
Improved Release of a Drug with Poor Water Solubility by Using Electrospun Water-Soluble Polymers as Carriers


Abstract
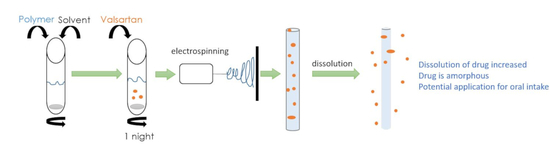
:
1. Introduction
2. Experimental
2.1. Materials
2.2. Solutions
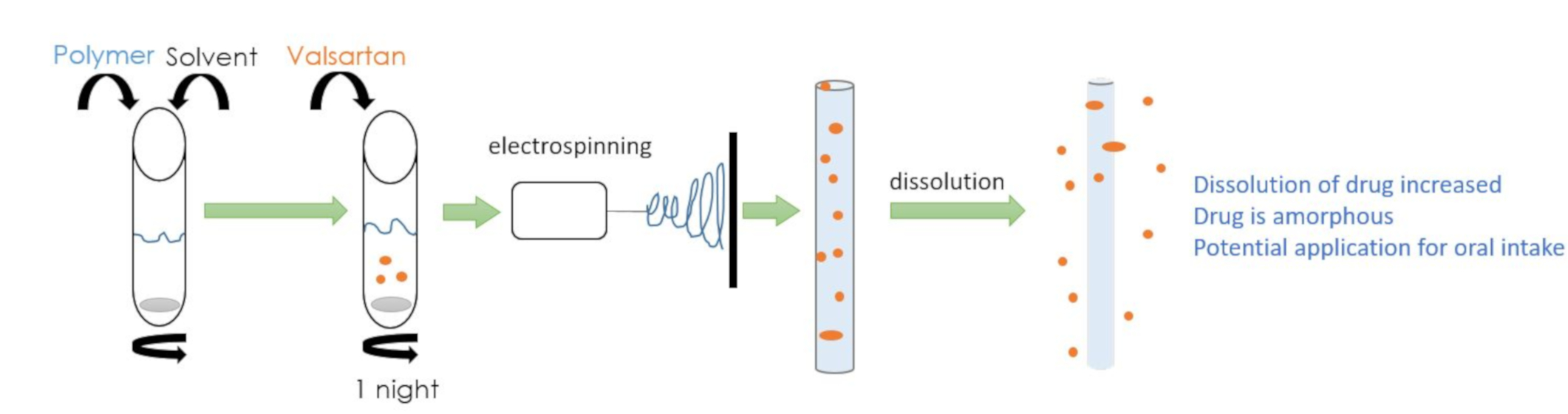
2.3. Electrospinning
2.4. Release Experiments
2.5. Characterization
3. Results and Discussion
3.1. Fiber Spinning, Parameters
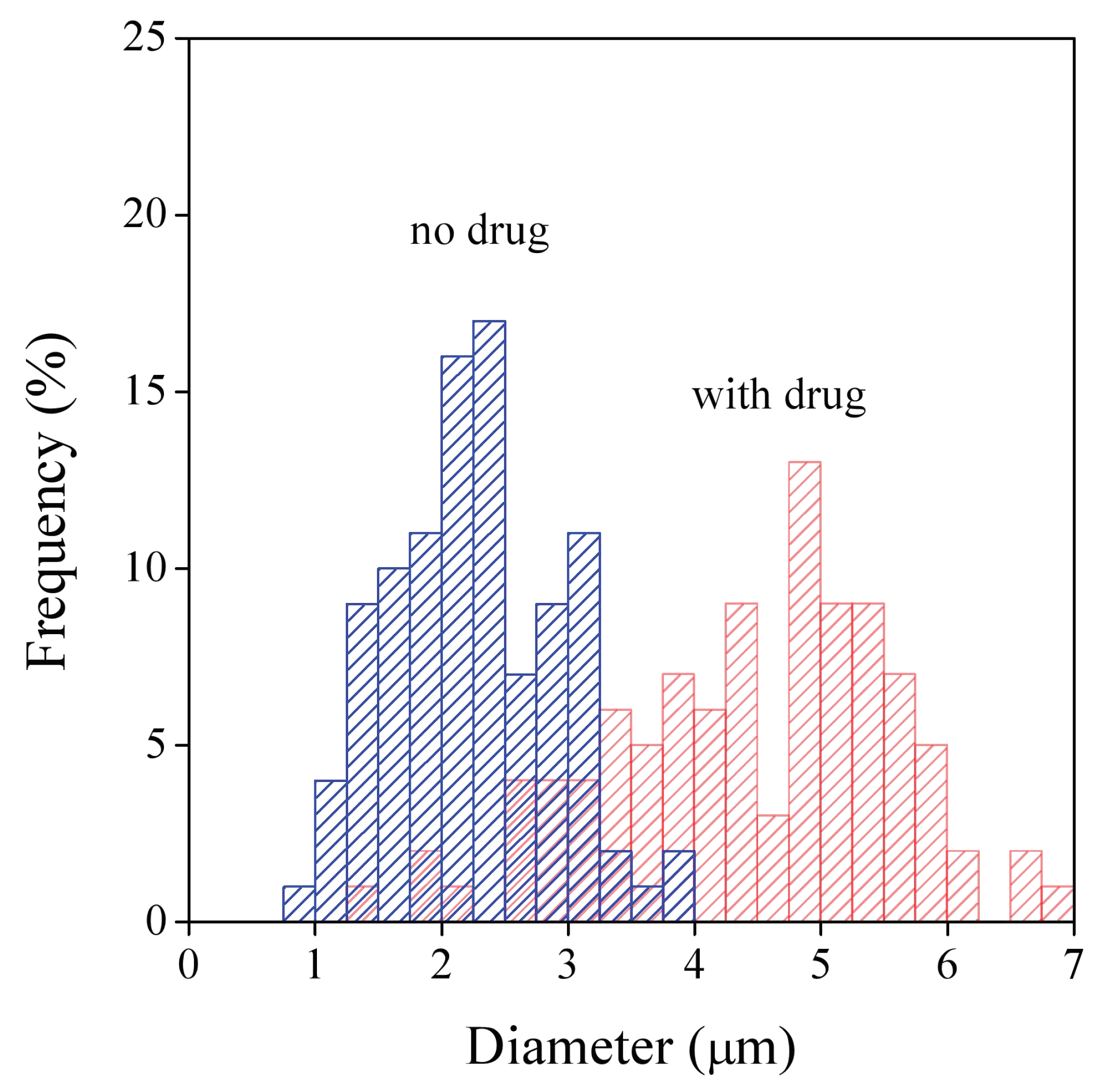
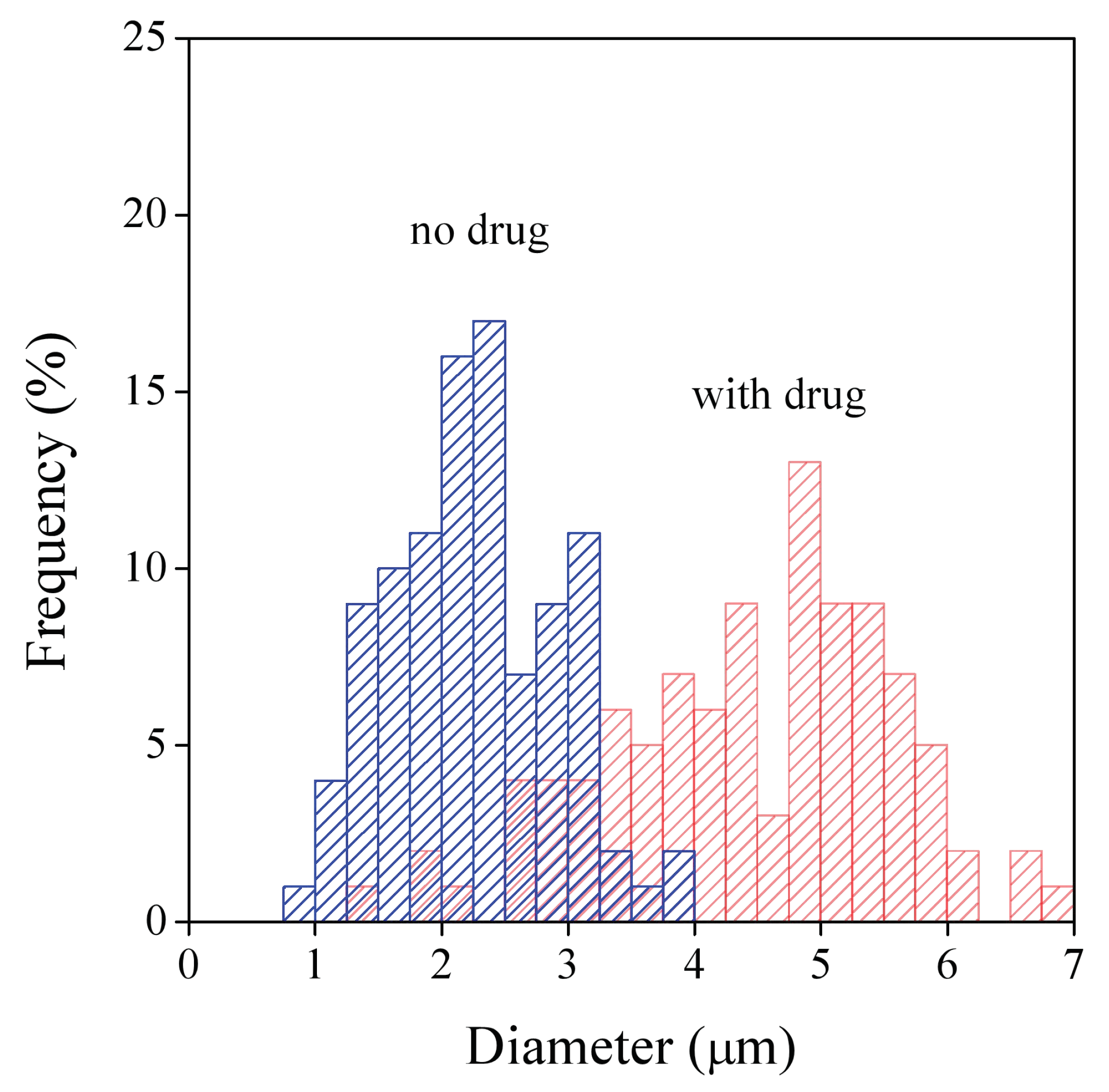
3.2. Fiber Characteristics
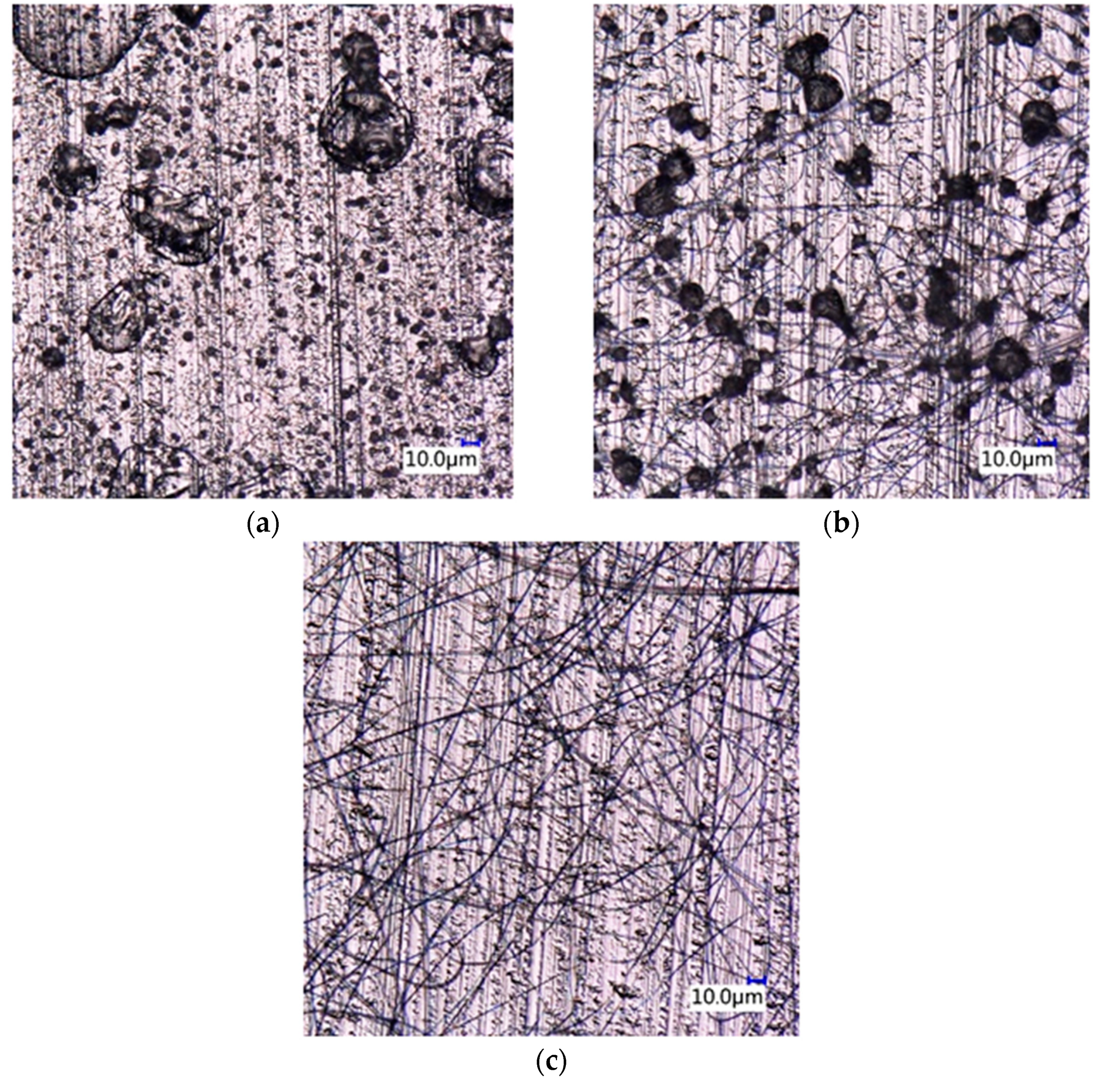
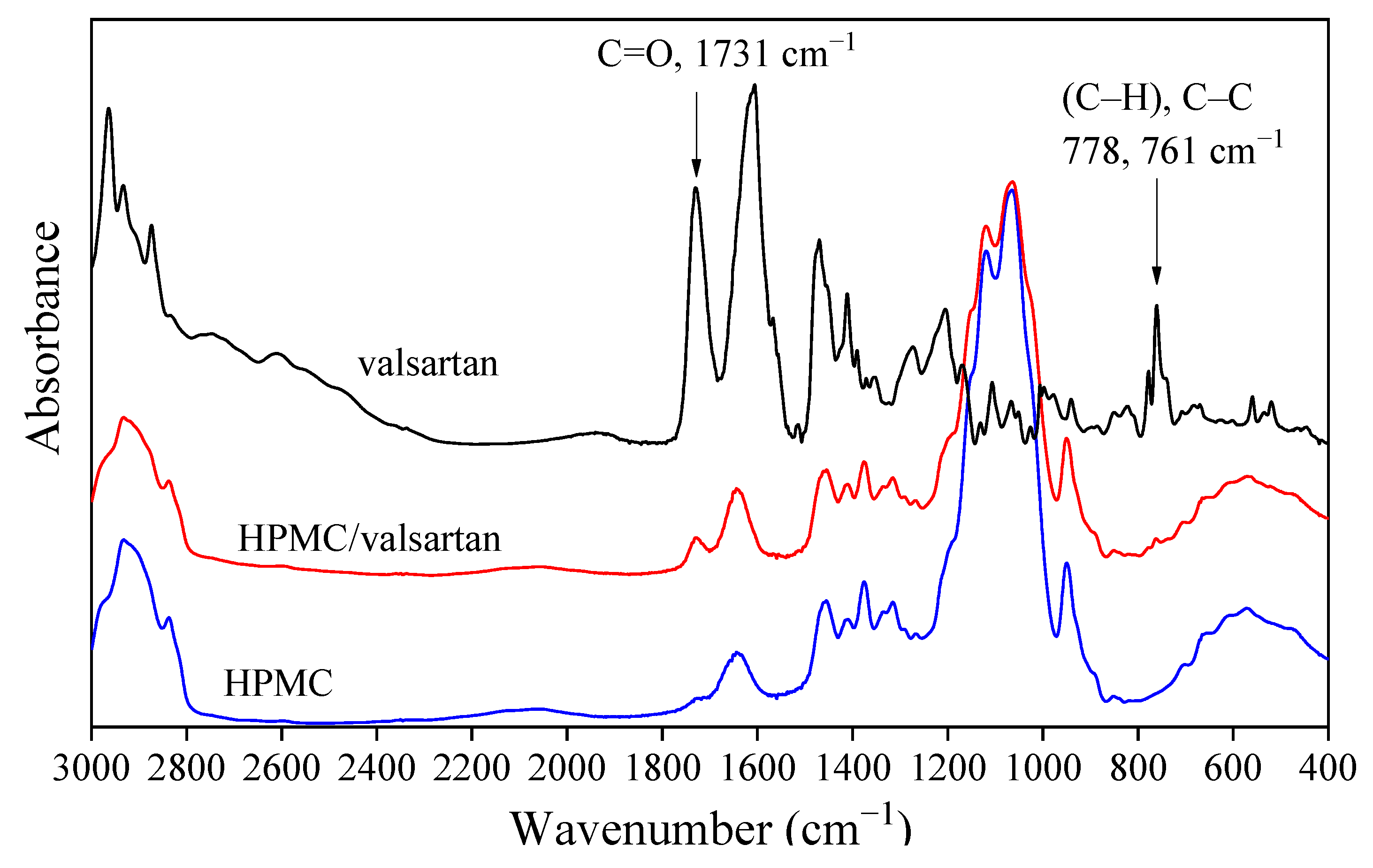
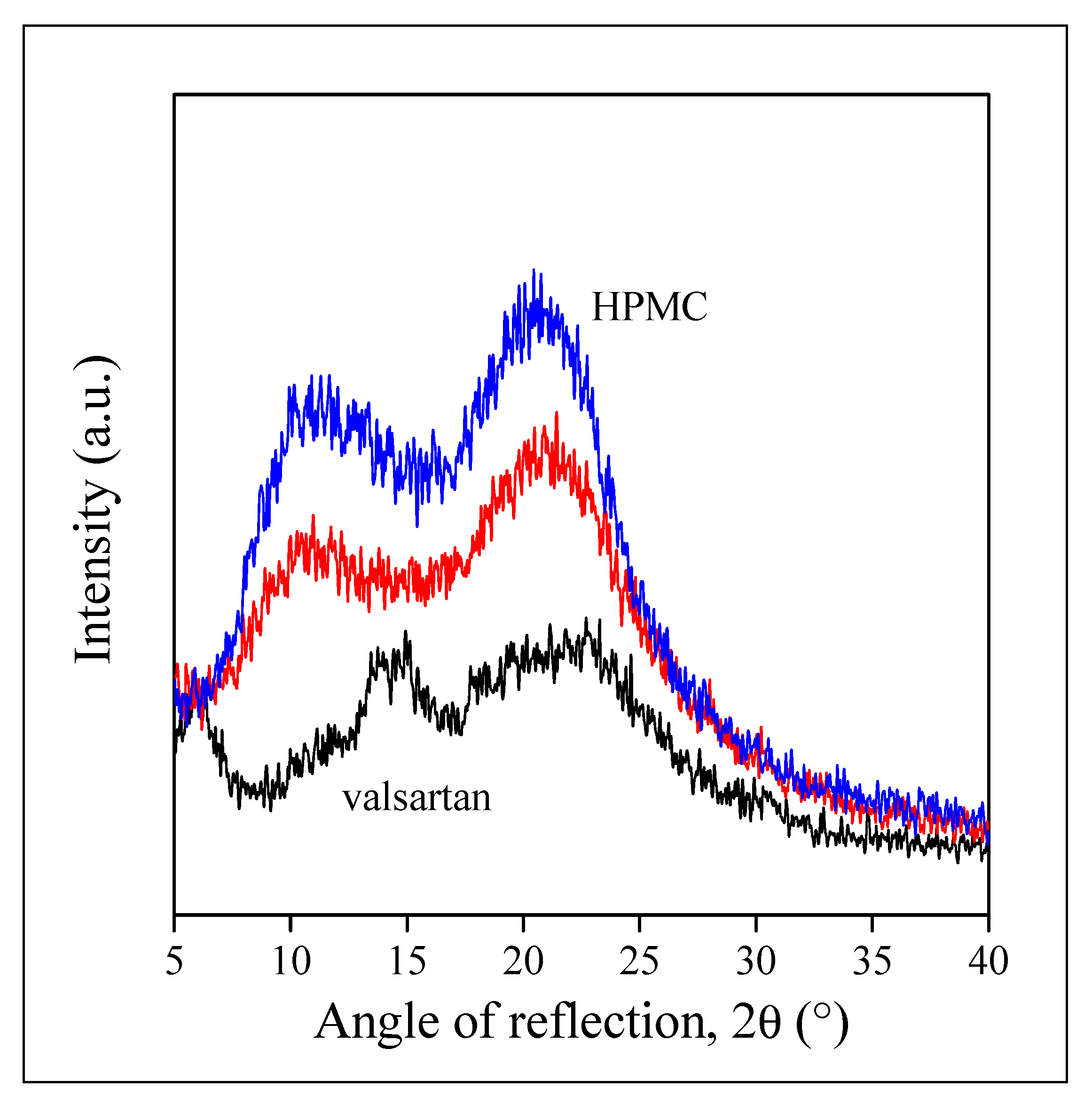
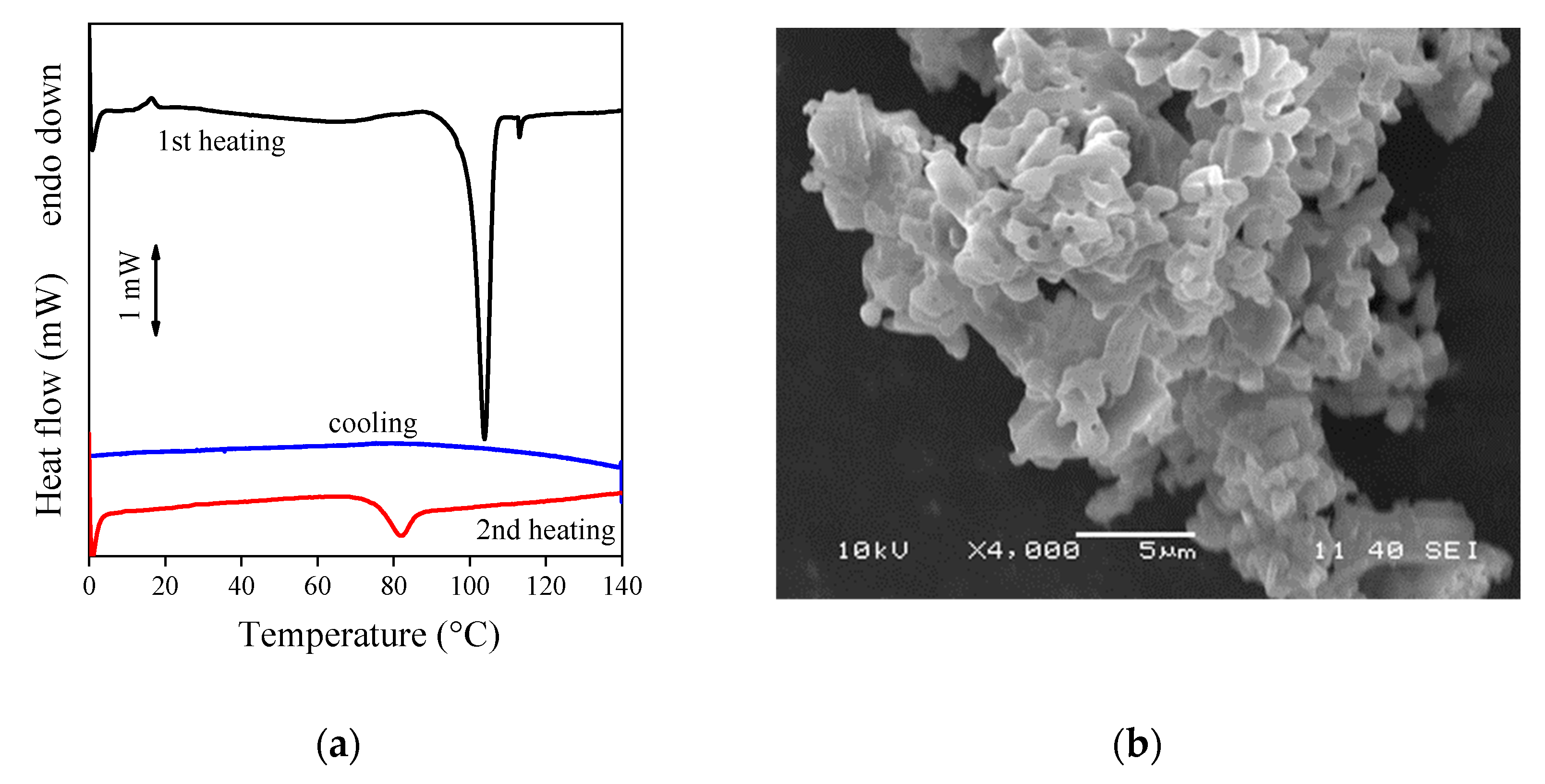
3.3. Drug Morphology, Location
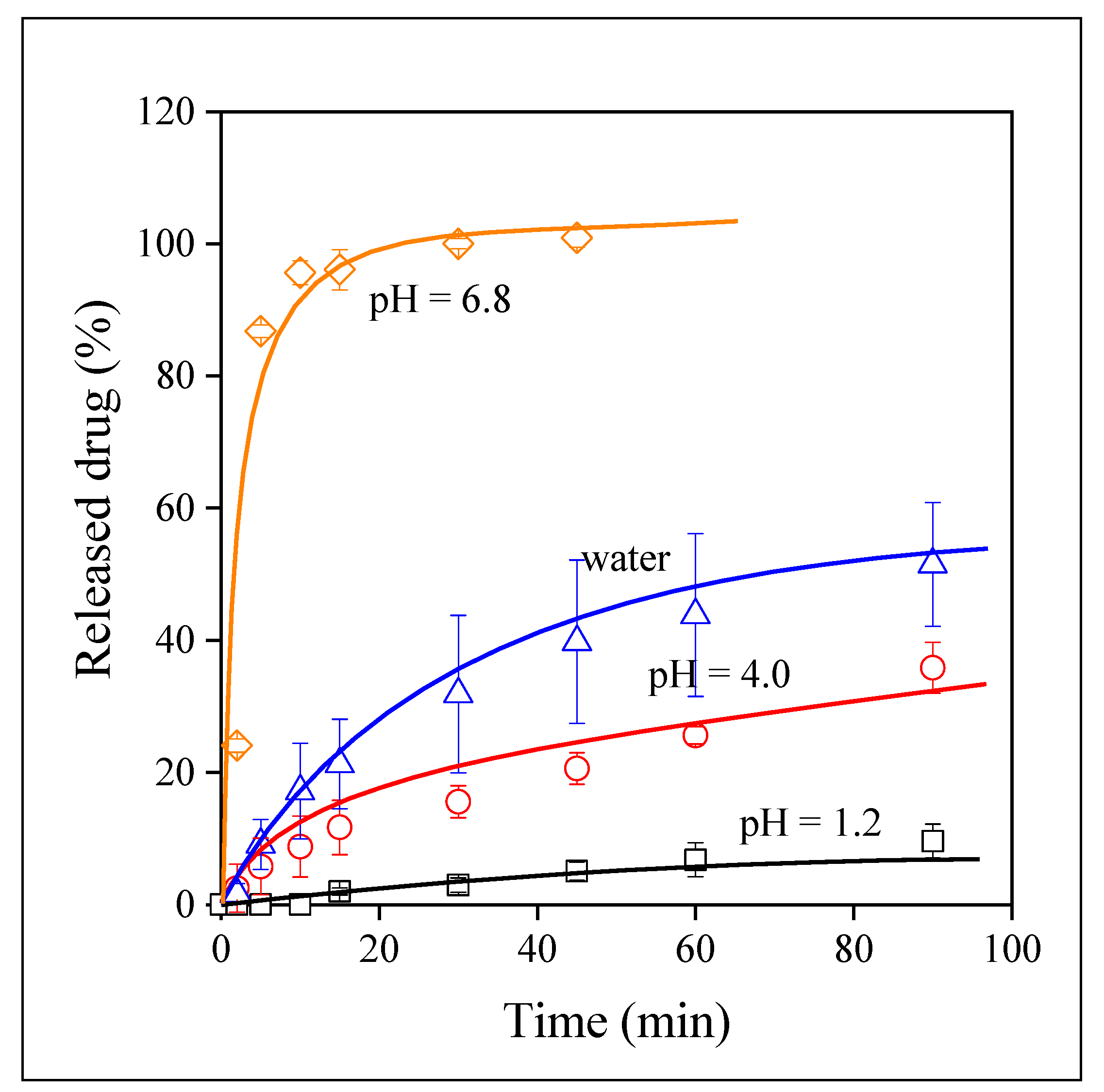
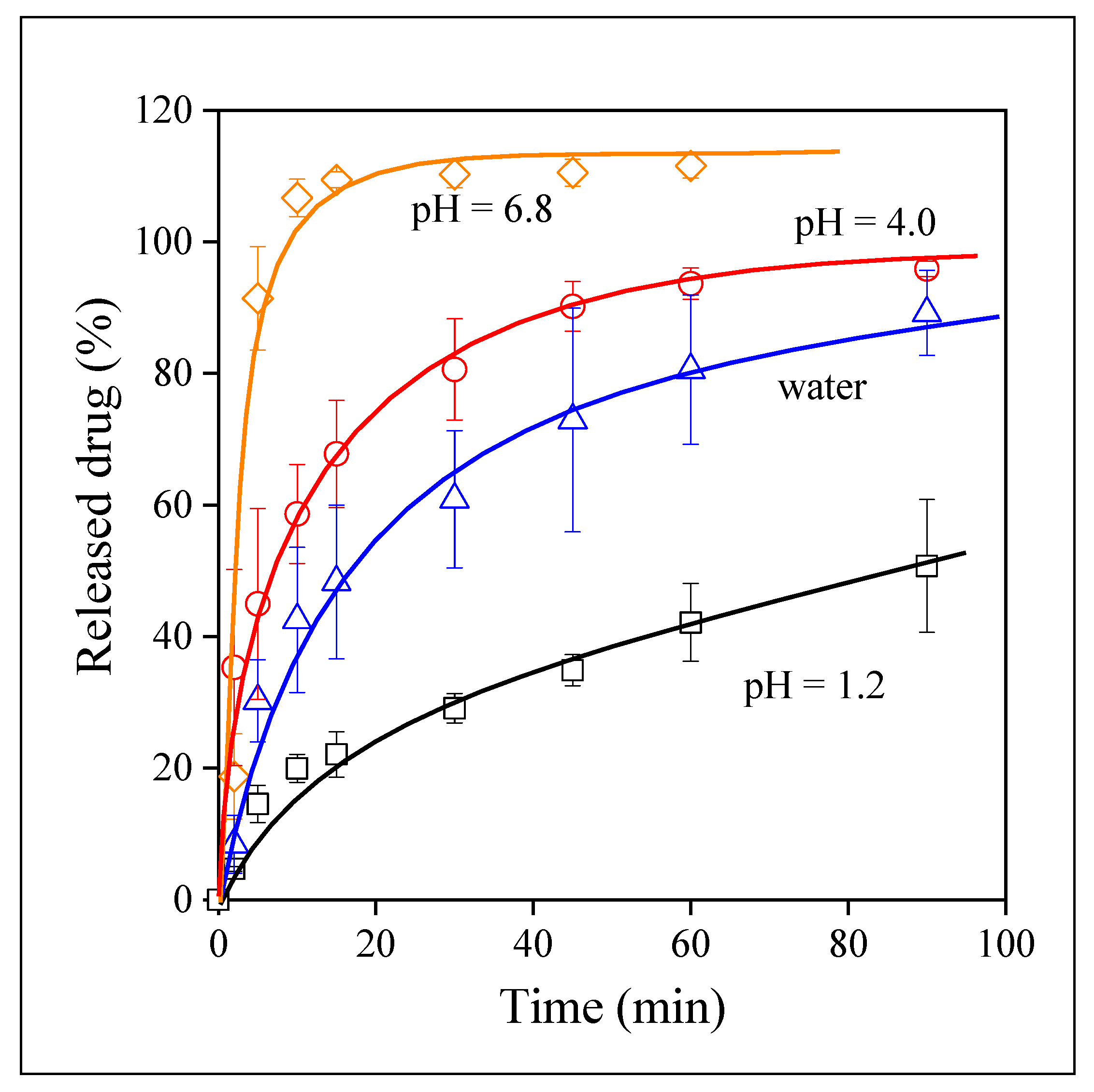
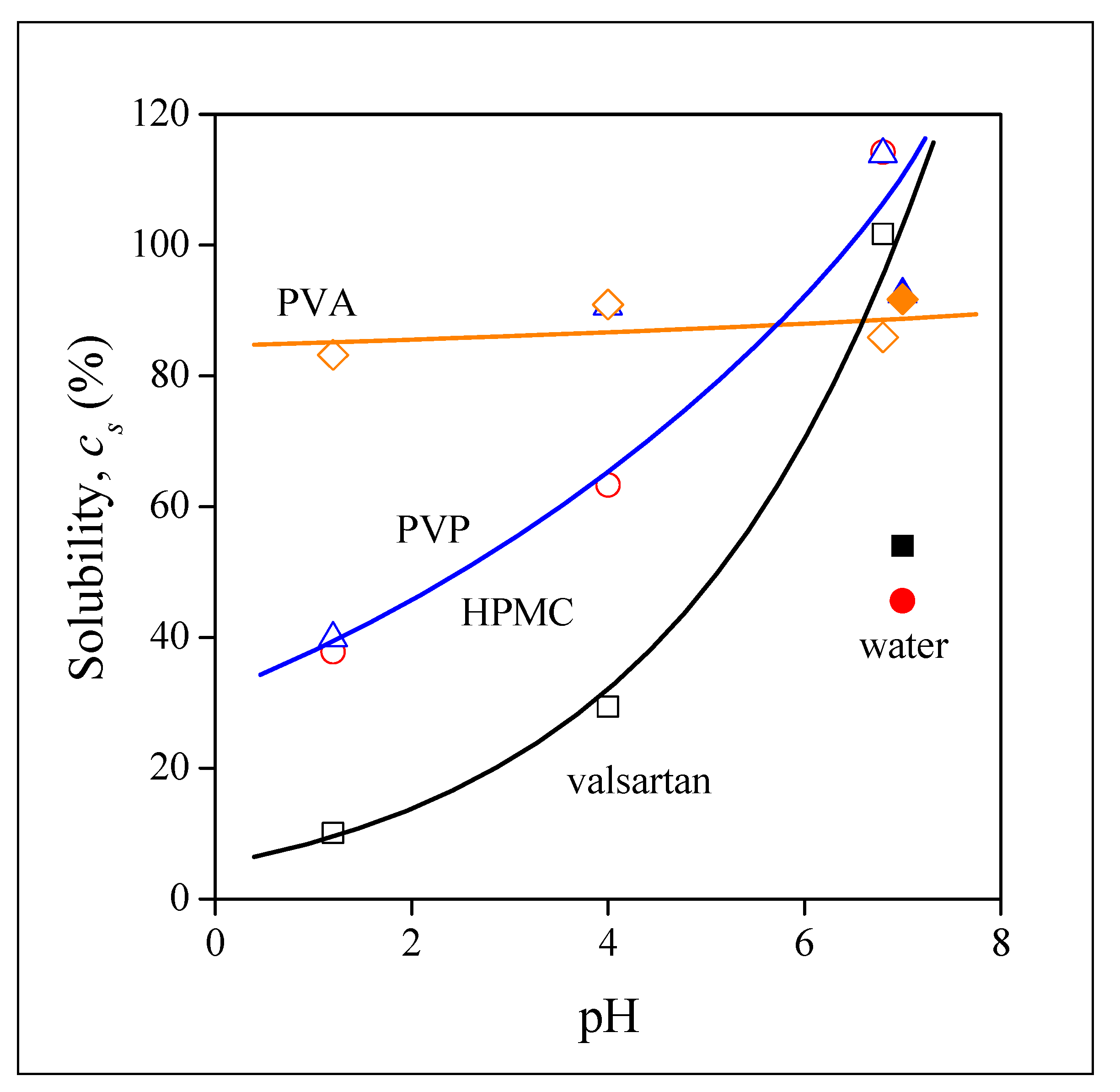
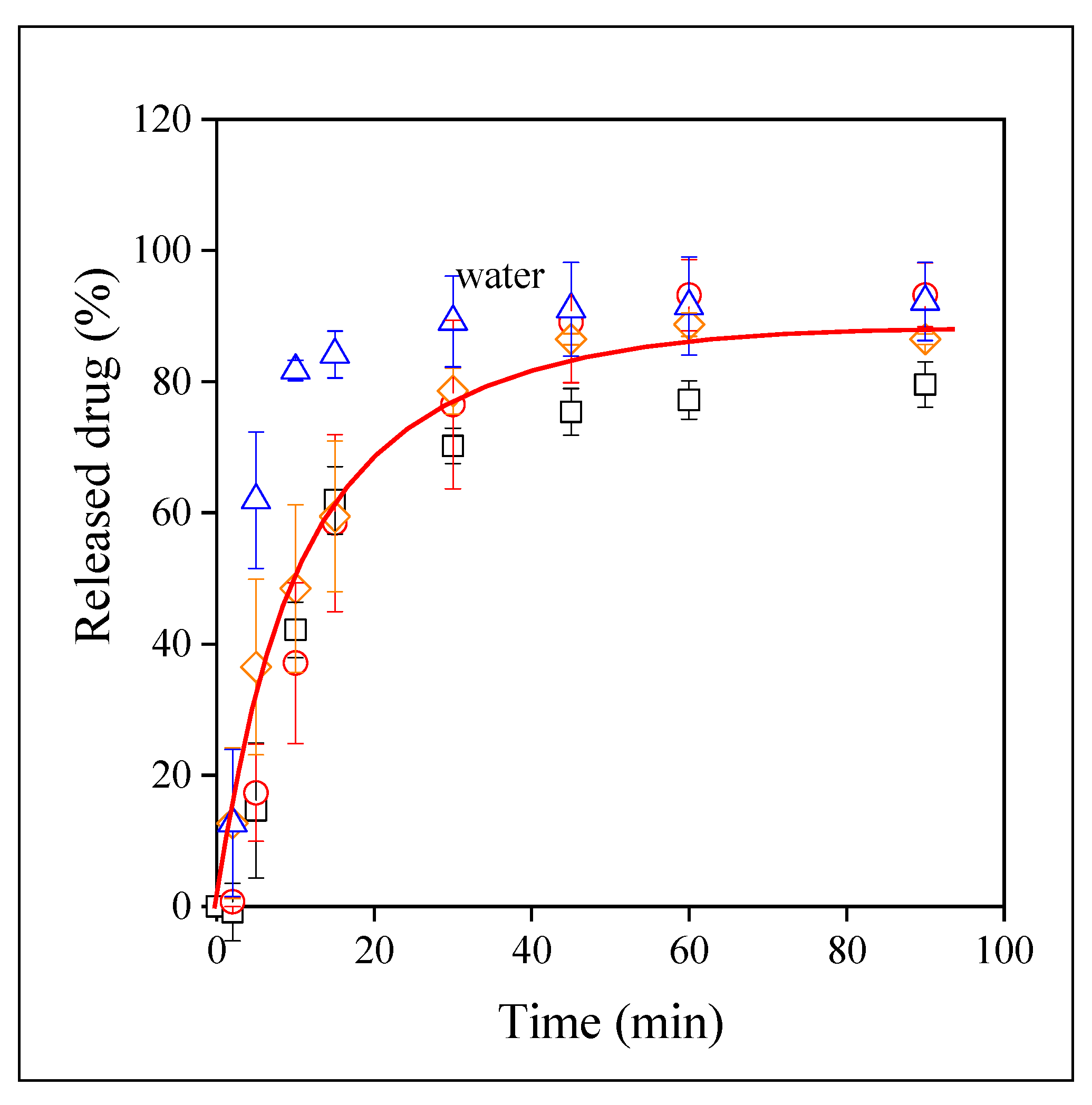
3.4. Drug Release
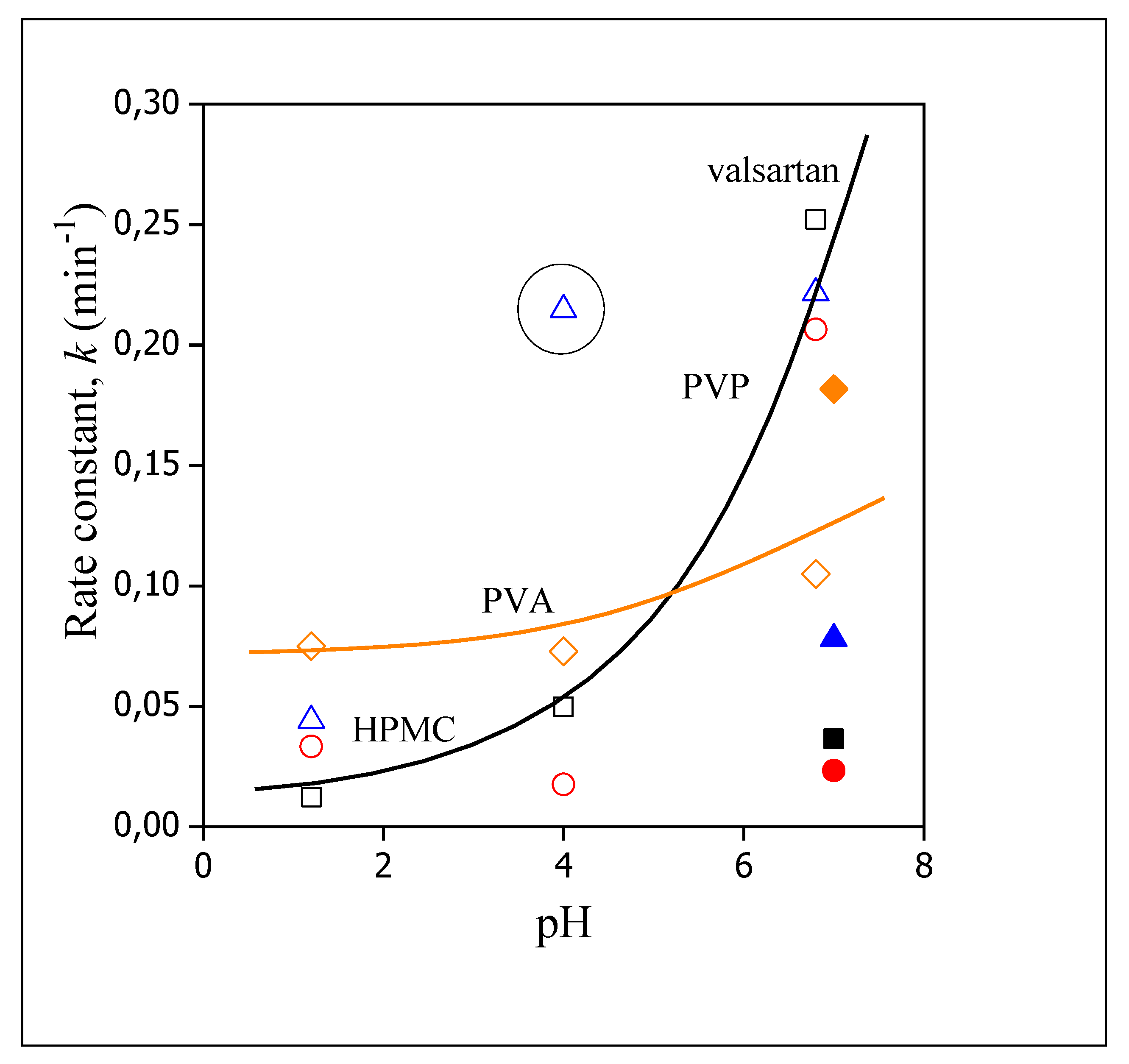
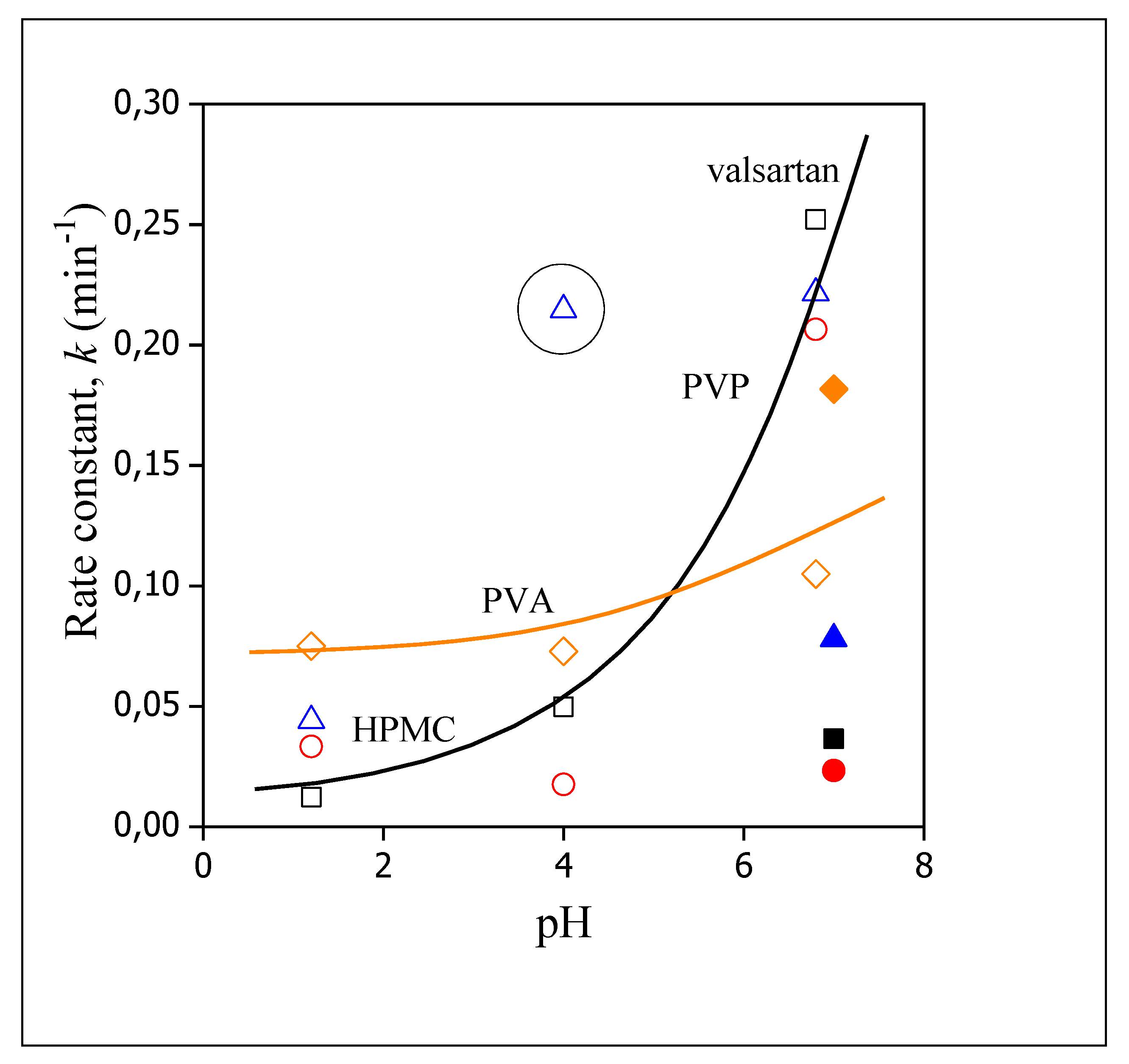
3.5. Release Rates, Mechanisms
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Indurkhya, A.; Patel, M.; Sharma, P.; Abed, S.N.; Shnoudeh, A.; Maheshwari, R.; Deb, P.K.; Tekade, R.K. Chapter 6—Influence of drug properties and routes of drug administration on the design of controlled release system. In Dosage Form Design Considerations; Tekade, R.K., Ed.; Academic Press: London, UK, 2018; Volume I, pp. 179–223. [Google Scholar]
- Sahoo, D.; Bandaru, R.; Samal, S.K.; Naik, R.; Kumar, P.; Kesharwani, P.; Dandela, R. Chapter 9—Oral drug delivery of nanomedicine. In Theory and Applications of Nonparenteral Nanomedicines; Kesharwani, P., Taurin, S., Greish, K., Eds.; Academic Press: London, UK, 2021; pp. 181–207. [Google Scholar]
- Rewatkar, P.; Kumeria, T.; Popat, A. Chapter 5—Size, shape and surface charge considerations of orally delivered nanomedicines. In Nanotechnology for Oral Drug Delivery; Martins, J.P., Santos, H.A., Eds.; Academic Press: London, UK, 2020; pp. 143–176. [Google Scholar]
- Koenigsknecht, M.J.; Baker, J.R.; Wen, B.; Frances, A.; Zhang, H.; Yu, A.; Zhao, T.; Tsume, Y.; Pai, M.P.; Bleske, B.E.; et al. In vivo dissolution and systemic absorption of immediate release ibuprofen in human gastrointestinal tract under fed and fasted conditions. Mol. Pharm. 2017, 14, 4295–4304. [Google Scholar] [CrossRef]
- Van den Abeele, J.; Brouwers, J.; Mattheus, R.; Tack, J.; Augustijns, P. Gastrointestinal behavior of weakly acidic BCS Class II drugs in man—Case study of diclofenac potassium. J. Pharm. Sci. 2016, 105, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Hens, B.; Brouwers, J.; Corsetti, M.; Augustijns, P. Gastrointestinal behavior of nano- and microsized fenofibrate: In vivo evaluation in man and in vitro simulation by assessment of the permeation potential. Eur. J. Pharm. Sci. 2015, 77, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Hens, B.; Corsetti, M.; Brouwers, J.; Augustijns, P. Gastrointestinal and systemic monitoring of posaconazole in humans after fasted and fed state administration of a solid dispersion. J. Pharm. Sci. 2016, 105, 2904–2912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brouwers, J.; Geboers, S.; Mols, R.; Tack, J.; Augustijns, P. Gastrointestinal behavior of itraconazole in humans—Part 1: Supersaturation from a solid dispersion and a cyclodextrin-based solution. Int. J. Pharm. 2017, 525, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Brouwers, J.; Tack, J.; Lammert, F.; Augustijns, P. Intraluminal drug and formulation behavior and integration in in vitro permeability estimation: A case study with amprenavir. J. Pharm. Sci. 2006, 95, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, R.B.; Conover, C.D.; Choe, Y.H. Poly(ethylene glycol) conjugated drugs and prodrugs: A comprehensive review. Crit. Rev. Ther. Drug Carr. Syst. 2000, 17, 62. [Google Scholar] [CrossRef]
- Choi, J.-S.; Jo, B.-W. Enhanced paclitaxel bioavailability after oral administration of pegylated paclitaxel prodrug for oral delivery in rats. Int. J. Prarm. 2004, 280, 221–227. [Google Scholar] [CrossRef]
- Blagden, N.; de Matas, M.; Gavan, P.T.; York, P. Crystal engineering of active pharmaceutical ingredients to improve solubility and dissolution rates. Adv. Drug Deliv. Rev. 2007, 59, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Serajuddin, A.T.M. Solid dispersion of poorly water-soluble drugs: Early promises, subsequent problems, and recent breakthroughs. J. Pharm. Sci. 1999, 88, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Marano, S.; Barker, S.A.; Raimi-Abraham, B.T.; Missaghi, S.; Rajabi-Siahboomi, A.; Craig, D.Q.M. Development of micro-fibrous solid dispersions of poorly water-soluble drugs in sucrose using temperature-controlled centrifugal spinning. Eur. J. Pharm. Biopharm. 2016, 103, 84–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Li, W.; Wang, G.; Qu, Y.-L.; Yu, D.-G. Electrospun 4th-generation solid dispersions of poorly water-soluble drug utilizing two different processes. J. Nanomat. 2018, 2018, 2012140. [Google Scholar] [CrossRef] [Green Version]
- Baghel, S.; Cathcart, H.; O’Reilly, N.J. Polymeric amorphous solid dispersions: A review of amorphization, crystallization, stabilization, solid-state characterization, and aqueous solubilization of Biopharmaceutical Classification System Class II Drugs. J. Pharm. Sci. 2016, 105, 2527–2544. [Google Scholar] [CrossRef] [Green Version]
- Loftsson, T.; Brewster, M.E. Pharmaceutical applications of cyclodextrins. 1. drug solubilization and stabilization. J. Pharm. Sci. 1996, 85, 1017–1025. [Google Scholar] [CrossRef]
- Rasenack, N.; Müller, B.W. Dissolution rate enhancement by in situ micronization of poorly water-soluble drugs. Pharm. Res. 2002, 19, 1894–1900. [Google Scholar] [CrossRef] [PubMed]
- Rasenack, N.; Müller, B.W. Micron-size drug particles: Common and novel micronization techniques. Pharm. Dev. Technol. 2004, 9, 1–13. [Google Scholar] [CrossRef]
- Loftsson, T.; Duchêne, D. Cyclodextrins and their pharmaceutical applications. Int. J. Pharm. 2007, 329, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-H.; Lee, S.-E.; Pyo, Y.-C.; Tran, P.; Park, J.-S. Solubility enhancement and application of cyclodextrins in local drug delivery. J. Pharm. Investig. 2020, 50, 17–27. [Google Scholar] [CrossRef]
- Prosapio, V.; Reverchon, E.; de Marco, I. Coprecipitation of Polyvinylpyrrolidone/β-Carotene by supercritical antisolvent processing. Ind. Eng. Chem. Res. 2015, 54, 11568–11575. [Google Scholar] [CrossRef]
- García-Casas, I.; Montes, A.; Pereyra, C.; Martínez de la Ossa, E.J. Generation of quercetin/cellulose acetate phthalate systems for delivery by supercritical antisolvent process. Eur. J. Pharm. Sci. 2017, 100, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Markham, A.; Goa, K.L. Valsartan. Drugs 1997, 54, 299–311. [Google Scholar] [CrossRef]
- Baek, I.-H.; Kim, J.-S.; Ha, E.-S.; Choo, G.-H.; Cho, W.; Hwang, S.-J.; Kim, M.-S. Oral absorption of a valsartan-loaded spray-dried emulsion based on hydroxypropylmethyl cellulose. Int. J. Biol. Macromol. 2014, 69, 222–228. [Google Scholar] [CrossRef]
- Pradhan, R.; Kim, S.Y.; Yong, C.S.; Kim, J.O. Preparation and characterization of spray-dried valsartan-loaded Eudragit® E PO solid dispersion microparticles. Asian J. Pharm. Sci. 2016, 11, 744–750. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.-D.; Sung, J.H.; Kim, K.K.; Kim, D.W.; Kim, J.O.; Lee, B.-J.; Yong, C.S.; Choi, H.-G. Novel valsartan-loaded solid dispersion with enhanced bioavailability and no crystalline changes. Int. J. Pharm. 2012, 422, 202–210. [Google Scholar] [CrossRef]
- Cappello, B.; Maio, C.D.; Iervolino, M.; Miro, A. Improvement of solubility and stability of valsartan by hydroxypropyl-\boldbeta-cyclodextrin. J. Incl. Phenom. Macrocycl. Chem. 2005, 54, 289. [Google Scholar] [CrossRef]
- Youn, Y.-S.; Oh, J.H.; Ahn, K.H.; Kim, M.; Kim, J.; Lee, Y.-W. Dissolution rate improvement of valsartan by low temperature recrystallization in compressed CO2: Prevention of excessive agglomeration. J. Supercrit. Fluids 2011, 59, 117–123. [Google Scholar] [CrossRef]
- Mamidi, N.; Delgadillo, R.M.V. Design, fabrication and drug release potential of dual stimuli-responsive composite hydrogel nanoparticle interfaces. Colloids Surf. B Biointerfaces 2021, 204, 111819. [Google Scholar] [CrossRef] [PubMed]
- Mohamady Hussein, M.A.; Guler, E.; Rayaman, E.; Cam, M.E.; Sahin, A.; Grinholc, M.; Sezgin Mansuroglu, D.; Sahin, Y.M.; Gunduz, O.; Muhammed, M.; et al. Dual-drug delivery of Ag-chitosan nanoparticles and phenytoin via core-shell PVA/PCL electrospun nanofibers. Carbohydr. Polym. 2021, 270, 118373. [Google Scholar] [CrossRef] [PubMed]
- Mamidi, N.; Delgadillo, R.M.V.; Castrejón, J.V. Unconventional and facile production of a stimuli-responsive multifunctional system for simultaneous drug delivery and environmental remediation. Environ. Sci. Nano 2021, 8, 2081–2097. [Google Scholar] [CrossRef]
- Mohammadinejad, R.; Madamsetty, V.S.; Kumar, A.; Varzandeh, M.; Dehshahri, A.; Zarrabi, A.; Sharififar, F.; Mohammadi, M.; Fahimipour, A.; Ramakrishna, S. Electrospun nanocarriers for delivering natural products for cancer therapy. Trends Food Sci. Technol. 2021, 118, 887–904. [Google Scholar] [CrossRef]
- Mamidi, N.; Zuníga, A.E.; Villela-Castrejón, J. Engineering and evaluation of forcespun functionalized carbon nano-onions reinforced poly (ε-caprolactone) composite nanofibers for pH-responsive drug release. Mater. Sci. Eng. C 2020, 112, 110928. [Google Scholar] [CrossRef] [PubMed]
- Mamidi, N.; Velasco Delgadillo, R.M.; Barrera, E.V. Covalently functionalized carbon nano-onions integrated gelatin methacryloyl nanocomposite hydrogel containing γ-cyclodextrin as drug carrier for high-performance pH-triggered drug release. Pharmaceuticals 2021, 14, 291. [Google Scholar] [CrossRef]
- Kwak, H.W.; Woo, H.; Kim, I.-C.; Lee, K.H. Fish gelatin nanofibers prevent drug crystallization and enable ultrafast delivery. RSC Adv. 2017, 7, 40411–40417. [Google Scholar] [CrossRef] [Green Version]
- Ilomuanya, M.O.; Okafor, P.S.; Amajuoyi, J.N.; Onyejekwe, J.C.; Okubanjo, O.O.; Adeosun, S.O.; Silva, B.O. Polylactic acid-based electrospun fiber and hyaluronic acid-valsartan hydrogel scaffold for chronic wound healing. Beni-Seuf Univ. J. Basic Appl. Sci. 2020, 9, 31. [Google Scholar] [CrossRef]
- Bukhary, H.; Williams, G.R.; Orlu, M. Electrospun fixed dose formulations of amlodipine besylate and valsartan. Int. J. Pharm. 2018, 549, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Cheng, S.; Lu, W.; Wang, Y.; Zhang, P.; Yao, Q. Electrospun fibers and their application in drug controlled release, biological dressings, tissue repair, and enzyme immobilization. RSC Adv. 2019, 9, 25712–25729. [Google Scholar] [CrossRef] [Green Version]
- Angel, N.; Guo, L.; Yan, F.; Wang, H.; Kong, L. Effect of processing parameters on the electrospinning of cellulose acetate studied by response surface methodology. J. Agric. Food Res. 2020, 2, 100015. [Google Scholar] [CrossRef]
- Budai-Szűcs, M.; Léber, A.; Cui, L.; Józó, M.; Vályi, P.; Burián, K.; Kirschweng, B.; Csányi, E.; Pukánszky, B. Electrospun PLA fibers containing metronidazole for periodontal disease. Drug Des. Devel. Ther. 2020, 14, 233–242. [Google Scholar] [CrossRef] [Green Version]
- Cui, L.; Molnár, J.R.; Budai-Szűcs, M.; Szécsényi, M.; Burián, K.; Vályi, P.; Berkó, S.; Pukánszky, B. Physical–chemical aspects of the preparation and drug release of electrospun scaffolds. Pharmaceutics 2021, 13, 1645. [Google Scholar] [CrossRef]
- Gomori, G. Preparation of buffers for use in enzyme studies. In Methods in Enzymology; Colowick, S.P., Ed.; Academic Press: Baltimore, MD, USA, 1955; Volume 1, pp. 138–146. [Google Scholar]
- Tran, T.T.-D.; Tran, P.H.-L.; Park, J.-B.; Lee, B.-J. Effects of solvents and crystallization conditions on the polymorphic behaviors and dissolution rates of valsartan. Arch. Pharm. Res. 2012, 35, 1223–1230. [Google Scholar] [CrossRef] [PubMed]
- Hattori, Y.; Haruna, Y.; Otsuka, M. Dissolution process analysis using model-free Noyes–Whitney integral equation. Colloids Surf. B 2013, 102, 227–231. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | Concentration (wt%) | Voltage (kV) | Feeding Rate (mL/h) | Collector Distance (mm) |
|---|---|---|---|---|
| HPMC | 10 | 20 | 5 | 100 |
| PVP | 40 | 20 | 7 | 125 |
| PVA a) | 15 | 15 | 0.8 | 140 |
| Polymer | Average Diameter (µm) | |
|---|---|---|
| No Drug | With Drug | |
| HPMC | 2.0 ± 1.0 | 1.5 ± 0.7 |
| PVP | 2.3 ± 0.7 | 4.4 ± 1.1 |
| PVA | 0.5 ± 0.1 | 0.7 ± 0.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Józó, M.; Simon, N.; Yi, L.; Móczó, J.; Pukánszky, B. Improved Release of a Drug with Poor Water Solubility by Using Electrospun Water-Soluble Polymers as Carriers. Pharmaceutics 2022, 14, 34. https://doi.org/10.3390/pharmaceutics14010034
Józó M, Simon N, Yi L, Móczó J, Pukánszky B. Improved Release of a Drug with Poor Water Solubility by Using Electrospun Water-Soluble Polymers as Carriers. Pharmaceutics. 2022; 14(1):34. https://doi.org/10.3390/pharmaceutics14010034
Chicago/Turabian StyleJózó, Muriel, Nóra Simon, Lan Yi, János Móczó, and Béla Pukánszky. 2022. "Improved Release of a Drug with Poor Water Solubility by Using Electrospun Water-Soluble Polymers as Carriers" Pharmaceutics 14, no. 1: 34. https://doi.org/10.3390/pharmaceutics14010034
APA StyleJózó, M., Simon, N., Yi, L., Móczó, J., & Pukánszky, B. (2022). Improved Release of a Drug with Poor Water Solubility by Using Electrospun Water-Soluble Polymers as Carriers. Pharmaceutics, 14(1), 34. https://doi.org/10.3390/pharmaceutics14010034