
Design of Oral Sustained-Release Pellets by Modeling and Simulation Approach to Improve Compliance for Repurposing Sobrerol


Abstract
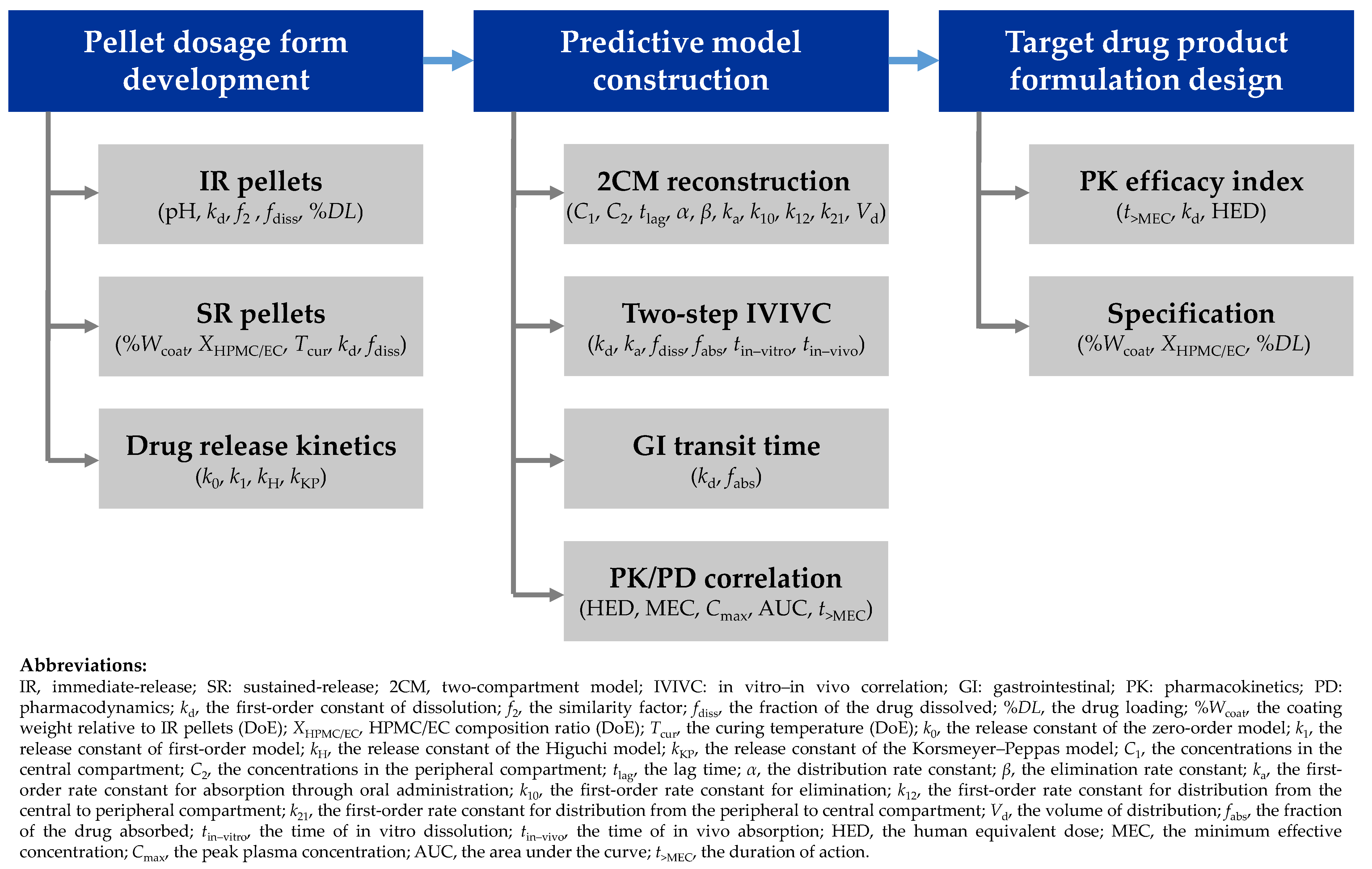
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Manufacturing
2.2.1. IR Pellets
2.2.2. SR Pellets
2.3. Characterization
2.3.1. Scanning Electron Microscopy (SEM)
2.3.2. High-Performance Liquid Chromatography (HPLC)
2.3.3. Dissolution Test
2.3.4. The Drug Release Mechanism
2.4. Establishing the Predictive Model
2.4.1. Reconstruction of the Sobrerol Pharmacokinetic Model
2.4.2. Exploring the IVIVC of Sobrerol
2.4.3. Consideration of the Gastrointestinal Transit Time
2.4.4. Finding the Pharmacokinetic Efficacy Index
3. Results and Discussion
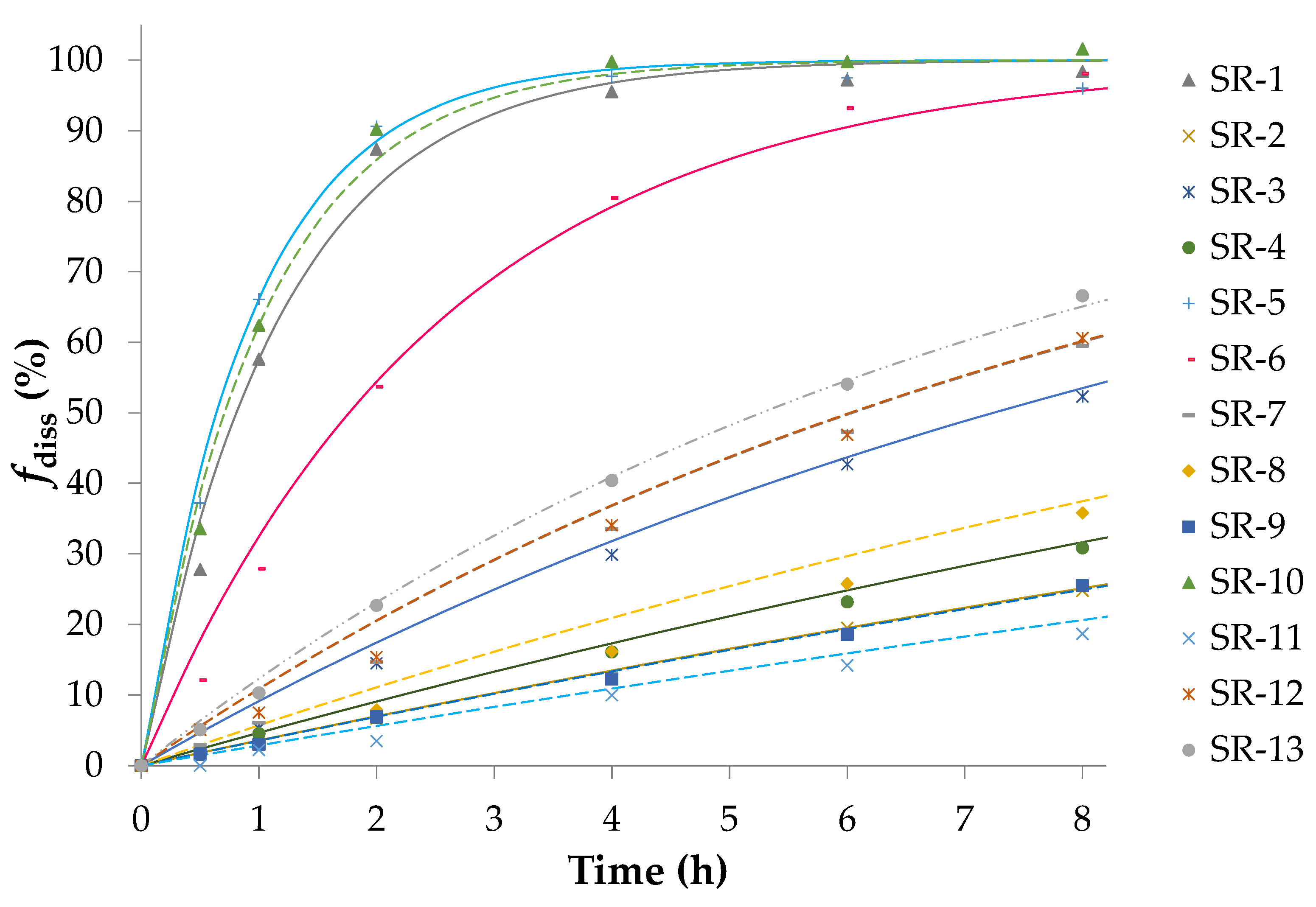
3.1. Preparation of IR/SR Pellets and Dissolution Study
3.2. Establishment of the Predictive Model
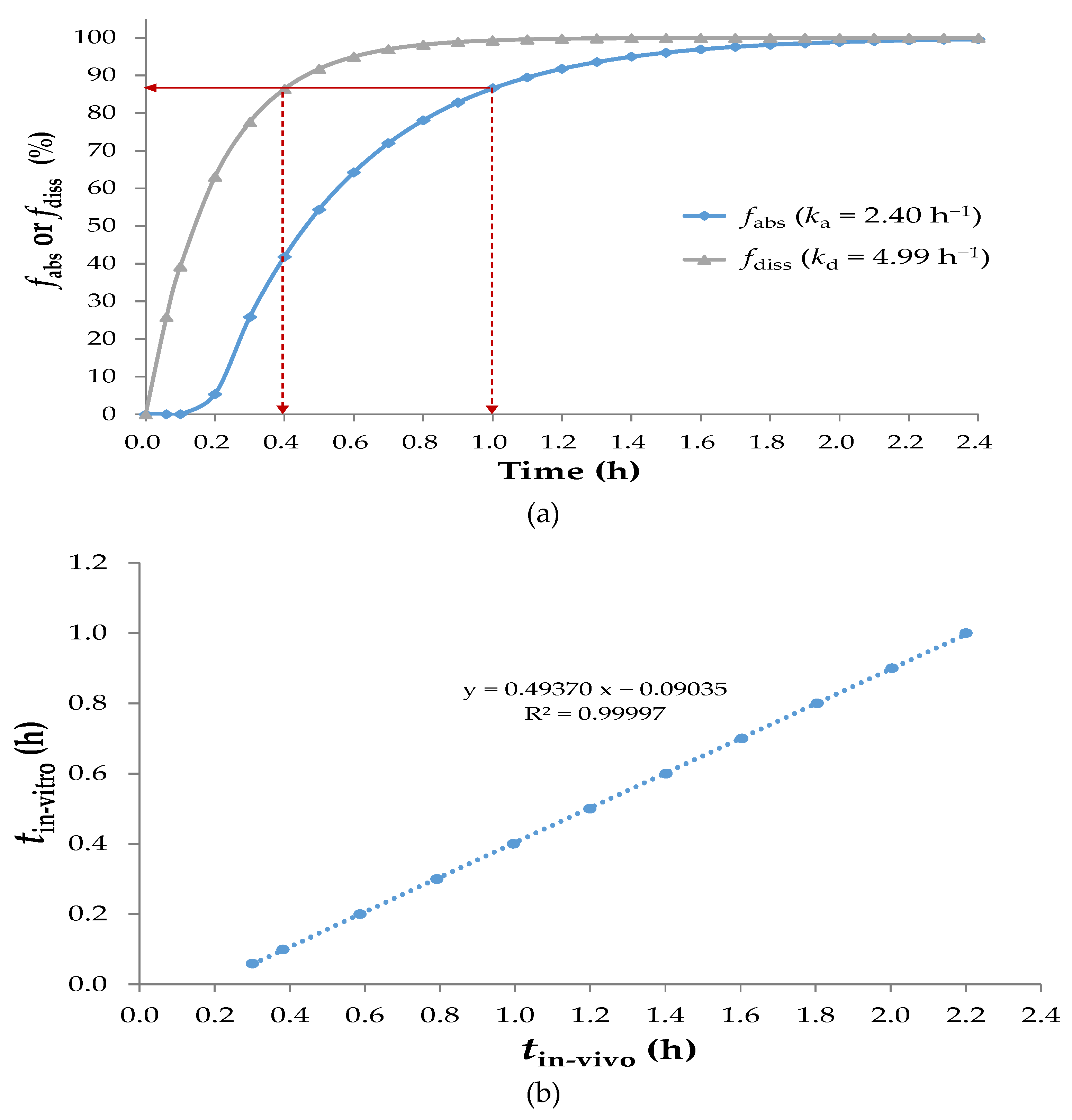
3.2.1. Reconstruction of the Sobrerol Pharmacokinetic Model with Lag Time
3.2.2. Time-Scaling IVIVC of Sobrerol
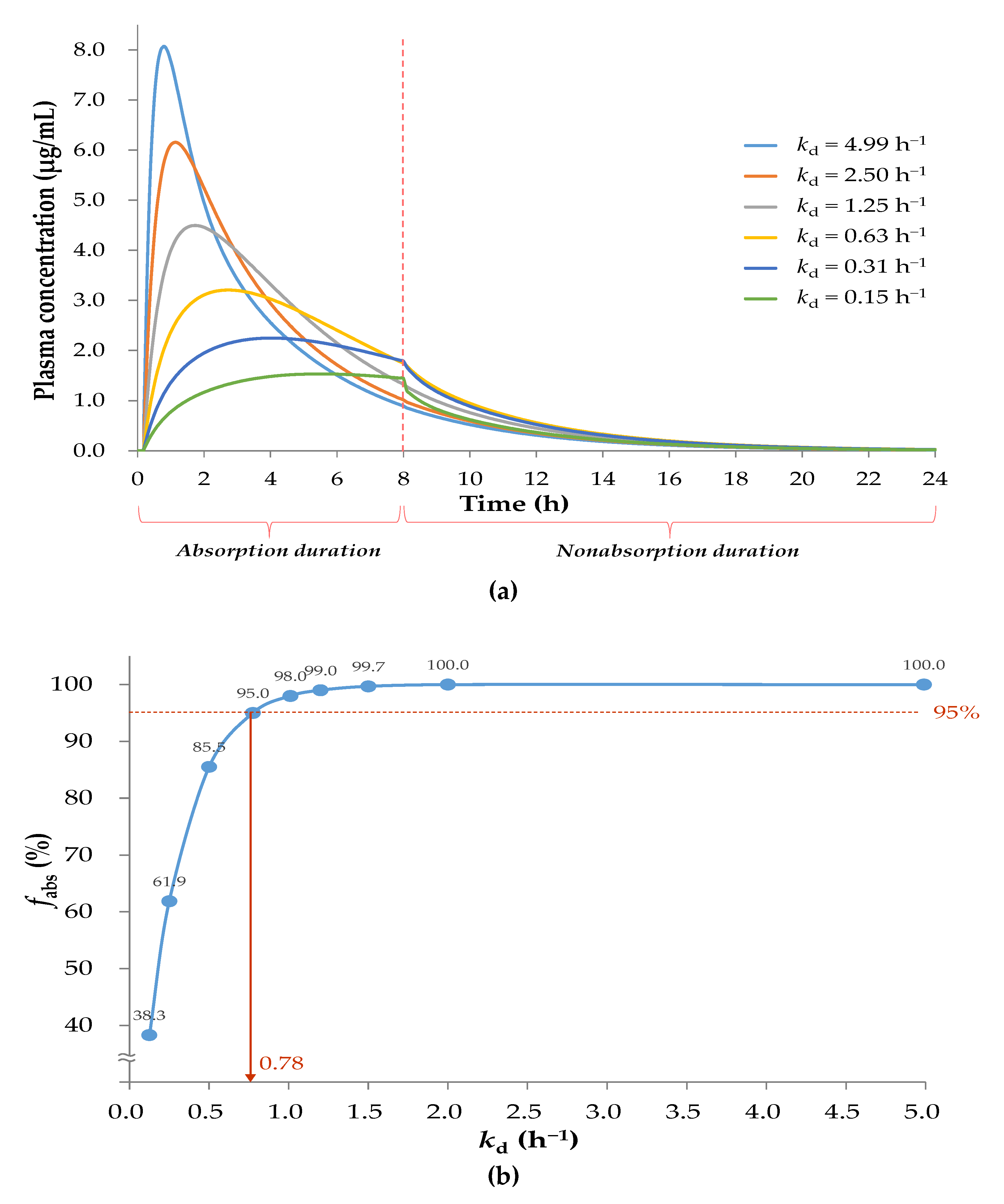
3.2.3. Pharmacokinetic Profiles Derived with Gastrointestinal Transit Time
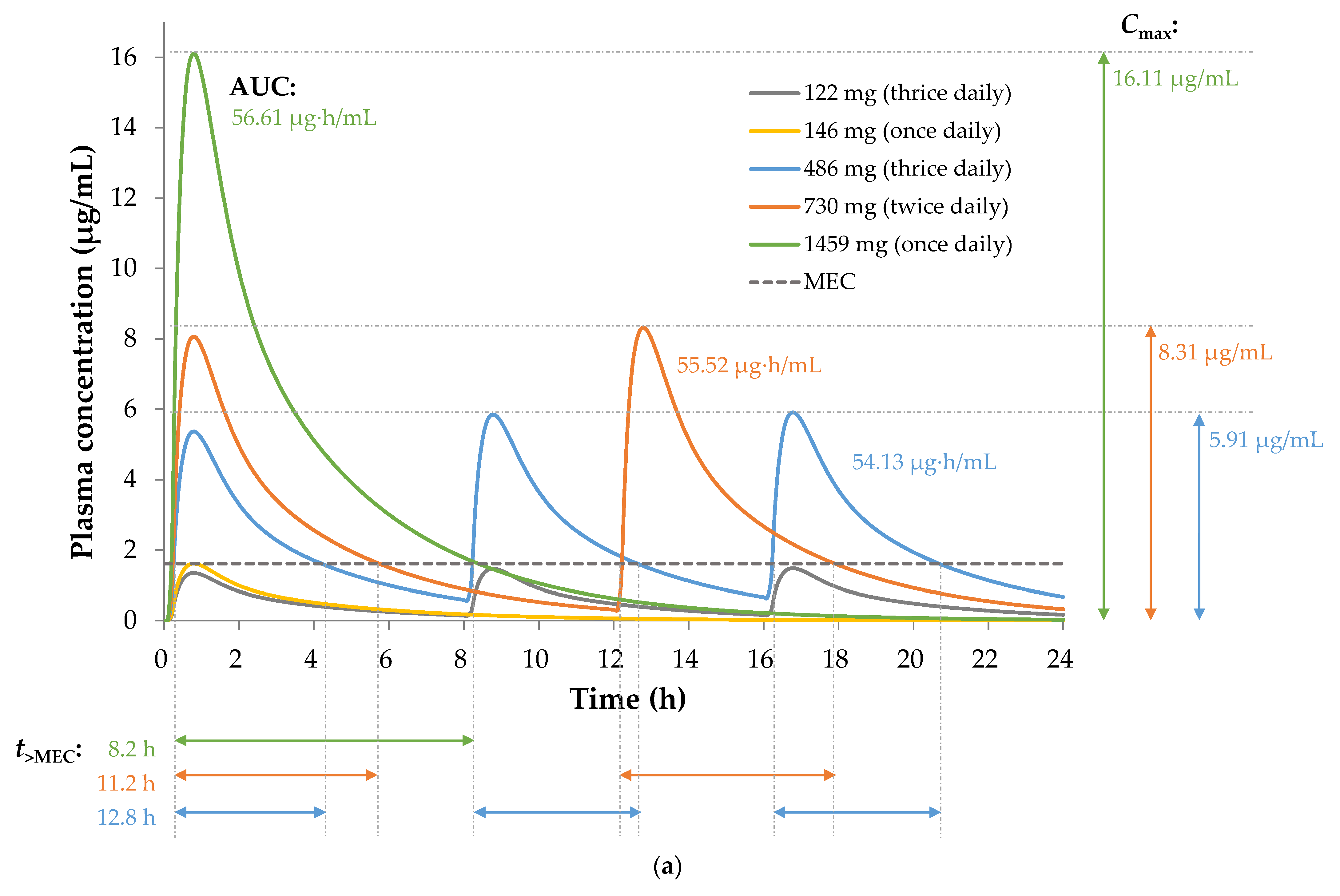
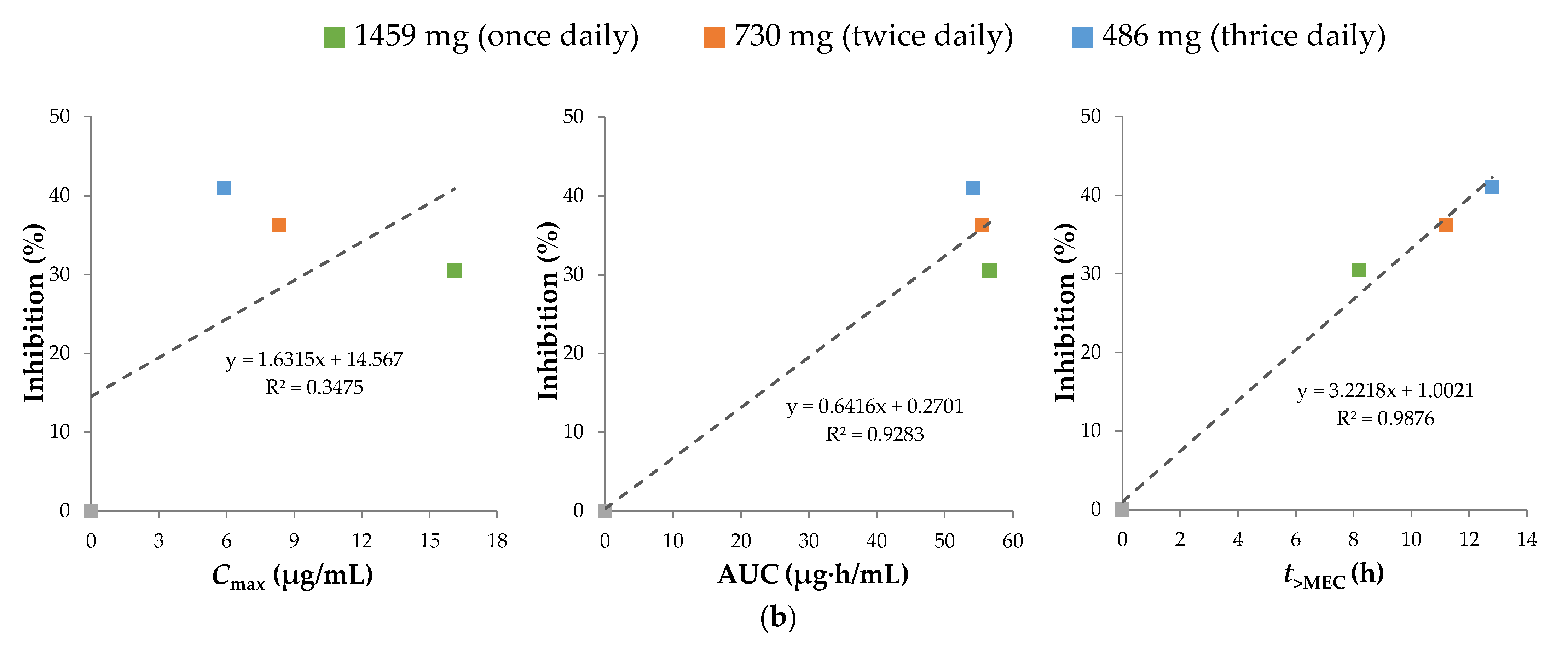
3.2.4. Pharmacokinetic Efficacy Index of Sobrerol
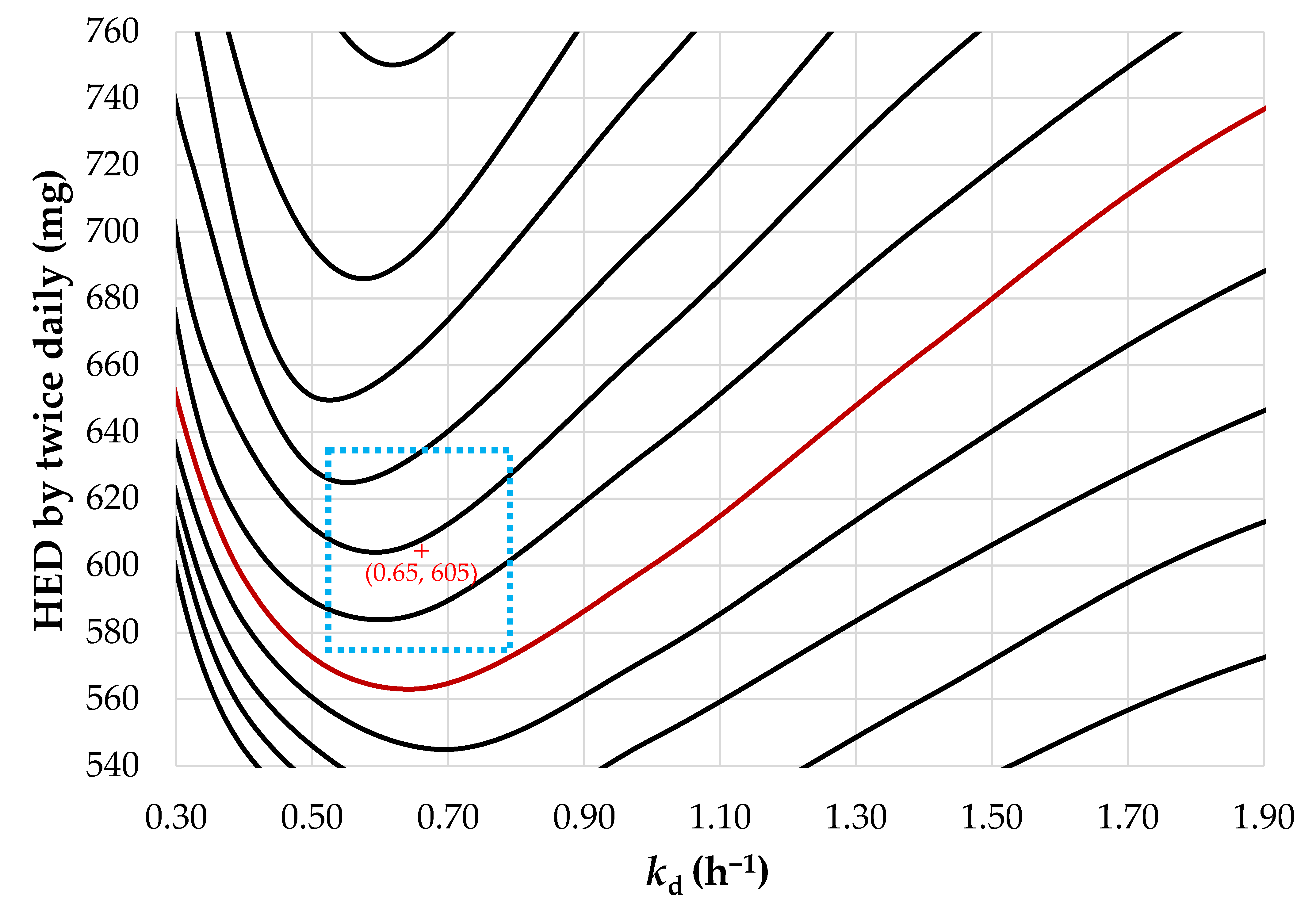
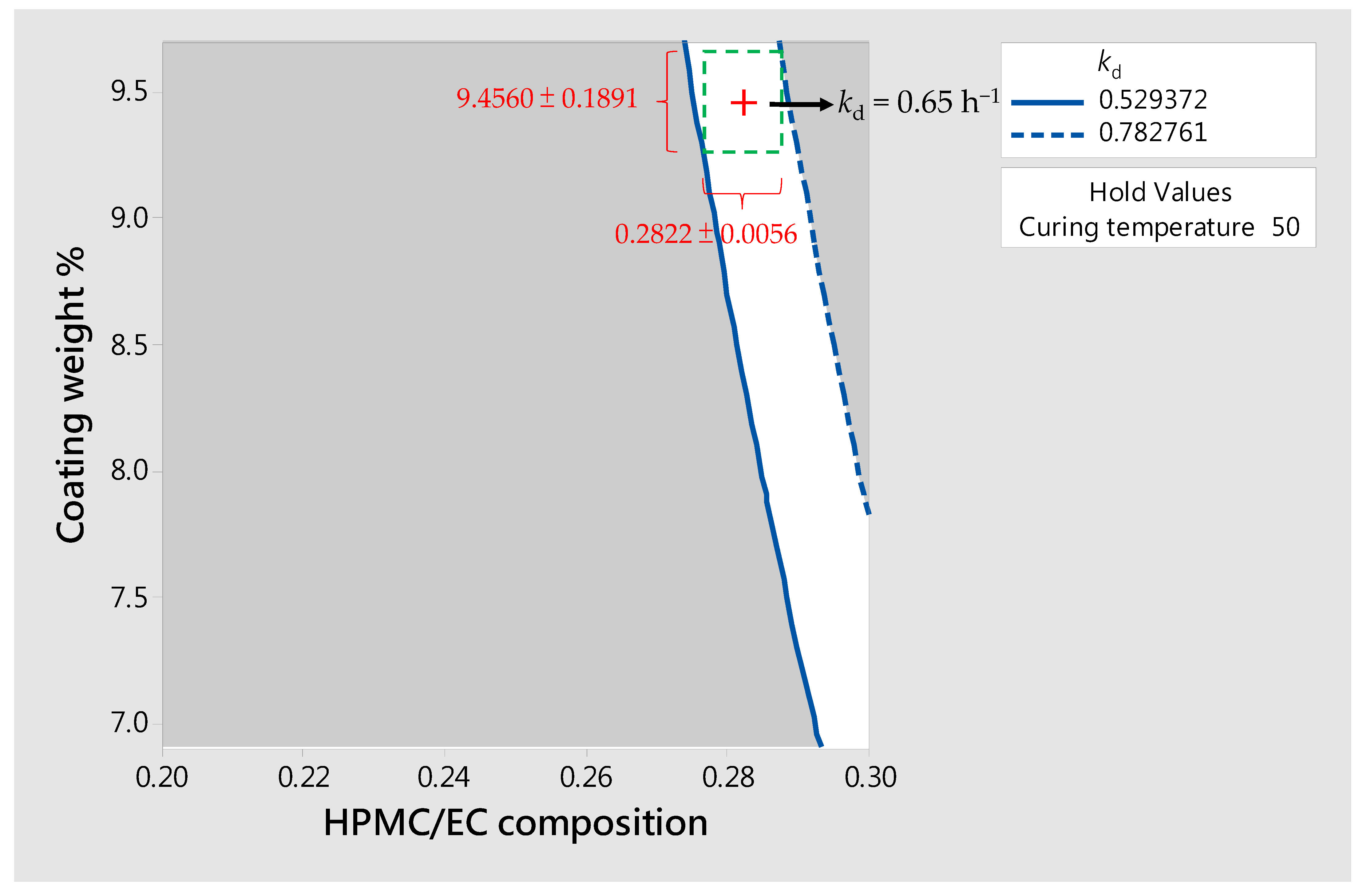
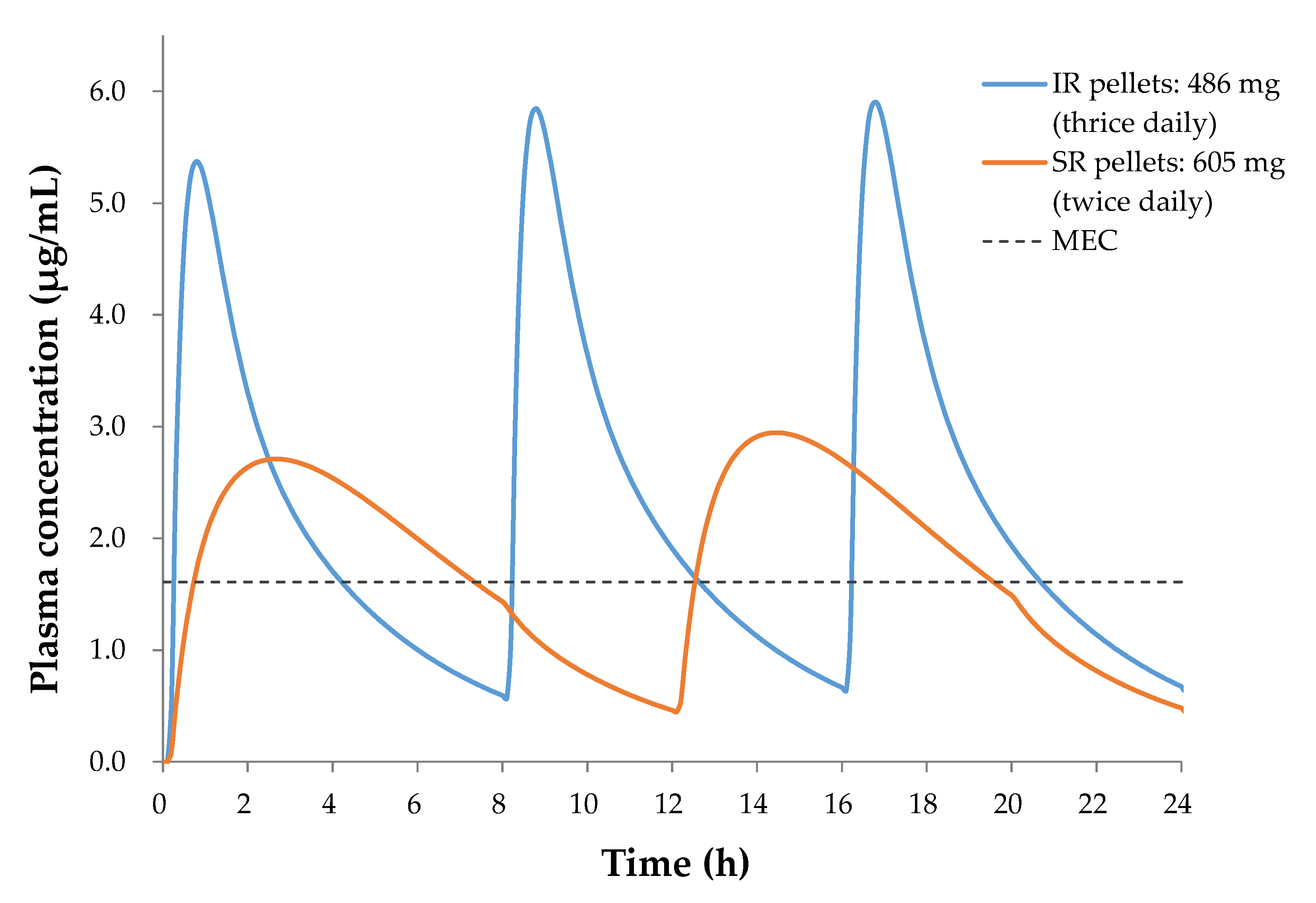
3.3. Design of SR Pellets
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Braga, P.C.; Allegra, L.; Bossi, R.; Scuri, R.; Castiglioni, C.L.; Romandini, S. Review on sobrerol as a muco-modifying drug: Experimental data and clinical findings in hypersecretory bronchopulmonary diseases. Int. J. Clin. Pharmacol. Res. 1987, 7, 381–400. [Google Scholar]
- Castiglioni, C.L.; Gramolini, C. Effect of long-term treatment with sobrerol on the exacerbations of chronic bronchitis. Respiration 1986, 50, 202–217. [Google Scholar] [CrossRef] [PubMed]
- Bellussi, L.; Manini, G.; Buccella, M.; Cacchi, R. Evaluation of the efficacy and safety of sobrerol granules in patients suffering from chronic rhinosinusitis. J. Int. Med. Res. 1990, 18, 454–459. [Google Scholar] [CrossRef]
- Scaglione, F.; Petrini, O. Mucoactive Agents in the Therapy of Upper Respiratory Airways Infections: Fair to Describe Them Just as Mucoactive? Clin. Med. Insights Ear Nose Throat 2019, 12, 1179550618821930. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.K.; Pan, I.H.; Wen, S.F.; Lin, M.N.; Lu, I.H.; Kuo, Z.K.; Lu, C.H.; Lee, T.C.; Yang, Y.Y.; Liaw, J.H. Method for Treating an Autoimmune Neurological Disease and/or Neurodegenerative Disease and Pharmaceutical Formulations for a Liquid Dosage form and a Controlled Release Dosage form. U.S. Patent Pub. No.: US20190000776A1, 3 January 2019. [Google Scholar]
- Ng, X.; Sadeghian, M.; Heales, S.; Hargreaves, I.P. Assessment of Mitochondrial Dysfunction in Experimental Autoimmune Encephalomyelitis (EAE) Models of Multiple Sclerosis. Int. J. Mol. Sci. 2019, 20, 4975. [Google Scholar] [CrossRef] [PubMed]
- Stavropoulos, F.; Georgiou, E.; Sargiannidou, I.; Kleopa, K.A. Dysregulation of Blood-Brain Barrier and Exacerbated Inflammatory Response in Cx47-Deficient Mice after Induction of EAE. Pharmaceuticals 2021, 14, 621. [Google Scholar] [CrossRef] [PubMed]
- Serra, A.; Fox, R.J. Dimethyl fumarate for relapsing MS. Neurol Clin. Pract. 2013, 3, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Spencer, C.M.; Crabtree-Hartman, E.C.; Lehmann-Horn, K.; Cree, B.A.; Zamvil, S.S. Reduction of CD8(+) T lymphocytes in multiple sclerosis patients treated with dimethyl fumarate. Neurol Neuroimmunol. Neuroinflamm. 2015, 2, e76. [Google Scholar] [CrossRef]
- Palugan, L.; Cerea, M.; Zema, L.; Gazzaniga, A.; Maroni, A. Coated pellets for oral colon delivery. J. Drug Deliv. Sci. Technol. 2015, 25, 1–15. [Google Scholar] [CrossRef]
- Abdul, S.; Chandewar, A.V.; Jaiswal, S.B. A flexible technology for modified-release drugs: Multiple-unit pellet system (MUPS). J. Control Release 2010, 147, 2–16. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Jin, X.; Sun, Y.; Xu, F.; Zhang, M. Production and investigation of sustained berberine pellet drug release system. Adv. Powder Technol. 2018, 29, 682–691. [Google Scholar] [CrossRef]
- Lopez, F.L.; Bowles, A.; Gul, M.O.; Clapham, D.; Ernest, T.B.; Tuleu, C. Effect of formulation variables on oral grittiness and preferences of multiparticulate formulations in adult volunteers. Eur. J. Pharm. Sci. 2016, 92, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Wadher, K.J.; Kakde, R.B.; Umekar, M.J. Study on sustained-release metformin hydrochloride from matrix tablet: Influence of hydrophilic polymers and in vitro evaluation. Int. J. Pharm. Investig. 2011, 1, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Albanez, R.; Nitz, M.; Taranto, O.P. Enteric coating process of diclofenac sodium pellets in a fluid bed coater with a wurster insert: Influence of process variables on coating performance and release profile. Adv. Powder Technol. 2013, 24, 659–666. [Google Scholar] [CrossRef]
- Dey, N.; Majumdar, S.; Rao, M. Multiparticulate drug delivery systems for controlled release. Trop. J. Pharm. Res. 2008, 7, 1067–1075. [Google Scholar] [CrossRef]
- Kushwah, V.; Arora, S.; Tamás Katona, M.; Modhave, D.; Fröhlich, E.; Paudel, A. On Absorption Modeling and Food Effect Prediction of Rivaroxaban, a BCS II Drug Orally Administered as an Immediate-Release Tablet. Pharmaceutics 2021, 13, 283. [Google Scholar] [CrossRef] [PubMed]
- Chavda, V.P.; Shah, D.; Tandel, H.; Soniwala, M. In Vitro–In Vivo Correlation (IVIVC): A Strategic Tool in Drug Product Development. J. Drug Formul. Dev. Prod. 2016, 3, 31–54. [Google Scholar]
- Sipos, B.; Szabó-Révész, P.; Csóka, I.; Pallagi, E.; Dobó, D.G.; Bélteky, P.; Kónya, Z.; Deák, Á.; Janovák, L.; Katona, G. Quality by Design Based Formulation Study of Meloxicam-Loaded Polymeric Micelles for Intranasal Administration. Pharmaceutics 2020, 12, 697. [Google Scholar] [CrossRef] [PubMed]
- Braga, P.C.; De Angelis, L.; Bossi, R.; Scaglione, F.; Scarpazza, G.; Allegra, L.; Fraschini, F. Pharmacokinetics of Sobrerol in chronic bronchitis. Comparison of serum and bronchial mucus levels. Eur. J. Clin. Pharm. 1983, 24, 209–215. [Google Scholar] [CrossRef]
- Yalkowsky, S.H.; He, Y.; Jain, P. Handbook of Aqueous Solubility Data, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2016; p. 731. [Google Scholar]
- Kambayashi, A.; Blume, H.; Dressman, J.B. Predicting the oral pharmacokinetic profiles of multiple-unit (pellet) dosage forms using a modeling and simulation approach coupled with biorelevant dissolution testing: Case example diclofenac sodium. Eur. J. Pharm. Biopharm. 2014, 87, 236–243. [Google Scholar] [CrossRef]
- Vlachou, M.; Karalis, V. An In Vitro–In Vivo Simulation Approach for the Prediction of Bioequivalence. Materials 2021, 14, 555. [Google Scholar] [CrossRef] [PubMed]
- Muley, S.; Nandgude, T.; Poddar, S. Extrusion–spheronization a promising pelletization technique: In-depth review. Asian J. Pharm. Sci. 2016, 11, 684–699. [Google Scholar] [CrossRef]
- Broesder, A.; Bircan, S.Y.; de Waard, A.B.; Eissens, A.C.; Frijlink, H.W.; Hinrichs, W.L.J. Formulation and In Vitro Evaluation of Pellets Containing Sulfasalazine and Caffeine to Verify Ileo-Colonic Drug Delivery. Pharmaceutics 2021, 13, 1985. [Google Scholar] [CrossRef] [PubMed]
- KFDA. The Korean Pharmacopoeia, 11th ed.; Korea Food and Drug Administration: Seoul, Korea, 2014; pp. 758–759.
- Jantratid, E.; De Maio, V.; Ronda, E.; Mattavelli, V.; Vertzoni, M.; Dressman, J.B. Application of biorelevant dissolution tests to the prediction of in vivo performance of diclofenac sodium from an oral modified-release pellet dosage form. Eur. J. Pharm. Sci. 2009, 37, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.; Lalwani, D.; Gollmer, S.; Injeti, E.; Sari, Y.; Nesamony, J. Development and evaluation of a calcium alginate based oral ceftriaxone sodium formulation. Prog. Biomater. 2016, 5, 117–133. [Google Scholar] [CrossRef]
- Puhl, D.L.; Funnell, J.L.; D’Amato, A.R.; Bao, J.; Zagorevski, D.V.; Pressman, Y.; Morone, D.; Haggerty, A.E.; Oudega, M.; Gilbert, R.J. Aligned Fingolimod-Releasing Electrospun Fibers Increase Dorsal Root Ganglia Neurite Extension and Decrease Schwann Cell Expression of Promyelinating Factors. Front. Bioeng. Biotechnol. 2020, 8, 937. [Google Scholar] [CrossRef]
- Venkatesh, G.M.; Stevens, P.J.; Lai, J.W. Development of orally disintegrating tablets comprising controlled-release multiparticulate beads. Drug Dev. Ind. Pharm. 2012, 38, 1428–1440. [Google Scholar] [CrossRef][Green Version]
- Sánchez-Dengra, B.; González-García, I.; González-Álvarez, M.; González-Álvarez, I.; Bermejo, M. Two-step in vitro-in vivo correlations: Deconvolution and convolution methods, which one gives the best predictability? Comparison with one-step approach. Eur. J. Pharm. Biopharm. 2021, 158, 185–197. [Google Scholar] [CrossRef]
- Figueroa-Campos, A.; Sánchez-Dengra, B.; Merino, V.; Dahan, A.; González-Álvarez, I.; García-Arieta, A.; González-Álvarez, M.; Bermejo, M. Candesartan Cilexetil In Vitro–In Vivo Correlation: Predictive Dissolution as a Development Tool. Pharmaceutics 2020, 12, 633. [Google Scholar] [CrossRef]
- Qureshi, S.A. In vitro-in vivo correlation (ivivc) and determining drug concentrations in blood from dissolution testing–a simple and practical approach. Open Drug Deliv. J. 2010, 4, 38–47. [Google Scholar] [CrossRef]
- Hua, S. Advances in oral drug delivery for regional targeting in the gastrointestinal tract-Influence of physiological, pathophysiological and pharmaceutical factors. Front. Pharm. 2020, 11, 524. [Google Scholar] [CrossRef]
- Bounda, G.A.; Zhou, W.; Wang, D.D.; Yu, F. Rhein Elicits In Vitro Cytotoxicity in Primary Human Liver HL-7702 Cells by Inducing Apoptosis through Mitochondria-Mediated Pathway. Evid. Based Complement. Altern. Med. 2015, 2015, 329831. [Google Scholar] [CrossRef]
- Gupta, P.K. Illustrated Toxicology: With Study Questions, 1st ed.; Academic Press: Cambridge, MA, USA, 2018; p. 86. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | Factor A 1 %Wcoat | Factor B 2 XHPMC/EC | Factor C 3 Tcur |
|---|---|---|---|
| SR-1 | 0 | +1 | +1 |
| SR-2 | +1 | −1 | 0 |
| SR-3 | +1 | 0 | +1 |
| SR-4 | −1 | −1 | 0 |
| SR-5 | 0 | +1 | −1 |
| SR-6 | −1 | +1 | 0 |
| SR-7 | 0 | 0 | 0 |
| SR-8 | +1 | 0 | −1 |
| SR-9 | 0 | −1 | −1 |
| SR-10 | +1 | +1 | 0 |
| SR-11 | 0 | −1 | +1 |
| SR-12 | −1 | 0 | +1 |
| SR-13 | −1 | 0 | −1 |
| EAE Animal Tests | Human Plasma Simulations | |||||||
|---|---|---|---|---|---|---|---|---|
| In Vivo Dose (mg/kg) | Frequency (Daily) | EAE Score 1,2 | HED 3 (mg) | Inhibition 4,5 (%) | Cmax (μg/mL) | AUC (μg·h/mL) | t>MEC (h) | |
| Vehicle | thrice | 4.00 ± 0.22 | - | - | - | - | - | |
| 25 | thrice | 3.70 ± 0.15 | 122 | N/S | 1.48 | 13.59 | 0.0 | |
| 30 | once | 3.38 ± 0.27 | 146 | N/S | 1.61 | 5.67 | 0.0 | |
| 100 | thrice | 2.36 ± 0.27 ** | 486 | 41.00 | 5.91 | 54.13 | 12.8 | |
| 150 | twice | 2.55 ± 0.27 ** | 730 | 36.25 | 8.31 | 55.52 | 11.2 | |
| 300 | once | 2.78 ± 0.25 *** | 1459 | 30.50 | 16.11 | 56.61 | 8.2 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, C.-H.; Huang, Y.-F.; Chu, I.-M. Design of Oral Sustained-Release Pellets by Modeling and Simulation Approach to Improve Compliance for Repurposing Sobrerol. Pharmaceutics 2022, 14, 167. https://doi.org/10.3390/pharmaceutics14010167
Lu C-H, Huang Y-F, Chu I-M. Design of Oral Sustained-Release Pellets by Modeling and Simulation Approach to Improve Compliance for Repurposing Sobrerol. Pharmaceutics. 2022; 14(1):167. https://doi.org/10.3390/pharmaceutics14010167
Chicago/Turabian StyleLu, Chu-Hsun, Yu-Feng Huang, and I-Ming Chu. 2022. "Design of Oral Sustained-Release Pellets by Modeling and Simulation Approach to Improve Compliance for Repurposing Sobrerol" Pharmaceutics 14, no. 1: 167. https://doi.org/10.3390/pharmaceutics14010167
APA StyleLu, C.-H., Huang, Y.-F., & Chu, I.-M. (2022). Design of Oral Sustained-Release Pellets by Modeling and Simulation Approach to Improve Compliance for Repurposing Sobrerol. Pharmaceutics, 14(1), 167. https://doi.org/10.3390/pharmaceutics14010167