
D-DOPA Is a Potent, Orally Bioavailable, Allosteric Inhibitor of Glutamate Carboxypeptidase II
 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Chemicals
2.2. GCPII Activity Assay
2.3. Metabolic Stability Assays
2.4. Pharmacokinetic Studies in Mice
2.5. Bioanalysis in Plasma and Brain
2.6. D-DOPA Target Engagement Studies
2.7. D-DOPA Mode of Inhibition Studies
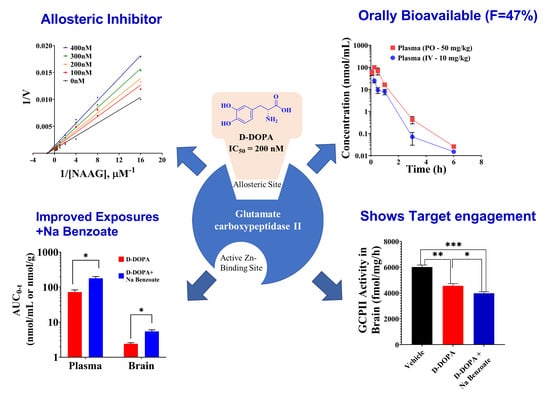
3. Results and Discussion
3.1. IC50 of Catechol-Based Scaffolds
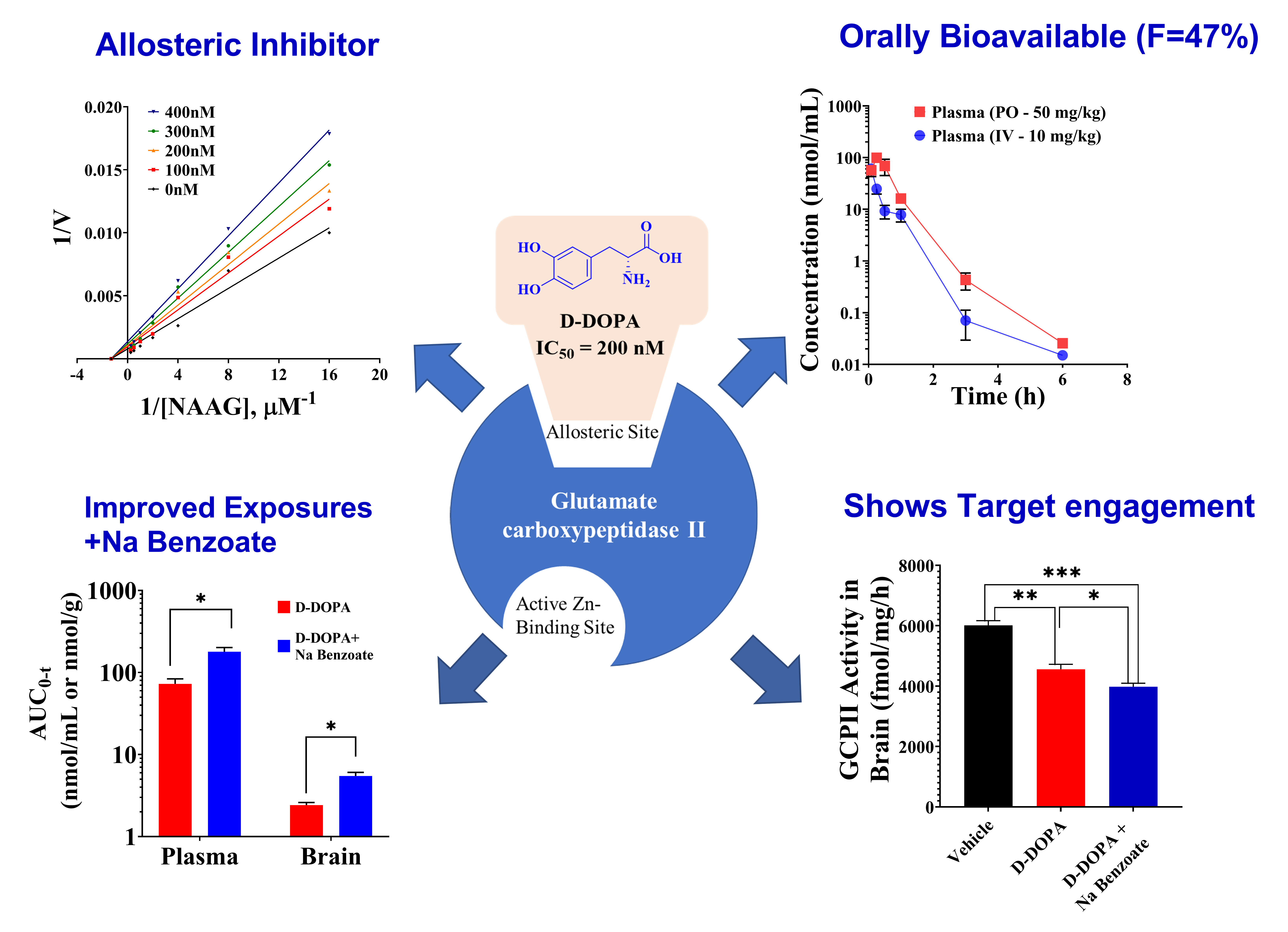
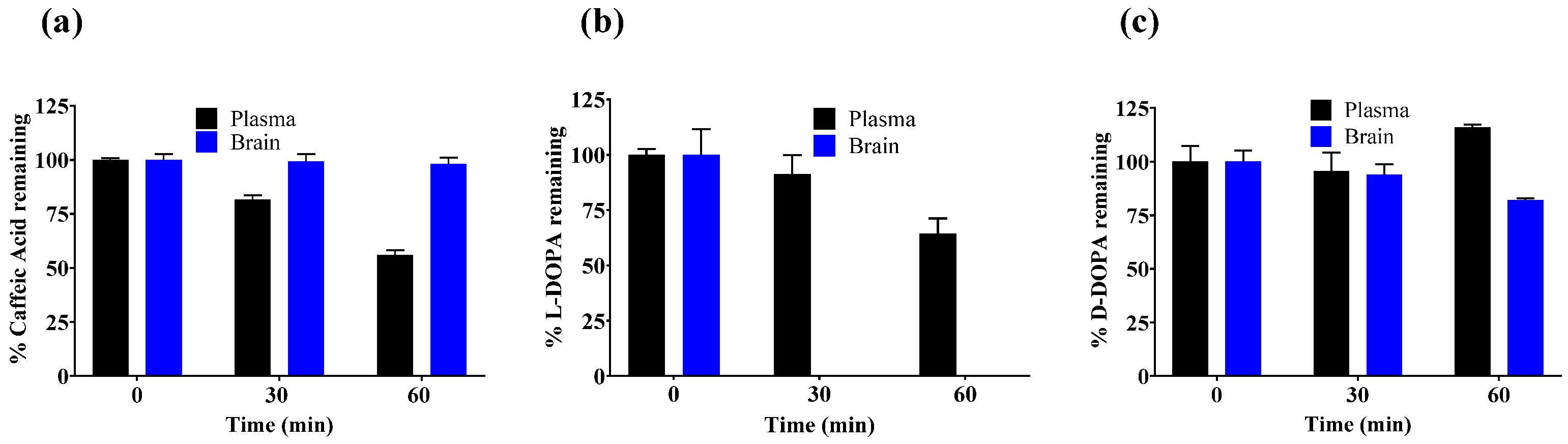
3.2. In Vitro Metabolic Stability of Caffeic Acid, L-DOPA, and D-DOPA
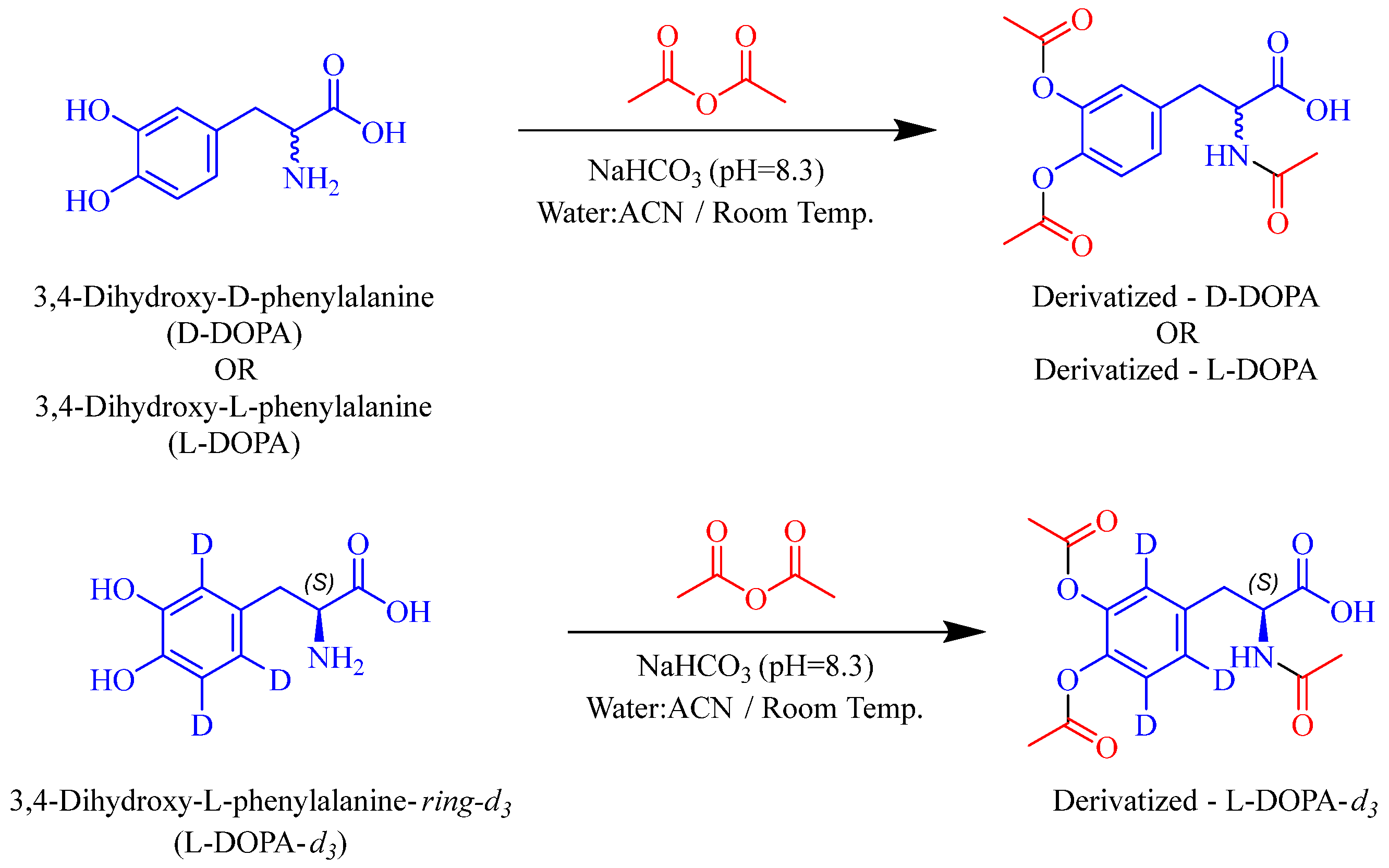
3.3. Bioanalytical Methods for Caffeic Acid, L-DOPA, and D-DOPA
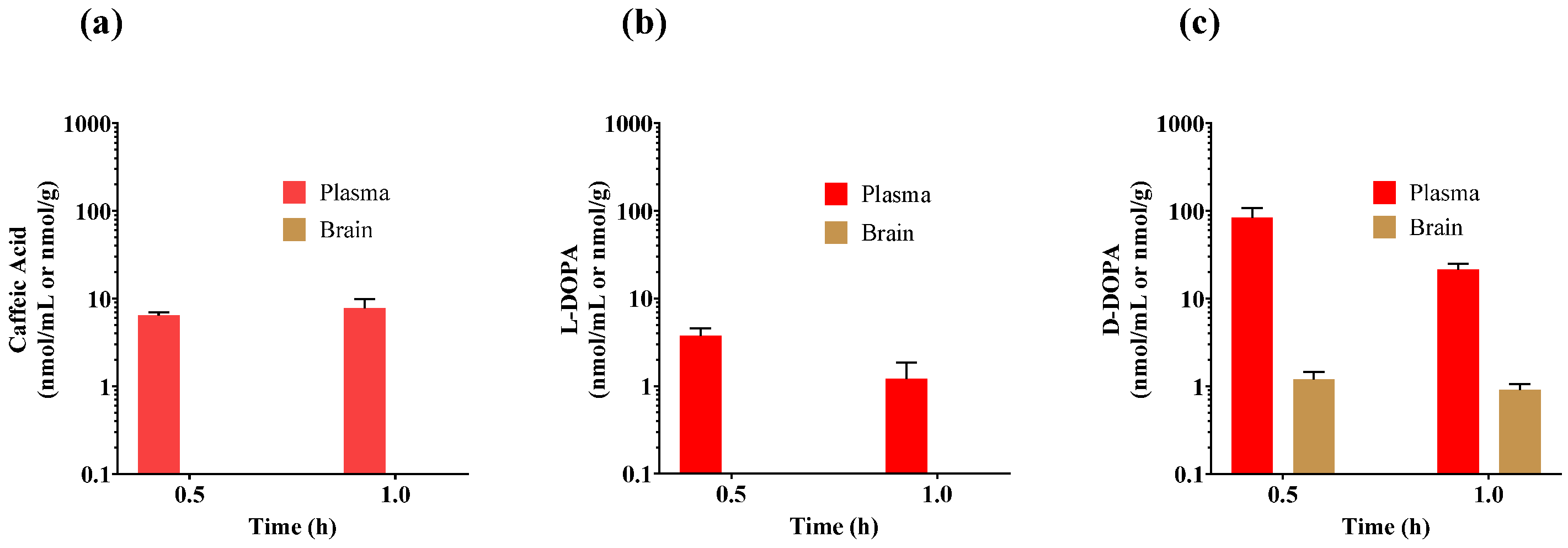
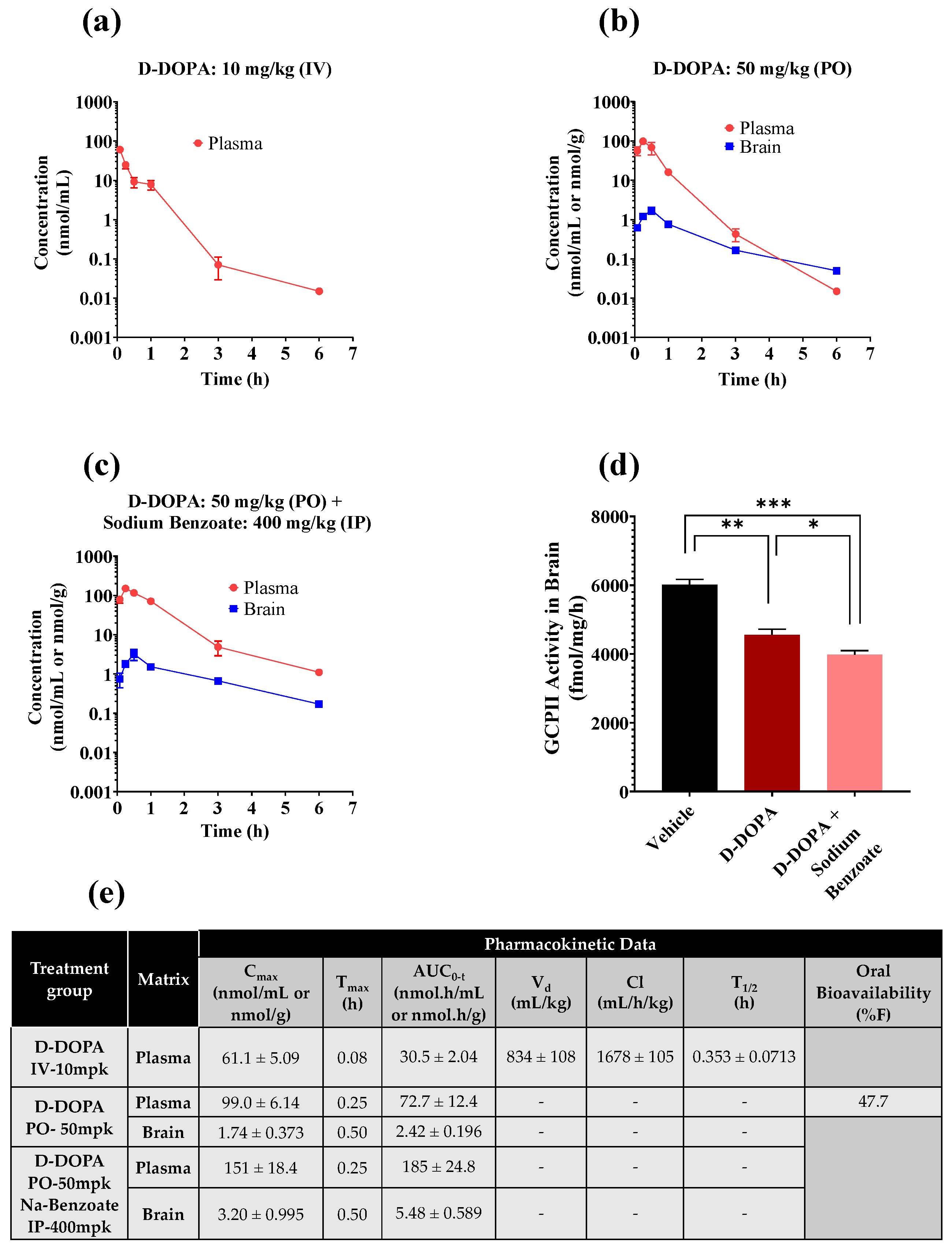
3.4. Initial Pharmacokinetic Studies of Caffeic Acid, L-DOPA, and D-DOPA in Mice
3.5. Pharmacokinetic and Target Engagement Studies of D-DOPA + Sodium Benzoate in Mice
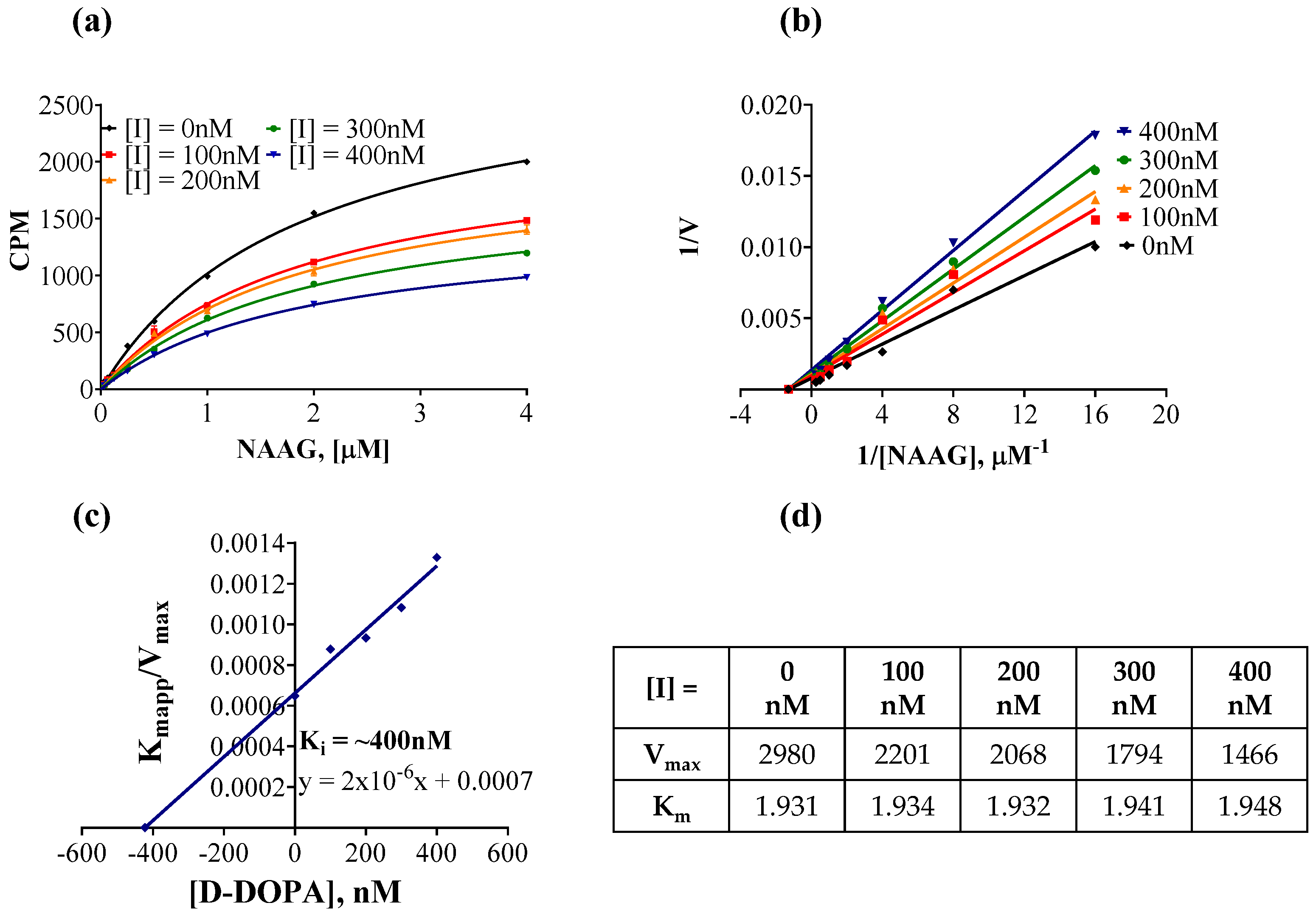
3.6. Characterization of D-DOPA’s Mode of GCPII Inhibition
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- O’Keefe, D.S.; Su, S.L.; Bacich, D.J.; Horiguchi, Y.; Luo, Y.; Powell, C.T.; Zandvliet, D.; Russell, P.J.; Molloy, P.L.; Nowak, N.J.; et al. Mapping, genomic organization and promoter analysis of the human prostate-specific membrane antigen gene. Biochim. Biophys. Acta 1998, 1443, 113–127. [Google Scholar] [CrossRef]
- Robinson, M.B.; Blakely, R.D.; Couto, R.; Coyle, J.T. Hydrolysis of the brain dipeptide N-acetyl-L-aspartyl-L-glutamate. Identification and characterization of a novel N-acetylated alpha-linked acidic dipeptidase activity from rat brain. J. Biol. Chem. 1987, 262, 14498–14506. [Google Scholar] [CrossRef]
- Chang, S.S.; Gaudin, P.B.; Reuter, V.E.; Heston, W.D. Prostate-specific membrane antigen: Present and future applications. Urology 2000, 55, 622–629. [Google Scholar] [CrossRef]
- Pinto, J.T.; Suffoletto, B.P.; Berzin, T.M.; Qiao, C.H.; Lin, S.; Tong, W.P.; May, F.; Mukherjee, B.; Heston, W.D. Prostate-specific membrane antigen: A novel folate hydrolase in human prostatic carcinoma cells. Clin. Cancer Res. 1996, 2, 1445–1451. [Google Scholar]
- Zhao, R.; Matherly, L.H.; Goldman, I.D. Membrane transporters and folate homeostasis: Intestinal absorption and transport into systemic compartments and tissues. Expert Rev. Mol. Med. 2009, 11, e4. [Google Scholar] [CrossRef]
- Barinka, C.; Rojas, C.; Slusher, B.; Pomper, M. Glutamate carboxypeptidase II in diagnosis and treatment of neurologic disorders and prostate cancer. Curr. Med. Chem. 2012, 19, 856–870. [Google Scholar] [CrossRef]
- Guilarte, T.R.; Hammoud, D.A.; McGlothan, J.L.; Caffo, B.S.; Foss, C.A.; Kozikowski, A.P.; Pomper, M.G. Dysregulation of glutamate carboxypeptidase II in psychiatric disease. Schizophr. Res. 2008, 99, 324–332. [Google Scholar] [CrossRef]
- Yang, S.; Datta, D.; Elizabeth, W.; Duque, A.; Morozov, Y.M.; Arellano, J.; Slusher, B.S.; Wang, M.; Arnsten, A.F.T. Inhibition of glutamate-carboxypeptidase-II in dorsolateral prefrontal cortex: Potential therapeutic target for neuroinflammatory cognitive disorders. Mol. Psychiatry 2022, 1–12. [Google Scholar] [CrossRef]
- Ferraris, D.V.; Shukla, K.; Tsukamoto, T. Structure-activity relationships of glutamate carboxypeptidase II (GCPII) inhibitors. Curr. Med. Chem. 2012, 19, 1282–1294. [Google Scholar] [CrossRef]
- Schmidt, L.H.; Heitkotter, B.; Schulze, A.B.; Schliemann, C.; Steinestel, K.; Trautmann, M.; Marra, A.; Hillejan, L.; Mohr, M.; Evers, G.; et al. Prostate specific membrane antigen (PSMA) expression in non-small cell lung cancer. PLoS ONE 2017, 12, e0186280. [Google Scholar] [CrossRef]
- Conway, R.E.; Petrovic, N.; Li, Z.; Heston, W.; Wu, D.; Shapiro, L.H. Prostate-specific membrane antigen regulates angiogenesis by modulating integrin signal transduction. Mol. Cell Biol. 2006, 26, 5310–5324. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Song, B.; Zhu, W.; Xu, X.; Gong, Q.Q.; Morando, C.; Dassopoulos, T.; Newberry, R.D.; Hunt, S.R.; Li, E. An ileal Crohn’s disease gene signature based on whole human genome expression profiles of disease unaffected ileal mucosal biopsies. PLoS ONE 2012, 7, e37139. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shachar, S.; Yanai, H.; Baram, L.; Elad, H.; Meirovithz, E.; Ofer, A.; Brazowski, E.; Tulchinsky, H.; Pasmanik-Chor, M.; Dotan, I. Gene expression profiles of ileal inflammatory bowel disease correlate with disease phenotype and advance understanding of its immunopathogenesis. Inflamm Bowel. Dis. 2013, 19, 2509–2521. [Google Scholar] [CrossRef] [PubMed]
- Noble, C.L.; Abbas, A.R.; Lees, C.W.; Cornelius, J.; Toy, K.; Modrusan, Z.; Clark, H.F.; Arnott, I.D.; Penman, I.D.; Satsangi, J.; et al. Characterization of intestinal gene expression profiles in Crohn’s disease by genome-wide microarray analysis. Inflamm Bowel. Dis. 2010, 16, 1717–1728. [Google Scholar] [CrossRef]
- Rais, R.; Jiang, W.; Zhai, H.; Wozniak, K.M.; Stathis, M.; Hollinger, K.R.; Thomas, A.G.; Rojas, C.; Vornov, J.J.; Marohn, M.; et al. FOLH1/GCPII is elevated in IBD patients, and its inhibition ameliorates murine IBD abnormalities. JCI Insight 2016, 1. [Google Scholar] [CrossRef]
- Evans, J.C.; Malhotra, M.; Cryan, J.F.; O’Driscoll, C.M. The therapeutic and diagnostic potential of the prostate specific membrane antigen/glutamate carboxypeptidase II (PSMA/GCPII) in cancer and neurological disease. Br. J. Pharmacol. 2016, 173, 3041–3079. [Google Scholar] [CrossRef]
- Vornov, J.J.; Hollinger, K.R.; Jackson, P.F.; Wozniak, K.M.; Farah, M.H.; Majer, P.; Rais, R.; Slusher, B.S. Chapter Nine-Still NAAG’ing After All These Years: The Continuing Pursuit of GCPII Inhibitors. In Advances in Pharmacology; Schwarcz, R., Ed.; Academic Press: Cambridge, MA, USA, 2016; Volume 76, pp. 215–255. [Google Scholar]
- Majer, P.; Jancarik, A.; Krecmerova, M.; Tichy, T.; Tenora, L.; Wozniak, K.; Wu, Y.; Pommier, E.; Ferraris, D.; Rais, R.; et al. Discovery of Orally Available Prodrugs of the Glutamate Carboxypeptidase II (GCPII) Inhibitor 2-Phosphonomethylpentanedioic Acid (2-PMPA). J. Med. Chem. 2016, 59, 2810–2819. [Google Scholar] [CrossRef]
- Ferraris, D.V.; Majer, P.; Ni, C.; Slusher, C.E.; Rais, R.; Wu, Y.; Wozniak, K.M.; Alt, J.; Rojas, C.; Slusher, B.S.; et al. delta-Thiolactones as prodrugs of thiol-based glutamate carboxypeptidase II (GCPII) inhibitors. J. Med. Chem. 2014, 57, 243–247. [Google Scholar] [CrossRef]
- Rais, R.; Vavra, J.; Tichy, T.; Dash, R.P.; Gadiano, A.J.; Tenora, L.; Monincova, L.; Barinka, C.; Alt, J.; Zimmermann, S.C.; et al. Discovery of a para-Acetoxy-benzyl Ester Prodrug of a Hydroxamate-Based Glutamate Carboxypeptidase II Inhibitor as Oral Therapy for Neuropathic Pain. J. Med. Chem. 2017, 60, 7799–7809. [Google Scholar] [CrossRef]
- Dash, R.P.; Tichy, T.; Veeravalli, V.; Lam, J.; Alt, J.; Wu, Y.; Tenora, L.; Majer, P.; Slusher, B.S.; Rais, R. Enhanced Oral Bioavailability of 2-(Phosphonomethyl)-pentanedioic Acid (2-PMPA) from its (5-Methyl-2-oxo-1,3-dioxol-4-yl)methyl (ODOL)-Based Prodrugs. Mol. Pharm. 2019, 16, 4292–4301. [Google Scholar] [CrossRef]
- Nedelcovych, M.; Dash, R.P.; Tenora, L.; Zimmermann, S.C.; Gadiano, A.J.; Garrett, C.; Alt, J.; Hollinger, K.R.; Pommier, E.; Jancarik, A.; et al. Enhanced Brain Delivery of 2-(Phosphonomethyl)pentanedioic Acid Following Intranasal Administration of Its gamma-Substituted Ester Prodrugs. Mol. Pharm. 2017, 14, 3248–3257. [Google Scholar] [CrossRef]
- Rais, R.; Wozniak, K.; Wu, Y.; Niwa, M.; Stathis, M.; Alt, J.; Giroux, M.; Sawa, A.; Rojas, C.; Slusher, B.S. Selective CNS Uptake of the GCP-II Inhibitor 2-PMPA following Intranasal Administration. PLoS ONE 2015, 10, e0131861. [Google Scholar] [CrossRef]
- Hollinger, K.R.; Sharma, A.; Tallon, C.; Lovell, L.; Thomas, A.G.; Zhu, X.; Wiseman, R.; Wu, Y.; Kambhampati, S.P.; Liaw, K.; et al. Dendrimer-2PMPA selectively blocks upregulated microglial GCPII activity and improves cognition in a mouse model of multiple sclerosis. Nanotheranostics 2022, 6, 126–142. [Google Scholar] [CrossRef]
- Arteaga Cabeza, O.; Zhang, Z.; Smith Khoury, E.; Sheldon, R.A.; Sharma, A.; Zhang, F.; Slusher, B.S.; Kannan, R.M.; Kannan, S.; Ferriero, D.M. Neuroprotective effects of a dendrimer-based glutamate carboxypeptidase inhibitor on superoxide dismutase transgenic mice after neonatal hypoxic-ischemic brain injury. Neurobiol Dis. 2021, 148, 105201. [Google Scholar] [CrossRef]
- Veldkamp, K.L.; Tubergen, P.J.; Swartz, M.A.; DeVries, J.T.; Tatko, C.D. Zinc binding with L-dopa peptides. Inorg. Chim. Acta 2017, 461, 120–126. [Google Scholar] [CrossRef]
- Rahman, F.; Nguyen, T.M.; Adekoya, O.A.; Campestre, C.; Tortorella, P.; Sylte, I.; Winberg, J.O. Inhibition of bacterial and human zinc-metalloproteases by bisphosphonate- and catechol-containing compounds. J. Enzym. Inhib. Med. Chem. 2021, 36, 819–830. [Google Scholar] [CrossRef]
- Kyriakou, S.; Mitsiogianni, M.; Mantso, T.; Cheung, W.; Todryk, S.; Veuger, S.; Pappa, A.; Tetard, D.; Panayiotidis, M.I. Anticancer activity of a novel methylated analogue of L-mimosine against an in vitro model of human malignant melanoma. Invest. New Drugs 2020, 38, 621–633. [Google Scholar] [CrossRef]
- Shchepin, R.; Moller, M.N.; Kim, H.Y.; Hatch, D.M.; Bartesaghi, S.; Kalyanaraman, B.; Radi, R.; Porter, N.A. Tyrosine-lipid peroxide adducts from radical termination: Para coupling and intramolecular Diels-Alder cyclization. J. Am. Chem. Soc. 2010, 132, 17490–17500. [Google Scholar] [CrossRef]
- Rojas, C.; Frazier, S.T.; Flanary, J.; Slusher, B.S. Kinetics and inhibition of glutamate carboxypeptidase II using a microplate assay. Anal. Biochem. 2002, 310, 50–54. [Google Scholar] [CrossRef]
- Zimmermann, S.C.; Tichy, T.; Vavra, J.; Dash, R.P.; Slusher, C.E.; Gadiano, A.J.; Wu, Y.; Jancarik, A.; Tenora, L.; Monincova, L.; et al. N-Substituted Prodrugs of Mebendazole Provide Improved Aqueous Solubility and Oral Bioavailability in Mice and Dogs. J. Med. Chem. 2018, 61, 3918–3929. [Google Scholar] [CrossRef]
- van Faassen, M.; Bischoff, R.; Eijkelenkamp, K.; de Jong, W.H.A.; van der Ley, C.P.; Kema, I.P. In Matrix Derivatization Combined with LC-MS/MS Results in Ultrasensitive Quantification of Plasma Free Metanephrines and Catecholamines. Anal. Chem. 2020, 92, 9072–9078. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, W.; Ma, X.; Chu, Y.; Li, S.; Guo, J.; Jia, Y.; Zhou, S.; Zhu, Y.; Liu, C. Simultaneous determination of caffeic acid and its major pharmacologically active metabolites in rat plasma by LC-MS/MS and its application in pharmacokinetic study. Biomed. Chromatogr. 2015, 29, 552–559. [Google Scholar] [CrossRef]
- Rahn, K.A.; Watkins, C.C.; Alt, J.; Rais, R.; Stathis, M.; Grishkan, I.; Crainiceau, C.M.; Pomper, M.G.; Rojas, C.; Pletnikov, M.V.; et al. Inhibition of glutamate carboxypeptidase II (GCPII) activity as a treatment for cognitive impairment in multiple sclerosis. Proc. Natl. Acad. Sci. USA 2012, 109, 20101–20106. [Google Scholar] [CrossRef]
- Maruyama, W.; Naoi, M.; Narabayashi, H. The metabolism of L-DOPA and L-threo-3,4-dihydroxyphenylserine and their effects on monoamines in the human brain: Analysis of the intraventricular fluid from parkinsonian patients. J. Neurol. Sci. 1996, 139, 141–148. [Google Scholar] [CrossRef]
- Meiser, J.; Weindl, D.; Hiller, K. Complexity of dopamine metabolism. Cell Commun. Signal. 2013, 11, 34. [Google Scholar] [CrossRef] [PubMed]
- Peaston, R.T.; Weinkove, C. Measurement of catecholamines and their metabolites. Ann. Clin. Biochem. 2004, 41, 17–38. [Google Scholar] [CrossRef]
- Li, W.L.; Rossi, D.T.; Fountain, S.T. Development and validation of a semi-automated method for L-dopa and dopamine in rat plasma using electrospray LC/MS/MS. J. Pharm. Biomed. 2000, 24, 325–333. [Google Scholar] [CrossRef]
- Igarashi, K.; Hotta, K.; Kasuya, F.; Abe, K.; Sakoda, S. Determination of cabergoline and L-dopa in human plasma using liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2003, 792, 55–61. [Google Scholar] [CrossRef]
- Zhou, Y.Z.; Alany, R.G.; Chuang, V.; Wen, J.Y. Studies of the Rate Constant of L-DOPA Oxidation and Decarboxylation by HPLC. Chromatographia 2012, 75, 597–606. [Google Scholar] [CrossRef]
- Cho, Y.A.; Park, S.; Seo, O.N.; Jeong, S.W.; Lee, W.-K.; Kim, C.Y.; Kim, S.T.; Cho, M.J.; Shin, S.C. Development and validation of an LC–ESI–MS/MS method for simultaneous determination of levodopa, dopamine, L-α-methyldopa and 3-O-methyldopa in rat plasma. J. Pharm. Investig. 2012, 42, 361–368. [Google Scholar] [CrossRef]
- Lv, L.; Jiang, W.Z.; Zhou, S.Y.; Huang, X.Z.; Shi, X.X.; Lv, C.; Wu, L.L.; Xu, C.Y. LC-MS-MS Simultaneous Determination of l-Dopa and Its Prodrug l-Dopa n-Pentyl Ester Hydrochloride in Rat Plasma. Chromatographia 2010, 72, 239–243. [Google Scholar] [CrossRef]
- Wang, S.J.; Zeng, J.; Yang, B.K.; Zhong, Y.M. Bioavailability of caffeic acid in rats and its absorption properties in the Caco-2 cell model. Pharm. Biol. 2014, 52, 1150–1157. [Google Scholar] [CrossRef] [PubMed]
- de Vries, M.H.; Hamelijnck, M.A.; Hofman, G.A.; Koster, A.S.; Noordhoek, J. Decarboxylation of L-dopa in the rat isolated vascularly perfused small intestine: Contribution to systemic elimination and dose-dependent first pass effect. J. Pharm. Pharmacol. 1992, 44, 311–314. [Google Scholar] [CrossRef]
- Vieira-Coelho, M.A.; Soares-da-Silva, P. Dopamine formation, from its immediate precursor 3,4-dihydroxyphenylalanine, along the rat digestive tract. Fundam. Clin. Pharmacol. 1993, 7, 235–243. [Google Scholar] [CrossRef]
- Muller, T. Pharmacokinetics and pharmacodynamics of levodopa/carbidopa cotherapies for Parkinson’s disease. Expert Opin. Drug Metab. Toxicol. 2020, 16, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.; Jenner, P.; Marsden, C.D. Peripheral pharmacokinetic handling and metabolism of L-dopa in the rat: The effect of route of administration and carbidopa pretreatment. J. Pharm. Pharmacol. 1991, 43, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Diederich, C.; Milakofsky, L.; Hare, T.A.; Hofford, J.M.; Dadmarz, M.; Vogel, W.H. Effects of L-DOPA/carbidopa administration on the levels of L-DOPA, other amino acids and related compounds in the plasma, brain and heart of the rat. Pharmacology 1997, 55, 109–116. [Google Scholar] [CrossRef]
- Muddapu, V.R.; Vijayakumar, K.; Ramakrishnan, K.; Chakravarthy, V.S. A Multi-Scale Computational Model of Levodopa-Induced Toxicity in Parkinson’s Disease. Front. Neurosci. 2022, 16, 797127. [Google Scholar] [CrossRef]
- Asanuma, M.; Miyazaki, I.; Ogawa, N. Dopamine- or L-DOPA-induced neurotoxicity: The role of dopamine quinone formation and tyrosinase in a model of Parkinson’s disease. Neurotox. Res. 2003, 5, 165–176. [Google Scholar] [CrossRef]
- Alexander, T.; Sortwell, C.E.; Sladek, C.D.; Roth, R.H.; Steece-Collier, K. Comparison of neurotoxicity following repeated administration of l-dopa, d-dopa and dopamine to embryonic mesencephalic dopamine neurons in cultures derived from Fisher 344 and Sprague-Dawley donors. Cell Transplant. 1997, 6, 309–315. [Google Scholar] [CrossRef]
- Abbott, A. Levodopa: The story so far. Nature 2010, 466, S6–S7. [Google Scholar] [CrossRef] [PubMed]
- Hoon, M.; Petzer, J.P.; Viljoen, F.; Petzer, A. The Design and Evaluation of an l-Dopa-Lazabemide Prodrug for the Treatment of Parkinson’s Disease. Molecules 2017, 22, 2076. [Google Scholar] [CrossRef] [PubMed]
- Moses, J.; Siddiqui, A.; Silverman, P.B. Sodium benzoate differentially blocks circling induced by D-and L-dopa in the hemi-parkinsonian rat. Neurosci. Lett. 1996, 218, 145–148. [Google Scholar] [CrossRef]
- Wu, M.; Zhou, X.J.; Konno, R.; Wang, Y.X. D-dopa is unidirectionally converted to L-dopa by D-amino acid oxidase, followed by dopa transaminase. Clin. Exp. Pharmacol. Physiol. 2006, 33, 1042–1046. [Google Scholar] [CrossRef] [PubMed]
- Karoum, F.; Freed, W.J.; Chuang, L.W.; Cannon-Spoor, E.; Wyatt, R.J.; Costa, E. D-dopa and L-dopa similarly elevate brain dopamine and produce turning behavior in rats. Brain Res. 1988, 440, 190–194. [Google Scholar] [CrossRef]
- Rais, R.; Thomas, A.G.; Wozniak, K.; Wu, Y.; Jaaro-Peled, H.; Sawa, A.; Strick, C.A.; Engle, S.J.; Brandon, N.J.; Rojas, C.; et al. Pharmacokinetics of oral D-serine in D-amino acid oxidase knockout mice. Drug Metab. Dispos. 2012, 40, 2067–2073. [Google Scholar] [CrossRef]
- Ferraris, D.; Duvall, B.; Ko, Y.S.; Thomas, A.G.; Rojas, C.; Majer, P.; Hashimoto, K.; Tsukamoto, T. Synthesis and biological evaluation of D-amino acid oxidase inhibitors. J. Med. Chem. 2008, 51, 3357–3359. [Google Scholar] [CrossRef]
- van der Post, J.P.; de Visser, S.J.; de Kam, M.L.; Woelfler, M.; Hilt, D.C.; Vornov, J.; Burak, E.S.; Bortey, E.; Slusher, B.S.; Limsakun, T.; et al. The central nervous system effects, pharmacokinetics and safety of the NAALADase-inhibitor GPI 5693. Br. J. Clin. Pharmacol. 2005, 60, 128–136. [Google Scholar] [CrossRef]
- Vornov, J.J.; Wozniak, K.M.; Wu, Y.; Rojas, C.; Rais, R.; Slusher, B.S. Pharmacokinetics and pharmacodynamics of the glutamate carboxypeptidase II inhibitor 2-MPPA show prolonged alleviation of neuropathic pain through an indirect mechanism. J. Pharmacol. Exp. Ther. 2013, 346, 406–413. [Google Scholar] [CrossRef]
- Rais, R.; Hoover, R.; Wozniak, K.; Rudek, M.A.; Tsukamoto, T.; Alt, J.; Rojas, C.; Slusher, B.S. Reversible disulfide formation of the glutamate carboxypeptidase II inhibitor E2072 results in prolonged systemic exposures in vivo. Drug Metab. Dispos. 2012, 40, 2315–2323. [Google Scholar] [CrossRef]
- Sala, M.; Hollinger, K.R.; Thomas, A.G.; Dash, R.P.; Tallon, C.; Veeravalli, V.; Lovell, L.; Kogler, M.; Hrebabecky, H.; Prochazkova, E.; et al. Novel Human Neutral Sphingomyelinase 2 Inhibitors as Potential Therapeutics for Alzheimer’s Disease. J. Med. Chem. 2020, 63, 6028–6056. [Google Scholar] [CrossRef] [PubMed]
- Jackson, P.F.; Tays, K.L.; Maclin, K.M.; Ko, Y.S.; Li, W.; Vitharana, D.; Tsukamoto, T.; Stoermer, D.; Lu, X.C.; Wozniak, K.; et al. Design and pharmacological activity of phosphinic acid based NAALADase inhibitors. J. Med. Chem. 2001, 44, 4170–4175. [Google Scholar] [CrossRef]
- Jackson, P.F.; Cole, D.C.; Slusher, B.S.; Stetz, S.L.; Ross, L.E.; Donzanti, B.A.; Trainor, D.A. Design, synthesis, and biological activity of a potent inhibitor of the neuropeptidase N-acetylated alpha-linked acidic dipeptidase. J. Med. Chem. 1996, 39, 619–622. [Google Scholar] [CrossRef] [PubMed]
- Majer, P.; Jackson, P.F.; Delahanty, G.; Grella, B.S.; Ko, Y.S.; Li, W.; Liu, Q.; Maclin, K.M.; Polakova, J.; Shaffer, K.A.; et al. Synthesis and biological evaluation of thiol-based inhibitors of glutamate carboxypeptidase II: Discovery of an orally active GCP II inhibitor. J. Med. Chem. 2003, 46, 1989–1996. [Google Scholar] [CrossRef] [PubMed]
- Kozikowski, A.P.; Zhang, J.; Nan, F.; Petukhov, P.A.; Grajkowska, E.; Wroblewski, J.T.; Yamamoto, T.; Bzdega, T.; Wroblewska, B.; Neale, J.H. Synthesis of urea-based inhibitors as active site probes of glutamate carboxypeptidase II: Efficacy as analgesic agents. J. Med. Chem. 2004, 47, 1729–1738. [Google Scholar] [CrossRef] [PubMed]
- Stoermer, D.; Liu, Q.; Hall, M.R.; Flanary, J.M.; Thomas, A.G.; Rojas, C.; Slusher, B.S.; Tsukamoto, T. Synthesis and biological evaluation of hydroxamate-Based inhibitors of glutamate carboxypeptidase II. Bioorg Med. Chem. Lett. 2003, 13, 2097–2100. [Google Scholar] [CrossRef]
- Parellada, J.; Suarez, G.; Guinea, M. Inhibition of zinc metallopeptidases by flavonoids and related phenolic compounds: Structure-activity relationships. J. Enzym. Inhib. 1998, 13, 347–359. [Google Scholar] [CrossRef]
- Rahn, K.A.; Slusher, B.S.; Kaplin, A.I. Glutamate in CNS neurodegeneration and cognition and its regulation by GCPII inhibition. Curr Med. Chem. 2012, 19, 1335–1345. [Google Scholar] [CrossRef]
- Slusher, B.S.; Vornov, J.J.; Thomas, A.G.; Hurn, P.D.; Harukuni, I.; Bhardwaj, A.; Traystman, R.J.; Robinson, M.B.; Britton, P.; Lu, X.C.; et al. Selective inhibition of NAALADase, which converts NAAG to glutamate, reduces ischemic brain injury. Nat. Med. 1999, 5, 1396–1402. [Google Scholar] [CrossRef]
- Yamamoto, T.; Nozaki-Taguchi, N.; Sakashita, Y.; Inagaki, T. Inhibition of spinal N-acetylated-alpha-linked acidic dipeptidase produces an antinociceptive effect in the rat formalin test. Neuroscience 2001, 102, 473–479. [Google Scholar] [CrossRef]
- Yamamoto, T.; Nozaki-Taguchi, N.; Sakashita, Y. Spinal N-acetyl-alpha-linked acidic dipeptidase (NAALADase) inhibition attenuates mechanical allodynia induced by paw carrageenan injection in the rat. Brain Res. 2001, 909, 138–144. [Google Scholar] [CrossRef]
- Chen, S.R.; Wozniak, K.M.; Slusher, B.S.; Pan, H.L. Effect of 2-(phosphono-methyl)-pentanedioic acid on allodynia and afferent ectopic discharges in a rat model of neuropathic pain. J. Pharmacol. Exp. Ther. 2002, 300, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, K.J.; Sen, S.; Matthews, E.A.; Flatters, S.L.; Wozniak, K.M.; Slusher, B.S.; Dickenson, A.H. Effects of GCP-II inhibition on responses of dorsal horn neurones after inflammation and neuropathy: An electrophysiological study in the rat. Neuropeptides 2003, 37, 298–306. [Google Scholar] [CrossRef]
- Ghadge, G.D.; Slusher, B.S.; Bodner, A.; Canto, M.D.; Wozniak, K.; Thomas, A.G.; Rojas, C.; Tsukamoto, T.; Majer, P.; Miller, R.J.; et al. Glutamate carboxypeptidase II inhibition protects motor neurons from death in familial amyotrophic lateral sclerosis models. Proc. Natl. Acad Sci. USA 2003, 100, 9554–9559. [Google Scholar] [CrossRef] [PubMed]
- Ghose, S.; Chin, R.; Gallegos, A.; Roberts, R.; Coyle, J.; Tamminga, C. Localization of NAAG-related gene expression deficits to the anterior hippocampus in schizophrenia. Schizophr. Res. 2009, 111, 131–137. [Google Scholar] [CrossRef][Green Version]
- Vornov, J.J.; Peters, D.; Nedelcovych, M.; Hollinger, K.; Rais, R.; Slusher, B.S. Looking for Drugs in All the Wrong Places: Use of GCPII Inhibitors Outside the Brain. Neurochem. Res. 2020, 45, 1256–1267. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Structure | IC50 (μM) | Compound | Structure | IC50 (μM) |
|---|---|---|---|---|---|
| 1 (L-DOPA) |  | 0.6 | 6 |  | 20 |
| 2 (Tyr) |  | 100 | 7 (Dopamine) |  | >100 |
| 3 |  | >100 | 8 |  | 4 |
| 4 |  | >100 | 9 (Caffeic Acid) |  | 0.3 |
| 5 |  | >100 | 10 (D-DOPA) |  | 0.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gori, S.S.; Thomas, A.G.; Pal, A.; Wiseman, R.; Ferraris, D.V.; Gao, R.-d.; Wu, Y.; Alt, J.; Tsukamoto, T.; Slusher, B.S.; et al. D-DOPA Is a Potent, Orally Bioavailable, Allosteric Inhibitor of Glutamate Carboxypeptidase II. Pharmaceutics 2022, 14, 2018. https://doi.org/10.3390/pharmaceutics14102018
Gori SS, Thomas AG, Pal A, Wiseman R, Ferraris DV, Gao R-d, Wu Y, Alt J, Tsukamoto T, Slusher BS, et al. D-DOPA Is a Potent, Orally Bioavailable, Allosteric Inhibitor of Glutamate Carboxypeptidase II. Pharmaceutics. 2022; 14(10):2018. https://doi.org/10.3390/pharmaceutics14102018
Chicago/Turabian StyleGori, Sadakatali S., Ajit G. Thomas, Arindom Pal, Robyn Wiseman, Dana V. Ferraris, Run-duo Gao, Ying Wu, Jesse Alt, Takashi Tsukamoto, Barbara S. Slusher, and et al. 2022. "D-DOPA Is a Potent, Orally Bioavailable, Allosteric Inhibitor of Glutamate Carboxypeptidase II" Pharmaceutics 14, no. 10: 2018. https://doi.org/10.3390/pharmaceutics14102018
APA StyleGori, S. S., Thomas, A. G., Pal, A., Wiseman, R., Ferraris, D. V., Gao, R.-d., Wu, Y., Alt, J., Tsukamoto, T., Slusher, B. S., & Rais, R. (2022). D-DOPA Is a Potent, Orally Bioavailable, Allosteric Inhibitor of Glutamate Carboxypeptidase II. Pharmaceutics, 14(10), 2018. https://doi.org/10.3390/pharmaceutics14102018