Sun, X. Optimization of activity localization of quinoline derivatives: Design, synthesis, and dual evaluation of biological activity for potential antitumor and antibacterial agents. Bioorg. Chem. 2020, 99, 103837. [Google Scholar] [CrossRef]
- Mishra, S.; Salahuddin, R.K.; Majumder, A.; Kumar, A.; Singh, C.; Tiglani, D. Updates on Synthesis and Biological Activities of Quinoline Derivatives: A Review. Int. J. Pharm. Sci. Rev. Res. 2021, 13, 3941–3960. [Google Scholar]
- Goyal, S.; Binnington, B.; McCarthy, S.D.; Desmaële, D.; Férrié, L.; Figadère, B.; Loiseau, P.M.; Branch, D.R. Inhibition of in vitro Ebola infection by anti-parasitic quinoline derivatives. F1000Research 2020, 9, 268. [Google Scholar] [CrossRef]
- Verma, C.; Quraishi, M.; Ebenso, E.E. Quinoline and its derivatives as corrosion inhibitors: A review. Surfaces Interfaces 2020, 21, 100634. [Google Scholar] [CrossRef]
- Kayamba, F.; Malimabe, T.; Ademola, I.K.; Pooe, O.J.; Kushwaha, N.D.; Mahlalela, M.; van Zyl, R.L.; Gordon, M.; Mudau, P.T.; Zininga, T.; et al. Design and synthesis of quinoline-pyrimidine inspired hybrids as potential plasmodial inhibitors. Eur. J. Med. Chem. 2021, 217, 113330. [Google Scholar] [CrossRef] [PubMed]
- Rao, K.V.; Cullen, W.P. Streptonigrin, an Antitumor Substance. I. Isolation and Characterization. Antibiot. Annu. 1959, 7, 950–953. [Google Scholar]
- Chirigos, M.A.; Pearson, J.W.; Papas, T.S.; Woods, W.A.; Wood, H.B., Jr.; Spahn, G. Effect of Three Strains of BeG Against a Murine Leukemia After Drug Therapy. Cancer Chemother. Rep. 1973, 57, 305–309. [Google Scholar]
- Balitz, D.M.; Bush, J.A.; Bradner, W.T.; Doyle, T.W.; O’Herron, F.A.; Nettleton, D.E. Isolation of lavendamycin. A new antibiotic from Streptomyces lavendulae. J. Antibiot. 1982, 35, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, T.; Min, J.; Thepchatri, P.; Du, Y.; Li, L.; Lewis, I.; Wilson, L.; Fu, H.; Chiosis, G.; Dingledine, R.; et al. Discovery of aminoquinolines as a new class of potent inhibitors of heat shock protein 90 (Hsp90): Synthesis, biology, and molecular modeling. Bioorg. Med. Chem. 2008, 16, 6903–6910. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Audisio, D.; Messaoudi, S.; Cegielkowski, L.; Peyrat, J.-F.; Brion, J.-D.; Methy-Gonnot, D.; Radanyi, C.; Renoir, J.-M.; Alami, M. Discovery and Biological Activity of 6BrCaQ as an Inhibitor of the Hsp90 Protein Folding Machinery. Chem. Med. Chem. 2011, 6, 804–815. [Google Scholar] [CrossRef]
- Nepali, K.; Kumar, S.; Huang, H.-L.; Kuo, F.-C.; Lee, C.-H.; Kuo, C.-C.; Yeh, T.-K.; Li, Y.-H.; Chang, J.-Y.; Liou, J.-P.; et al. 2-Aroylquinoline-5,8-diones as potent anticancer agents displaying tubulin and heat shock protein 90 (HSP90) inhibition. Org. Biomol. Chem. 2015, 14, 716–723. [Google Scholar] [CrossRef]
- Malayeri, S.O.; Abnous, K.; Arab, A.; Akaberi, M.; Mehri, S.; Zarghi, A.; Ghodsi, R. Design, Synthesis and Biological Evaluation of 7-(Aryl)-2,3-dihydro-[1,4] dioxino [2,3-g] Quinoline Derivatives as Potential Hsp90 Inhibitors and Anticancer Agents. Bioorg. Med. Chem. 2017, 25, 1294–1302. [Google Scholar] [CrossRef]
- Liang, C.; Hao, H.; Wu, X.; Li, Z.; Zhu, J.; Lu, C.; Shen, Y. Design and synthesis of N-(5-chloro-2,4-dihydroxybenzoyl)-(R)-1,2,3,4-tetrahydroisoquinoline-3-carboxamides as novel Hsp90 inhibitors. Eur. J. Med. Chem. 2016, 121, 272–282. [Google Scholar] [CrossRef]
- Nepali, K.; Lin, M.-H.; Chao, M.-W.; Peng, S.-J.; Hsu, K.-C.; Lin, T.E.; Chen, M.-C.; Lai, M.-J.; Pan, S.-L.; Liou, J.-P. Amide-tethered quinoline-resorcinol conjugates as a new class of HSP90 inhibitors suppressing the growth of prostate cancer cells. Bioorg. Chem. 2019, 91, 103119. [Google Scholar] [CrossRef]
- Relitti, N.; Saraswati, A.P.; Chemi, G.; Brindisi, M.; Brogi, S.; Herp, D.; Schmidtkunz, K.; Saccoccia, F.; Ruberti, G.; Ulivieri, C.; et al. Novel quinolone-based potent and selective HDAC6 inhibitors: Synthesis, molecular modeling studies and biological investigation. Eur. J. Med. Chem. 2021, 212, 112998. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Long, S.; Rakesh, K.P.; Zha, G.F. Structure-Activity Relationships (SAR) of Triazine Derivatives: Promising Antimicrobial Agents. Eur. J. Med. Chem. 2020, 185, 111804. [Google Scholar] [CrossRef] [PubMed]
- Gavade, S.N.; Markad, V.L.; Kodam, K.M.; Shingare, M.S.; Mane, D.V. Synthesis and Biological Evaluation of Novel 2, 4, 6-Triazine Derivatives as Antimicrobial Agents. Bioorg. Med. Chem. Lett. 2012, 22, 5075–5077. [Google Scholar] [CrossRef]
- Cascioferro, S.; Parrino, B.; Spanò, V.; Carbone, A.; Montalbano, A.; Barraja, P.; Diana, P.; Cirrincione, G. An overview on the recent developments of 1,2,4-triazine derivatives as anticancer compounds. Eur. J. Med. Chem. 2017, 142, 328–375. [Google Scholar] [CrossRef] [PubMed]
- Singla, P.; Luxami, V.; Paul, K. Triazine as a Promising Scaffold for its Versatile Biological Behavior. Eur. J. Med. Chem. 2015, 102, 39–57. [Google Scholar] [CrossRef]
- Patel, P.K.; Patel, R.V.; Mahajan, D.H.; Parikh, P.A.; Mehta, G.N.; Pannecouque, C.; Chikhalia, K.H. Different Heterocycles Functionalized s-Triazine Analogues: Design, Synthesis and In Vitro Antimicrobial, Antituberculosis, and Anti-HIV Assessment. J. Heterocycl. Chem. 2014, 51, 1641–1658. [Google Scholar] [CrossRef]
- Feldman, R.I.; Mintzer, B.; Zhu, D.; Wu, J.M.; Biroc, S.L.; Yuan, S.; Emayan, K.; Chang, Z.; Chen, D.; Arnaiz, D.O.; et al. Potent Triazolothione Inhibitor of Heat-Shock Protein-90. Oncol. Lett. 2009, 74, 43–50. [Google Scholar] [CrossRef]
- Lee, T.; Seo, Y.H. Targeting the hydrophobic region of Hsp90′s ATP binding pocket with novel 1,3,5-triazines. Bioorg. Med. Chem. Lett. 2013, 23, 6427–6431. [Google Scholar] [CrossRef]
- Miura, T.; Fukami, T.A.; Hasegawa, K.; Ono, N.; Suda, A.; Shindo, H.; Yoon, D.-O.; Kim, S.-J.; Na, Y.-J.; Aoki, Y.; et al. Lead generation of heat shock protein 90 inhibitors by a combination of fragment-based approach, virtual screening, and structure-based drug design. Bioorg. Med. Chem. Lett. 2011, 21, 5778–5783. [Google Scholar] [CrossRef]
- Suda, A.; Koyano, H.; Hayase, T.; Hada, K.; Kawasaki, K.-I.; Komiyama, S.; Hasegawa, K.; Fukami, T.A.; Sato, S.; Miura, T.; et al. Design and synthesis of novel macrocyclic 2-amino-6-arylpyrimidine Hsp90 inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 1136–1141. [Google Scholar] [CrossRef]
- Suda, A.; Kawasaki, K.-I.; Komiyama, S.; Isshiki, Y.; Yoon, D.-O.; Kim, S.-J.; Na, Y.-J.; Hasegawa, K.; Fukami, T.A.; Sato, S.; et al. Design and synthesis of 2-amino-6-(1H,3H-benzo[de]isochromen-6-yl)-1,3,5-triazines as novel Hsp90 inhibitors. Bioorg. Med. Chem. 2014, 22, 892–905. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Zhu, J.; Quan, H.; Wang, G.; Li, B.; Zhu, W.; Xie, C.; Lou, L. X66, a novel N-terminal heat shock protein 90 inhibitor, exerts antitumor effects without induction of heat shock response. Oncotarget 2016, 7, 29648–29663. [Google Scholar] [CrossRef] [PubMed]
- Ali, I.; Lone, M.; Al-Othman, Z.; Al-Warthan, A.; Sanagi, M. Heterocyclic Scaffolds: Centrality in Anticancer Drug Development. Curr. Drug Targets 2015, 16, 711–734. [Google Scholar] [CrossRef]
- Çalışkan, B.; Sinoplu, E.; İbiş, K.; Akhan Güzelcan, E.; Atalay, R.Ç.; Banoglu, E. Synthesis and Cellular Bioactivities of Novel Isoxazole Derivatives Incorporating an Arylpiperazine Moiety as Anticancer Agents. J. Enzym. Inhib. Med. Chem. 2018, 33, 1352–1361. [Google Scholar] [CrossRef] [PubMed]
- Shaik, A.; Bhandare, R.R.; Palleapati, K.; Nissankararao, S.; Kancharlapalli, V.; Shaik, S. Antimicrobial, Antioxidant, and Anticancer Activities of Some Novel Isoxazole Ring Containing Chalcone and Dihydropyrazole Derivatives. Molecules 2020, 25, 1047. [Google Scholar] [CrossRef] [PubMed]
- Barmade, M.A.; Murumkar, P.R.; Sharma, M.K.; Yadav, M.R. Medicinal Chemistry Perspective of Fused Isoxazole Derivatives. Curr. Top. Med. Chem. 2016, 16, 2863–2883. [Google Scholar] [CrossRef] [PubMed]
- Galenko, A.V.; Khlebnikov, A.F.; Novikov, M.S.; Pakalnis, V.V.; Rostovskii, N.V. Recent advances in isoxazole chemistry. Russ. Chem. Rev. 2015, 84, 335–377. [Google Scholar] [CrossRef]
- Sysak, A.; Obmińska-Mrukowicz, B. Isoxazole ring as a useful scaffold in a search for new therapeutic agents. Eur. J. Med. Chem. 2017, 137, 292–309. [Google Scholar] [CrossRef]
- Arya, G.C.; Kaur, K.; Jaitak, V. Isoxazole derivatives as anticancer agent: A review on synthetic strategies, mechanism of action and SAR studies. Eur. J. Med. Chem. 2021, 221, 113511. [Google Scholar] [CrossRef]
- Fernald, K.; Kurokawa, M. Evading apoptosis in cancer. Trends Cell Biol. 2013, 23, 620–633. [Google Scholar] [CrossRef]
- Tonks, N.K. Protein Tyrosine Phosphatases: From Genes, to Function, to Disease. Nat. Rev. Mol. Cell Biol. 2006, 7, 833–846. [Google Scholar] [CrossRef] [PubMed]
- Taldone, T.; Gozman, A.; Maharaj, R.; Chiosis, G. Targeting Hsp90: Small-molecule inhibitors and their clinical development. Curr. Opin. Pharmacol. 2008, 8, 370–374. [Google Scholar] [CrossRef] [PubMed]
- Sharp, S.Y.; Prodromou, C.; Boxall, K.; Powers, M.V.; Holmes, J.L.; Box, G.; Workman, P. Inhibition of The Heat Shock Protein 90 Molecular Chaperone in vitro and in vivo by Novel, Synthetic, Potent Resorcinylic Pyrazole/isoxazole Amide Analogues. Mol. Cancer Ther. 2007, 6, 1198–1211. [Google Scholar] [CrossRef] [PubMed]
- Eccles, S.A.; Massey, A.; Raynaud, F.I.; Sharp, S.Y.; Box, G.; Valenti, M.; Patterson, L.; de Haven Brandon, A.; Gowan, S.; Boxall, F.; et al. NVP-AUY922: A Novel Heat Shock Protein 90 Inhibitor Active against Xenograft Tumor Growth, Angiogenesis, and Metastasis. Cancer Res. 2008, 68, 2850–2860. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Shen, A.; Li, J.; Shi, F.; Chen, W.; Ren, J.; Liu, H.; Xu, Y.; Wang, X.; Yang, X.; et al. Discovery of potent N-(isoxazol-5-yl)amides as HSP90 inhibitors. Eur. J. Med. Chem. 2014, 87, 765–781. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Lin, C.; Qin, X.; Dong, X.; Tu, Z.; Tang, F.; Chen, C.; Zhang, J. Synthesis and biological evaluation of 3,5-disubstituted-4-alkynylisoxozales as a novel class of HSP90 inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 3129–3134. [Google Scholar] [CrossRef]
- Shi, W.; Hu, J.; Bao, N.; Li, D.; Chen, L.; Sun, J. Design, synthesis and cytotoxic activities of scopoletin-isoxazole and scopoletin-pyrazole hybrids. Bioorg. Med. Chem. Lett. 2017, 27, 147–151. [Google Scholar] [CrossRef]
- Abbasi, M.; Sadeghi-Aliabadi, H.; Amanlou, M. Prediction of New Hsp90 Inhibitors Based on 3, 4-Isoxazolediamide Scaffold Using QSAR Study, Molecular Docking and Molecular Dynamic Simulation. Daru J. Pharm. Sci. 2017, 25, 17. [Google Scholar] [CrossRef]
- Abbasi, M.; Sadeghi-Aliabadi, H.; Amanlou, M. 3D-QSAR, Molecular Docking, and Molecular Dynamic Simulations for Prediction of New Hsp90 Inhibitors Based on Isoxazole Scaffold. J. Biomol. Struct. Dyn. 2018, 36, 1463–1478. [Google Scholar] [CrossRef]
- Kaushik, S.; Sanawar, R.; Lekshmi, A.; Chandrasekhar, L.; Nair, M.; Bhatnagar, S.; Santhoshkumar, T.R. ER alpha selective chromone, isoxazolylchromones, induces ROS-mediated cell death without autophagy. Chem. Biol. Drug Des. 2019, 1352–1367. [Google Scholar]
- Jung, J.; Kwon, J.; Hong, S.; Moon, A.-N.; Jeong, J.; Kwon, S.; Kim, J.-A.; Lee, M.; Lee, H.; Lee, J.H.; et al. Discovery of novel heat shock protein (Hsp90) inhibitors based on luminespib with potent antitumor activity. Bioorg. Med. Chem. Lett. 2020, 30, 127165. [Google Scholar] [CrossRef] [PubMed]
- Aissa, I.; Abdelkafi-Koubaa, Z.; Chouaïb, K.; Jalouli, M.; Assel, A.; Romdhane, A.; Harrath, A.H.; Marrakchi, N.; Ben Jannet, H. Glioblastoma-specific anticancer activity of newly synthetized 3,5-disubstituted isoxazole and 1,4-disubstituted triazole-linked tyrosol conjugates. Bioorg. Chem. 2021, 114, 105071. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, M.; Kumar, N.; Baldi, A.; Chandra, R.; Babu, M.A.; Madan, J. Chloro and bromo-pyrazole curcumin Knoevenagel condensates augmented anticancer activity against human cervical cancer cells: Design, synthesis, in silico docking and in vitro cytotoxicity analysis. J. Biomol. Struct. Dyn. 2020, 38, 200–218. [Google Scholar] [CrossRef] [PubMed]
- Rai, G.; Urban, D.J.; Mott, B.T.; Hu, X.; Yang, S.-M.; Benavides, G.A.; Johnson, M.S.; Squadrito, G.L.; Brimacombe, K.R.; Lee, T.D.; et al. Pyrazole-Based Lactate Dehydrogenase Inhibitors with Optimized Cell Activity and Pharmacokinetic Properties. J. Med. Chem. 2020, 63, 10984–11011. [Google Scholar] [CrossRef]
- Azimi, F.; Azizian, H.; Najafi, M.; Hassanzadeh, F.; Sadeghi-Aliabadi, H.; Ghasemi, J.B.; Mahdavi, M. Design and Synthesis of Novel Quinazolinone-Pyrazole Derivatives as Potential α-Glucosidase Inhibitors: Structure-activity Relationship, Molecular Modeling and Kinetic Study. Bioorg. Chem. 2021, 114, 105127–105134. [Google Scholar] [CrossRef]
- Abbasi, M.; Amanlou, M.; Aghaei, M.; Bakherad, M.; Doosti, R.; Sadeghi-Aliabadi, H. New heat shock protein (Hsp90) inhibitors, designed by pharmacophore modeling and virtual screening: Synthesis, biological evaluation and molecular dynamics studies. J. Biomol. Struct. Dyn. 2020, 38, 3462–3473. [Google Scholar] [CrossRef]
- Bennani, F.E.; Doudach, L.; Cherrah, Y.; Ramli, Y.; Karrouchi, K.; Ansar, M.; Faouzi, M.E.A. Overview of recent developments of pyrazole derivatives as an anticancer agent in different cell line. Bioorg. Chem. 2020, 97, 103470. [Google Scholar] [CrossRef]
- Bai, S.-Y.; Dai, X.; Zhao, B.-X.; Miao, J.-Y. Discovery of a novel fluorescent HSP90 inhibitor and its anti-lung cancer effect. RSC Adv. 2014, 4, 19887–19890. [Google Scholar] [CrossRef]
- Abbasi, M.; Sadeghi-Aliabadi, H.; Hassanzadeh, F.; Amanlou, M. Prediction of dual agents as an activator of mutant p53 and inhibitor of Hsp90 by docking, molecular dynamic simulation and virtual screening. J. Mol. Graph. Model. 2015, 61, 186–195. [Google Scholar] [CrossRef]
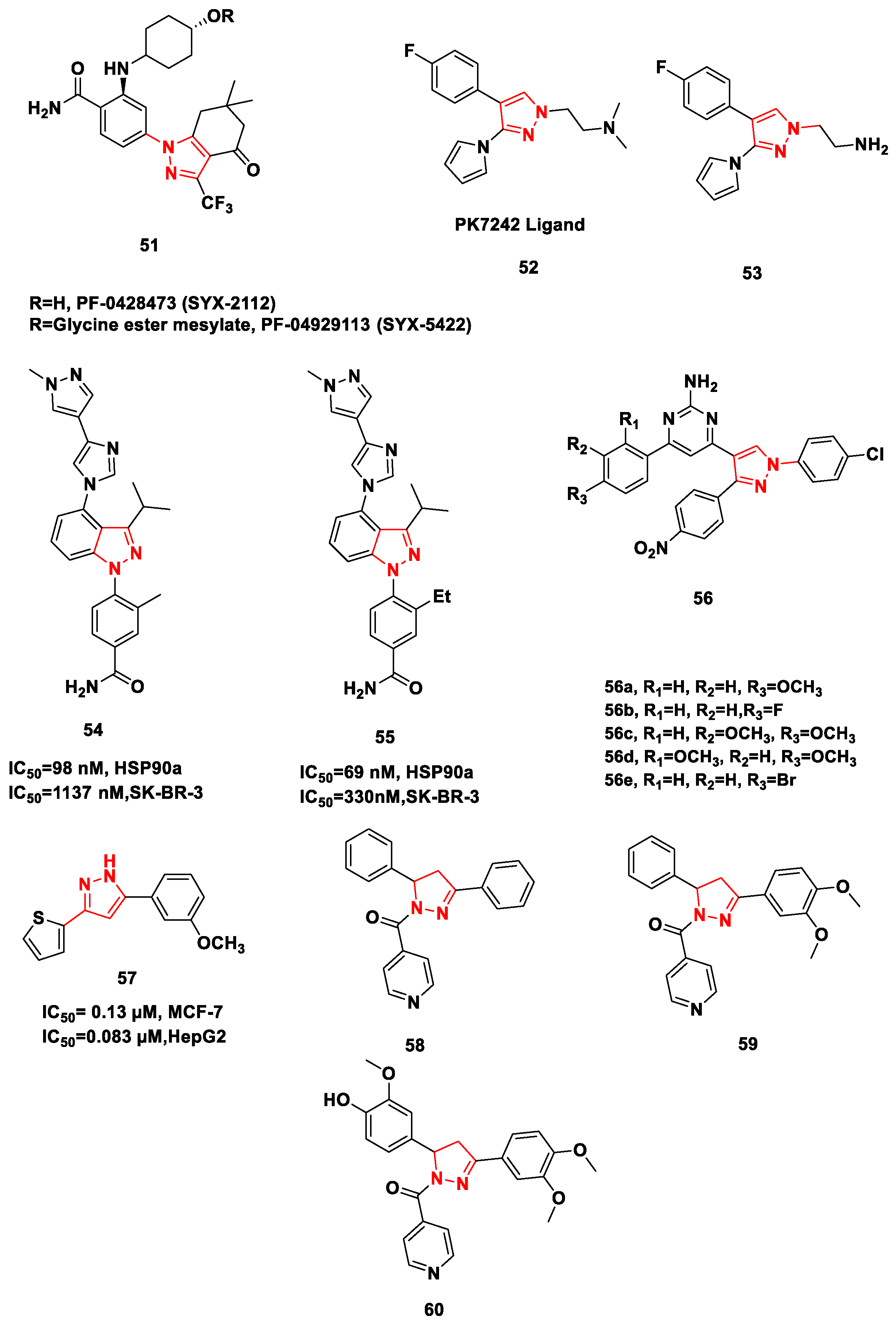
- Uno, T.; Kawai, Y.; Yamashita, S.; Oshiumi, H.; Yoshimura, C.; Mizutani, T.; Kitade, M. Discovery of 3-Ethyl-4-(3-isopropyl-4-(4-(1-methyl-1 H-pyrazol-4-yl)-1 H-imidazol-1-yl)-1 H-pyrazolo [3,4-b] pyridin-1-yl) benzamide (TAS-116) as a Potent, Selective, and Orally Available Hsp90 Inhibitor. J. Med. Chem. 2018, 62, 531–551. [Google Scholar] [CrossRef]
- Mettu, A.; Talla, V.; Bajaj, D.M.; Subhashini, N.J. Design, Synthesis, Molecular Docking Studies of Novel Pyrazolyl 2-Aminopyrimidine Derivatives as Hsp90 Inhibitors. Arch. Pharm. 2019, 352, 1900063. [Google Scholar] [CrossRef] [PubMed]
- Mettu, A.; Talla, V.; Naikal, S.J.P. Novel anticancer Hsp90 inhibitor disubstituted pyrazolyl 2-aminopyrimidine compound 7t induces cell cycle arrest and apoptosis via mitochondrial pathway in MCF-7 cells. Bioorg. Med. Chem. Lett. 2020, 30, 127470. [Google Scholar] [CrossRef] [PubMed]
- Mohamady, S.; Ismail, M.; Mogheith, S.M.; Attia, Y.M.; Taylor, S.D. Discovery of 5-aryl-3-thiophen-2-yl-1H-pyrazoles as a new class of Hsp90 inhibitors in hepatocellular carcinoma. Bioorg. Chem. 2020, 94, 103433. [Google Scholar] [CrossRef] [PubMed]
- Kadasi, S.; Yerroju, R.; Gaddam, S.; Pullanagiri, N.; Chary, M.; Pingili, D.; Raghavendra, N.M. Discovery of N-Pyridoyl-Δ2-pyrazolines as Hsp90 Inhibitors. Arch. Der Pharm. 2020, 353, 190019. [Google Scholar] [CrossRef]
















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ardestani, M.; Khorsandi, Z.; Keshavarzipour, F.; Iravani, S.; Sadeghi-Aliabadi, H.; Varma, R.S. Heterocyclic Compounds as Hsp90 Inhibitors: A Perspective on Anticancer Applications. Pharmaceutics 2022, 14, 2220. https://doi.org/10.3390/pharmaceutics14102220
Ardestani M, Khorsandi Z, Keshavarzipour F, Iravani S, Sadeghi-Aliabadi H, Varma RS. Heterocyclic Compounds as Hsp90 Inhibitors: A Perspective on Anticancer Applications. Pharmaceutics. 2022; 14(10):2220. https://doi.org/10.3390/pharmaceutics14102220
Chicago/Turabian StyleArdestani, Mina, Zahra Khorsandi, Fariba Keshavarzipour, Siavash Iravani, Hojjat Sadeghi-Aliabadi, and Rajender S. Varma. 2022. "Heterocyclic Compounds as Hsp90 Inhibitors: A Perspective on Anticancer Applications" Pharmaceutics 14, no. 10: 2220. https://doi.org/10.3390/pharmaceutics14102220
APA StyleArdestani, M., Khorsandi, Z., Keshavarzipour, F., Iravani, S., Sadeghi-Aliabadi, H., & Varma, R. S. (2022). Heterocyclic Compounds as Hsp90 Inhibitors: A Perspective on Anticancer Applications. Pharmaceutics, 14(10), 2220. https://doi.org/10.3390/pharmaceutics14102220