
Imination of Microporous Chitosan Fibers—A Route to Biomaterials with “On Demand” Antimicrobial Activity and Biodegradation for Wound Dressings

 , ,
, ,  ,
,  and
and 
Abstract
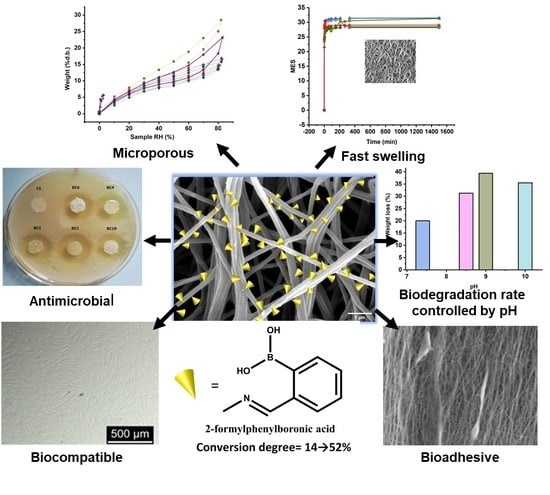
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental
2.1. Materials
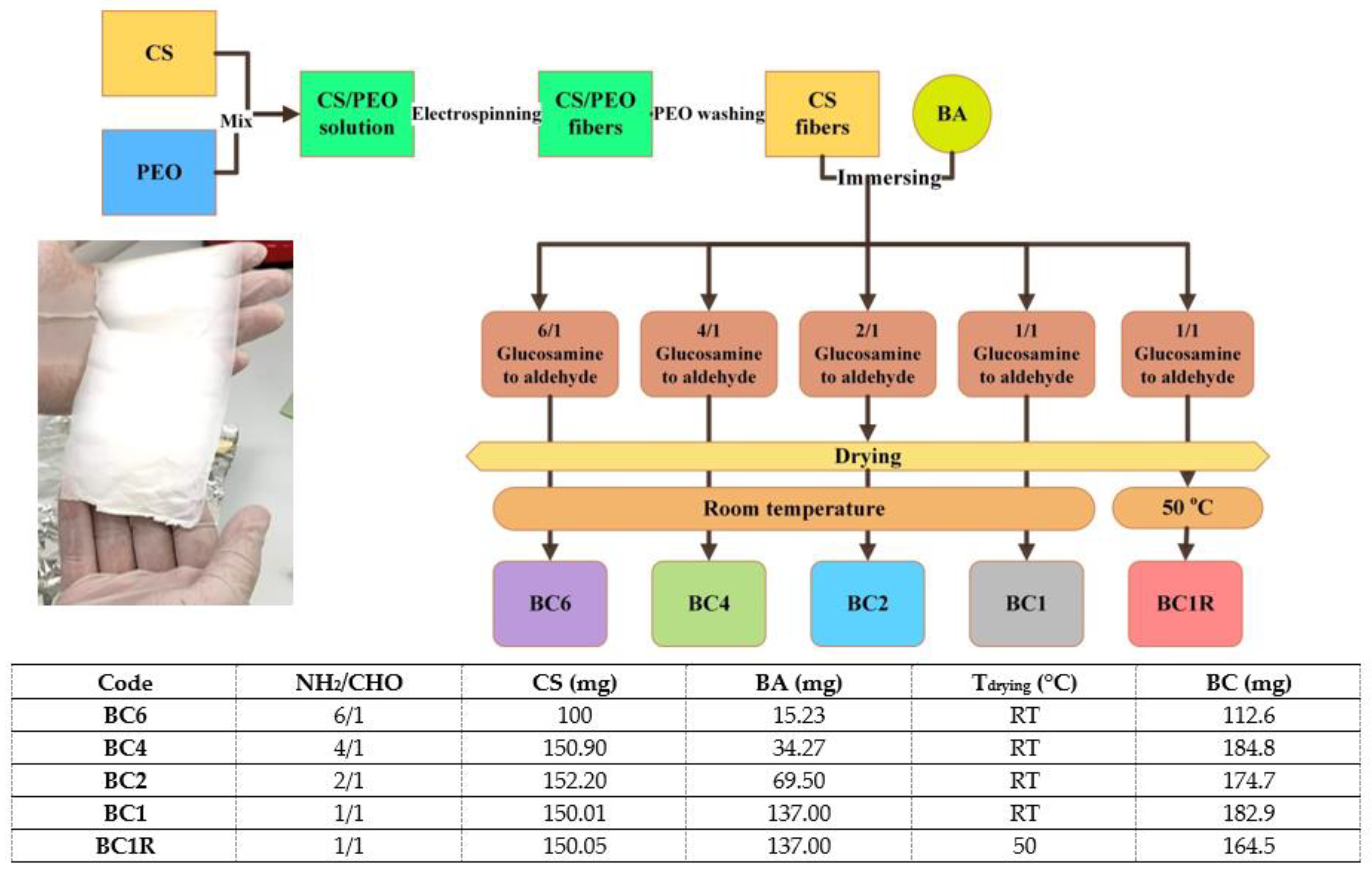
2.2. Fiber Preparation
- (1)
- Chitosan/poly (ethylene oxide) (CS/PEO) fibers were prepared by electrospinning a 2.1 g/mL solution of CS/PEO (2/1, w/w) in 80% acetic acid. The solution was loaded in a 5 mL syringe with a blunt needle with an inner diameter of 0.8 mm. The electrospinning was done at room temperature, applying a voltage of 7 kV, a tip-to-collector distance of 10 cm, a flow rate of 0.4 mL/h and a rotary drum collector speed of 800 rpm.
- (2)
- Microporous neat CS fibers were prepared by washing the PEO [20] from CS/PEO fibers with a 5% NaOH solution in order to remove the residual acetic acid, and then with distilled water to remove the PEO and to reach the neutral pH. Finally, the wet fibers were lyophilized in order to preserve the porosity gained by PEO washing.
- (3)
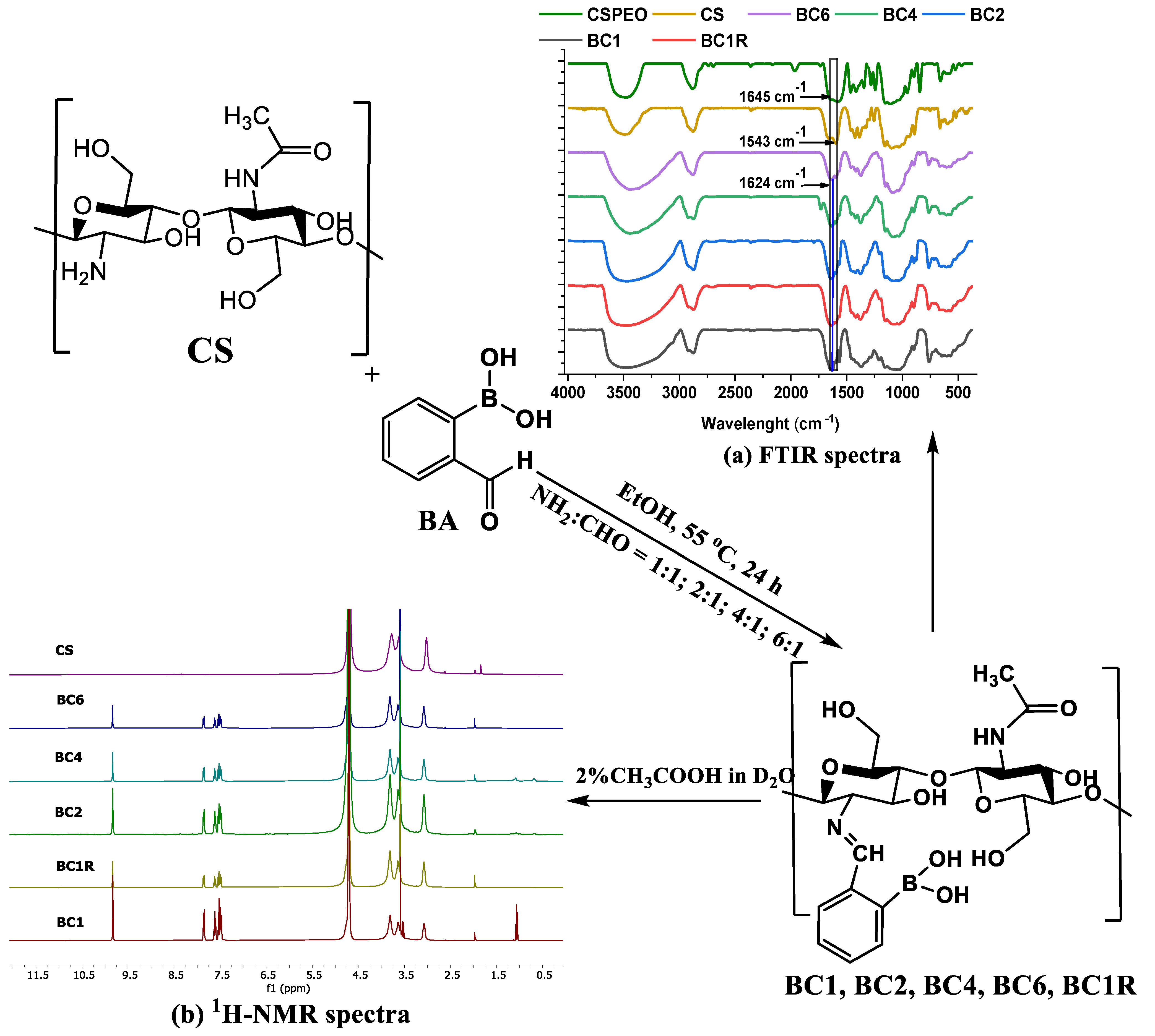
- The CS fibers were functionalized by imination reaction with 2-formylphenylboronic acid in a heterogeneous system, to give imino-chitosan fibers, coded BC. The fiber mat was immersed into a vessel containing 10 mL solution of aldehyde in ethanol and kept sealed at 55 °C, for 24 h. To obtain a series of BC fibers with different content of imine units, the molar ratio between glucosamine units of chitosan and 2-formylphenylboronic acid was varied from 1/1 up to 6/1 (Scheme 1). When the reaction time ended, the vessel was allowed to reach room temperature and then it was unsealed to allow the ethanol removal and fibers’ drying. Then, the fibers were washed with dry ethanol to remove the unreacted aldehyde and dried in atmospheric conditions. The sample with a 1/1 molar ratio of the functional groups was also prepared and rapidly dried at 50 °C after the reaction time passed. A representation of the preparation procedure and the sample codes is illustrated in Scheme 1.
2.3. Equipment and Measurements
3. Results and Discussion
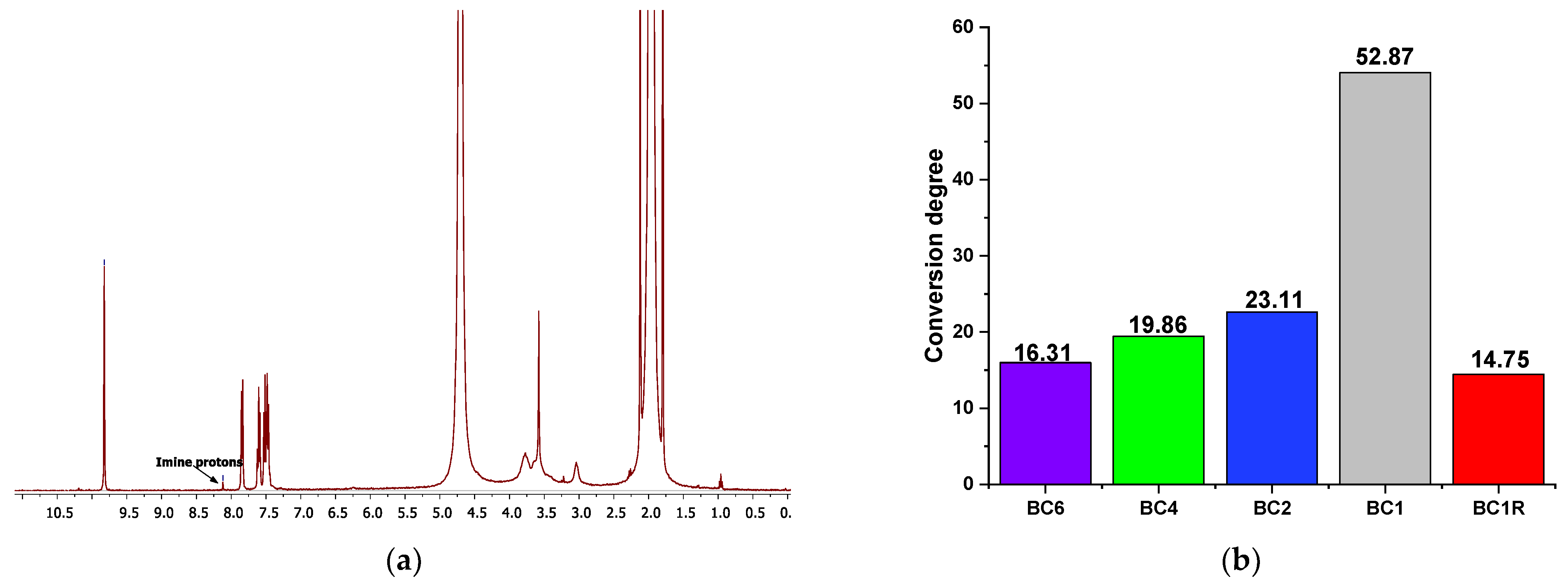
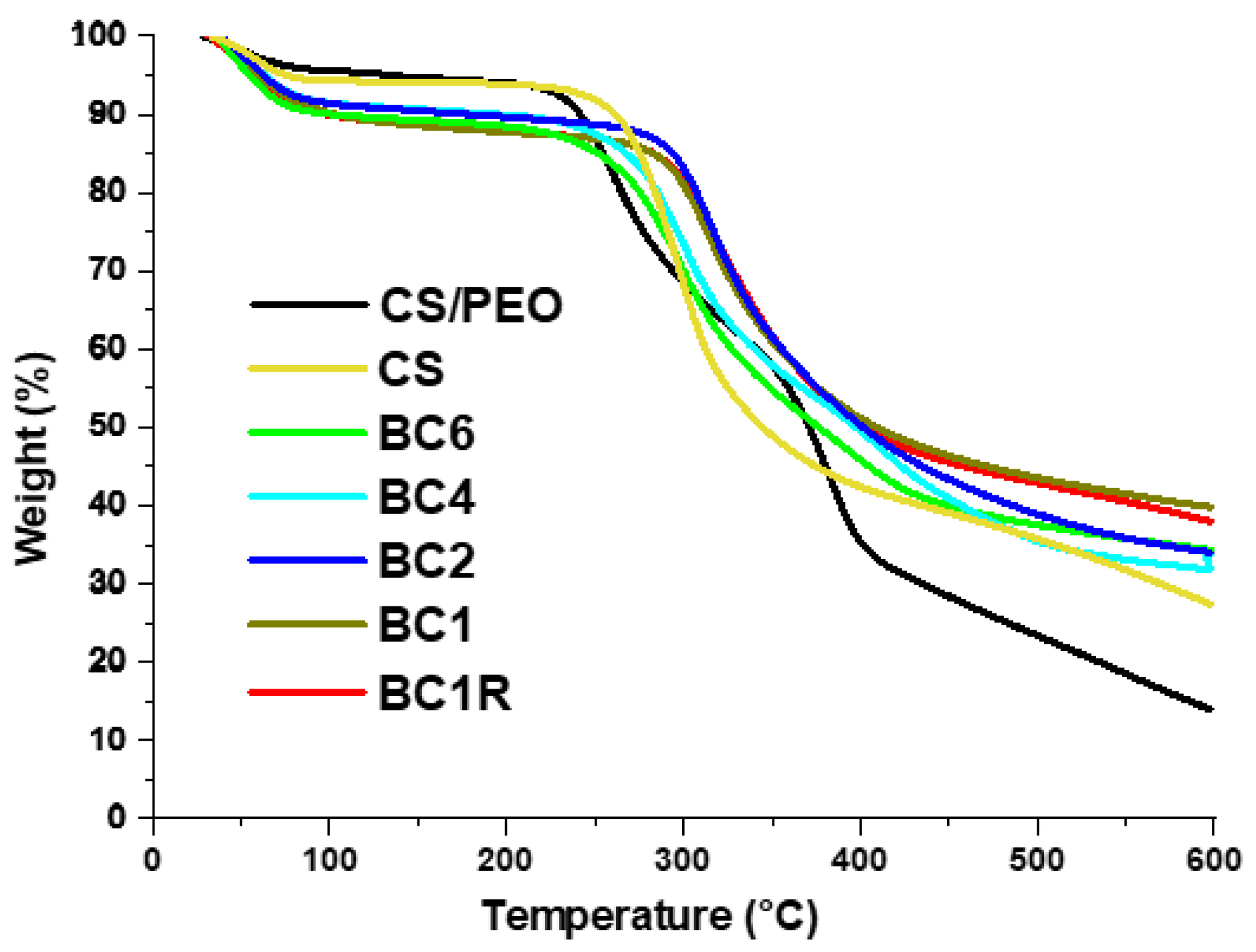
3.1. Structural Characterization of the Imino-Chitosan Fibers
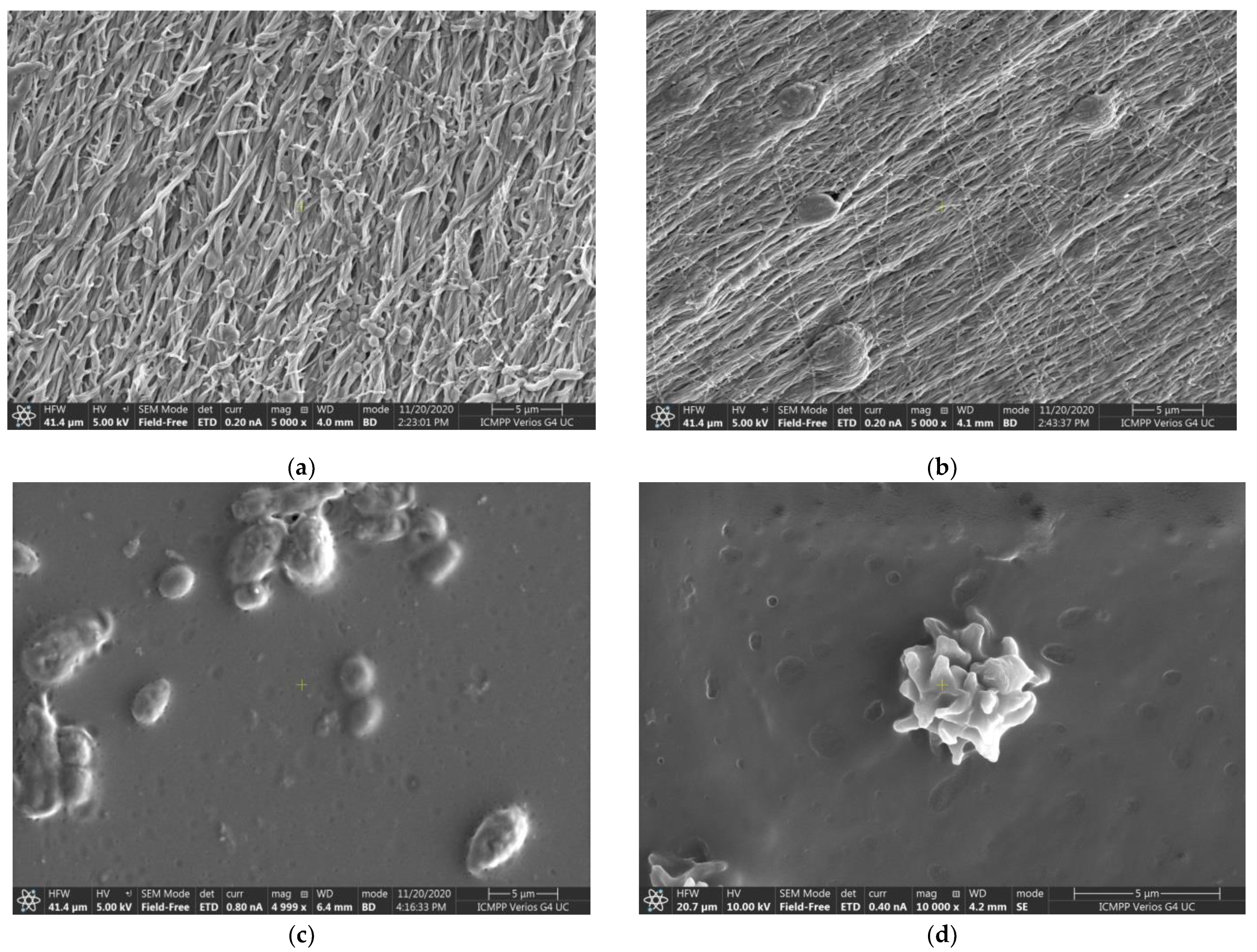
3.2. Fiber Mat Morphology
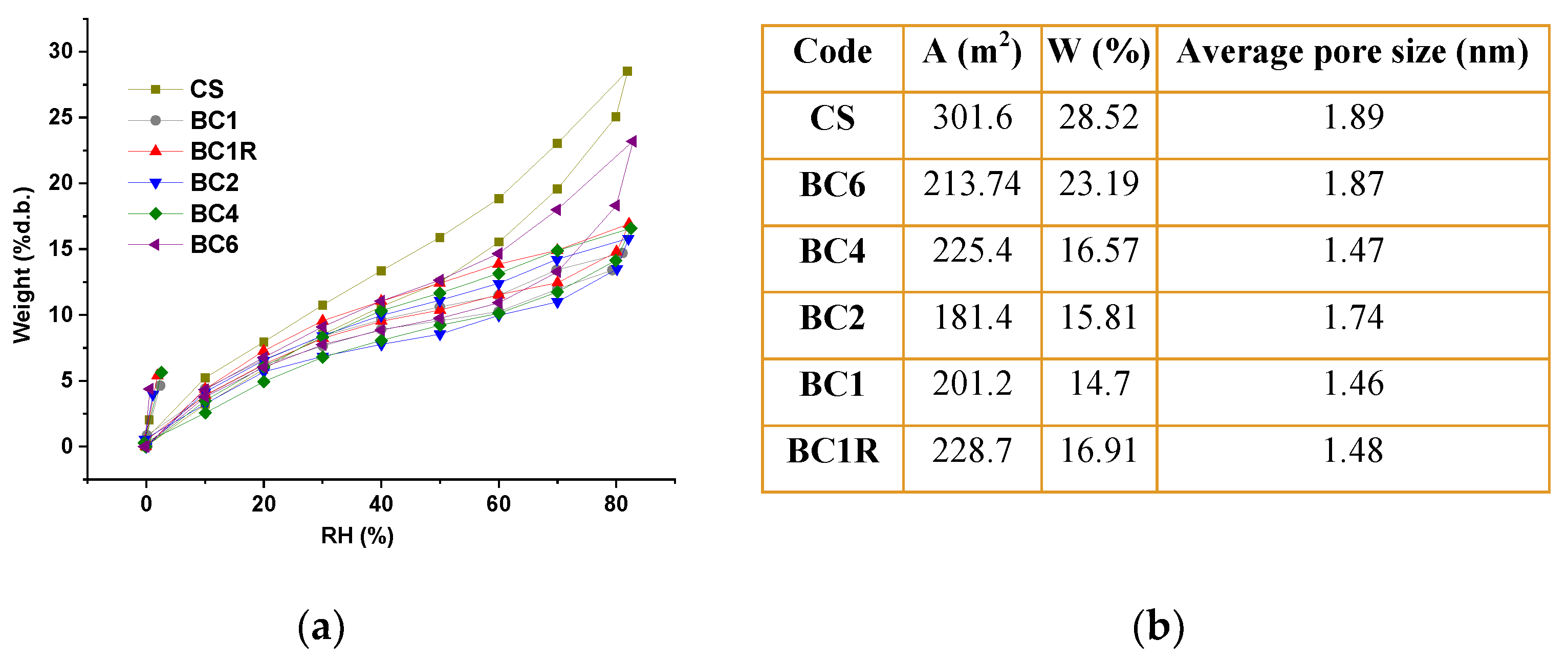
3.3. Swelling Behaviour
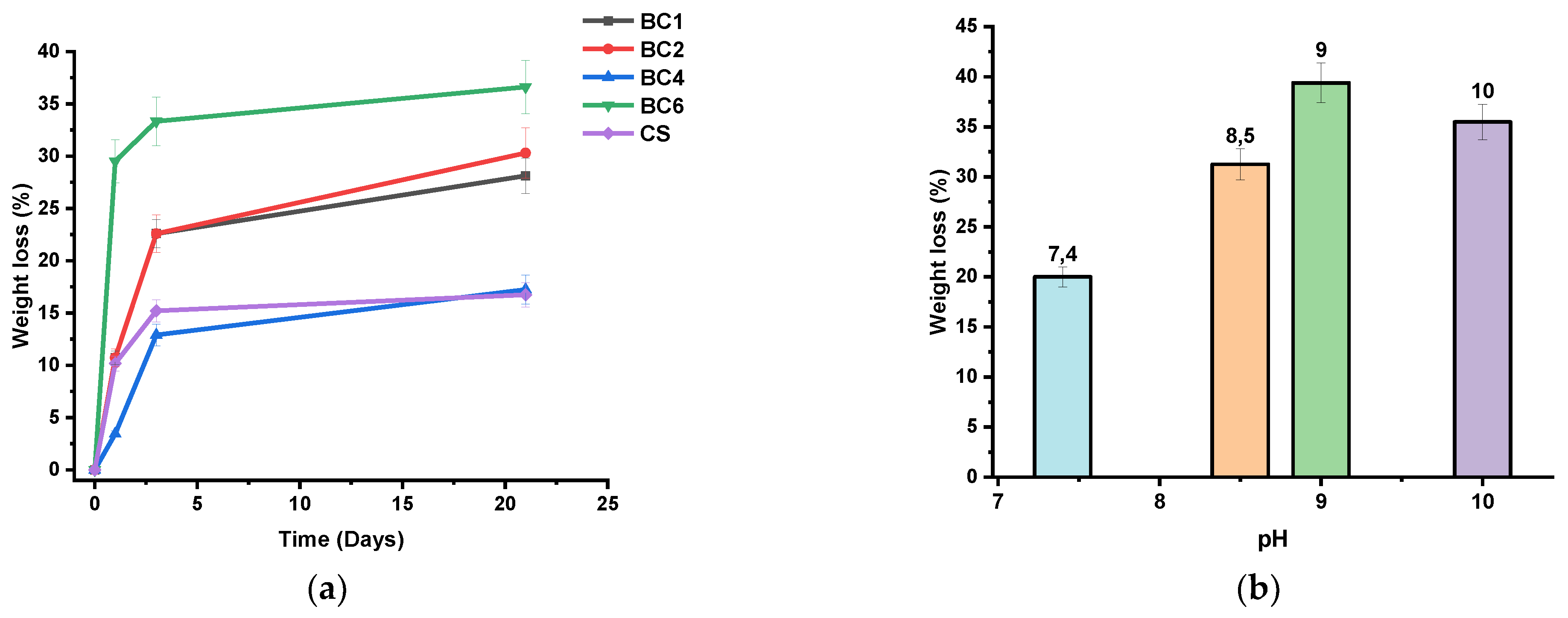
3.4. Fiber Mat Biodegradation
3.5. Antimicrobial Activity
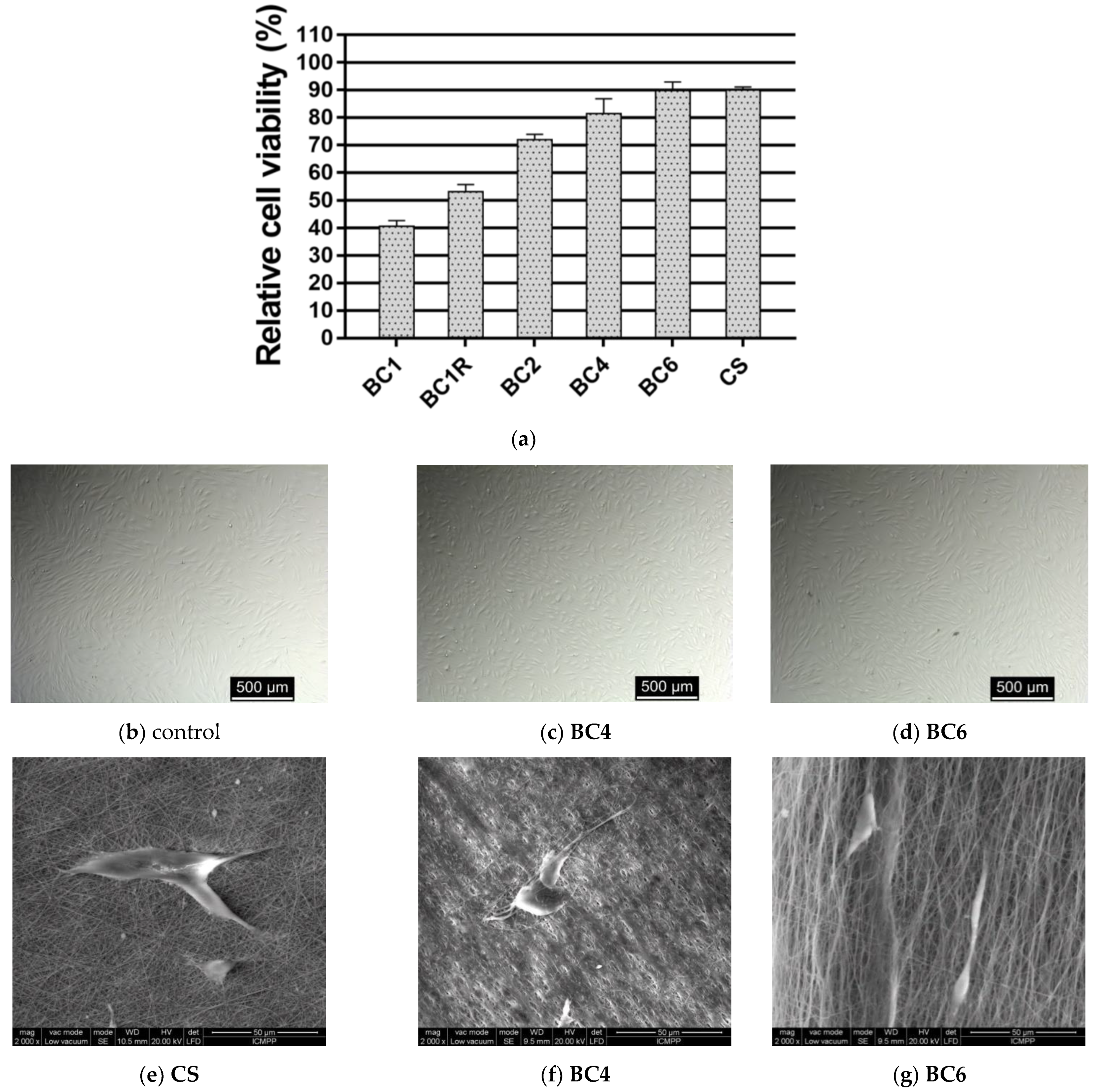
3.6. Biocompatibility
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stokes, M.A.R.; Johnson, W.D. Burns in the Third World: An unmet need. Ann. Burn. Fire Disasters 2017, 30, 243–246. [Google Scholar]
- Norbury, W.; Herndon, D.N.; Tanksley, J.; Jeschke, M.G.; Finnerty, C.C. Infection in Burns. Surg. Infect. 2016, 17, 250–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caló, E.; Khutoryanskiy, V.V. Biomedical applications of hydrogels: A review of patents and commercial products. Eur. Polym. J. 2015, 65, 252–267. [Google Scholar] [CrossRef] [Green Version]
- Kamińska, M.S.; Cybulska, A.M.; Skonieczna-Żydecka, K.; Augustyniuk, K.; Grochans, E.; Karakiewicz, B. Effectiveness of Hydrocolloid Dressings for Treating Pressure Ulcers in Adult Patients: A Systematic Review and Meta-Analysis. Int. J. Environ. Res. Public Health 2020, 17, 7881. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Zhang, H.; Yang, S.; Xi, Z.; Tang, T.; Yin, R.; Zhang, W. Electrospun PLGA membrane incorporated with andrographolide-loaded mesoporous silica nanoparticles for sustained antibacterial wound dressing. Nanomedicine 2018, 13, 2881–2899. [Google Scholar] [CrossRef] [PubMed]
- Lungu, R.; Anisiei, A.; Rosca, I.; Sandu, A.I.; Ailincai, D.; Marin, L. Double functionalization of chitosan based nanofibers towards biomaterials for wound healing. React. Funct. Polym. 2021, 167, 105028. [Google Scholar] [CrossRef]
- Iacob, A.T.; Drăgan, M.; Ionescu, O.M.; Profire, L.; Ficai, A.; Andronescu, E.; Confederat, L.G.; Lupașcu, D. An Overview of Biopolymeric Electrospun Nanofibers Based on Polysaccharides for Wound Healing Management. Pharmaceutics 2020, 12, 983. [Google Scholar] [CrossRef] [PubMed]
- Râpă, M.; Gaidau, C.; Mititelu-Tartau, L.; Berechet, M.D.; Berbecaru, A.C.; Rosca, I.; Chiriac, A.; Matei, E.; Predescu, A.M.; Predescu, C. Bioactive Collagen Hydrolysate-Chitosan/Essential Oil Electrospun Nanofibers Designed for Medical Wound Dressings. Pharmaceutics 2021, 13, 1939. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.; Luo, Y.; Ke, C.; Qiu, H.; Wang, W.; Zhu, Y.; Hou, R.; Xu, L.; Wu, S. Chitosan-Based Functional Materials for Skin Wound Repair: Mechanisms and Applications. Front. Bioeng. Biotechnol. 2021, 9, 111. [Google Scholar] [CrossRef] [PubMed]
- Hashemikia, S.; Farhangpazhouh, F.; Parsa, M.; Hasan, M.; Hassanzadeh, A.; Hamidi, M. Fabrication of ciprofloxacin-loaded chitosan/polyethylene oxide/silica nanofibers for wound dressing application: In Vitro and In Vivo evaluations. Int. J. Pharm. 2021, 597, 120313. [Google Scholar] [CrossRef]
- Hu, C.; Long, L.; Cao, J.; Zhang, S.; Wang, Y. Dual-crosslinked mussel-inspired smart hydrogels with enhanced antibacterial and angiogenic properties for chronic infected diabetic wound treatment via pH-responsive quick cargo release. Chem. Eng. J. 2021, 411, 128564. [Google Scholar] [CrossRef]
- Schulte-Werning, L.V.; Murugaiah, A.; Singh, B.; Johannessen, M.; Engstad, R.I.; Škalko-Basnet, N.; Holsæter, A.M. Multifunctional Nanofibrous Dressing with Antimicrobial and Anti-Inflammatory Properties Prepared by Needle-Free Electrospinning. Pharmaceutics 2021, 13, 1527. [Google Scholar] [CrossRef]
- Shi, R.; Niu, Y.; Gong, M.; Ye, J.; Tian, W.; Zhang, L. Antimicrobial gelatin-based elastomer nanocomposite membrane loaded with ciprofloxacin and polymyxin B sulfate in halloysite nanotubes for wound dressing. Mater. Sci. Eng. C 2018, 87, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Wang, Y.; Huang, Z.; Wang, X.; Chen, L.; Zhang, Y.; Zhang, L. On-Demand Dissolvable Self-Healing Hydrogel Based on Carboxymethyl Chitosan and Cellulose Nanocrystal for Deep Partial Thickness Burn Wound Healing. ACS Appl. Mater. Interfaces 2018, 10, 41076–41088. [Google Scholar] [CrossRef] [PubMed]
- Alazab, M.; Mitchell, G.R.; Davis, F.J.; Mohan, S.D. Sustainable Electrospinning of Nanoscale Fibres. Procedia Manuf. 2017, 12, 66–78. [Google Scholar] [CrossRef]
- Anisiei, A.; Oancea, F.; Marin, L. Electrospinning of chitosan-based nanofibers: From design to prospective applications. Rev. Chem. Eng. 2021. [Google Scholar] [CrossRef]
- Baumann, A.; Tuerck, D.; Prabhu, S.; Dickmann, L.; Sims, J. Pharmacokinetics, metabolism and distribution of PEGs and PEGylated proteins: Quo vadis? Drug Discov. Today 2014, 19, 1623–1631. [Google Scholar] [CrossRef] [PubMed]
- Halima, N.B. Poly(vinyl alcohol): Review of its promising applications and insights into biodegradation. RSC Adv. 2016, 6, 39823. [Google Scholar] [CrossRef]
- Jayakumar, R.; Prabaharan, M.; Sudheesh Kumar, P.T.; Nair, S.V.; Tamura, H. Biomaterials based on chitin and chitosan in wound dressing applications. Biotechnol. Adv. 2011, 29, 322–337. [Google Scholar] [CrossRef] [PubMed]
- Marin, L.; Dragoi, B.; Olaru, N.; Perju, E.; Coroaba, A.; Doroftei, F.; Scavia, G.; Destri, S.; Zappia, S.; Porzio, W. Nanoporous furfuryl-imine-chitosan fibers as a new pathway towards eco-materials for CO2 adsorption. Eur. Polym. J. 2019, 120, 109214. [Google Scholar] [CrossRef]
- Marin, L.; Simionescu, B.; Barboiu, M. Imino-chitosan biodynamers. Chem. Commun. 2012, 48, 8778. [Google Scholar] [CrossRef] [PubMed]
- Dragan, E.S.; Cazacu, M.; Nistor, A. Ionic organic/inorganic materials. III. Stimuli responsive hybrid hydrogels based on oligo(N,N-dimethylaminoethylmethacrylate) and chloroalkyl-functionalized siloxanes. J. Polym. Sci. Part A Polym. Chem. 2009, 47, 6801–6813. [Google Scholar] [CrossRef]
- Addinsoft. XLSTAT Statistical and Data Analysis Solution. 2020. Available online: https://www.xlstat.com (accessed on 17 May 2021).
- dos Santos, A.E.A.; dos Santos, F.V.; Freitas, K.M.; Pimenta, L.P.S.; de Oliveira Andrade, L.; Marinho, T.A.; de Avelar, G.F.; da Silva, A.B.; Ferreira, R.V. Cellulose acetate nanofibers loaded with crude annatto extract: Preparation, characterization, and in vivo evaluation for potential wound healing applications. Mater. Sci. Eng. C 2021, 118, 111322. [Google Scholar] [CrossRef]
- Ailincai, D.; Marin, L.; Morariu, S.; Mares, M.; Bostanaru, A.-C.; Pinteala, M.; Simionescu, B.C.; Barboiu, M. Dual crosslinked iminoboronate-chitosan hydrogels with strong antifungal activity against Candida planktonic yeasts and biofilms. Carbohydr. Polym. 2016, 152, 306–316. [Google Scholar] [CrossRef]
- Anisiei, A.; Bostanaru, A.C.; Mares, M.; Marin, L. Imination of Chitosan Nanofibers in a Heterogeneous System. Synthesis Optimization and Impact on Fiber Morphology. Cellul. Chem. Technol. 2021, 55, 785–793. [Google Scholar] [CrossRef]
- Fernandes Queiroz, M.; Melo, K.; Sabry, D.; Sassaki, G.; Rocha, H. Does the Use of Chitosan Contribute to Oxalate Kidney Stone Formation? Mar. Drugs 2014, 13, 141–158. [Google Scholar] [CrossRef]
- Adamczyk-Woźniak, A.; Cyrański, M.K.; Frączak, B.T.; Lewandowska, A.; Madura, I.D.; Sporzyński, A. Imino- and aminomethylphenylboronic acids: Stabilizing effect of hydrogen bonds. Tetrahedron 2012, 68, 3761–3767. [Google Scholar] [CrossRef]
- He, M.; Lehn, J.-M. Time-Dependent Switching of Constitutional Dynamic Libraries and Networks from Kinetic to Thermodynamic Distributions. J. Am. Chem. Soc. 2019, 141, 18560–18569. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Pham, Q.; Yu, R.; Petit, E.; Li, S.; Barboiu, M. Dynamic Hydrogels Based on Double Imine Connections and Application for Delivery of Fluorouracil. Front. Chem. 2020, 8, 739. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Han, W.; Chen, H.; Tu, M.; Zeng, Z.; Shi, Y.; Cha, Z.; Zhou, C. Preparation, structure and crystallinity of chitosan nano-fibers by a solid–liquid phase separation technique. Carbohydr. Polym. 2001, 83, 1541–1546. [Google Scholar] [CrossRef]
- Sing, K.S.W.; Williams, R.T. Physisorption Hysteresis Loops and the Characterization of Nanoporous Materials. Adsorpt. Sci. Technol. 2004, 22, 773–782. [Google Scholar] [CrossRef]
- Bonardd, S.; Schmidt, M.; Saavedra-Torres, M.; Leiva, A.; Radic, D.; Saldías, C. Thermal and morphological behavior of chitosan/PEO blends containing gold nanoparticles. Experimental and theoretical studies. Carbohydr. Polym. 2016, 144, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Marin, L.; Ailincai, D.; Mares, M.; Paslaru, E.; Cristea, M.; Nica, V.; Simionescu, B.C. Imino-chitosan biopolymeric films. Obtaining, self-assembling, surface and antimicrobial properties. Carbohydr. Polym. 2015, 117, 762–770. [Google Scholar] [CrossRef] [PubMed]
- Corazzari, I.; Nisticò, R.; Turci, F.; Faga, M.G.; Franzoso, F.; Tabasso, S.; Magnacca, G. Advanced physico-chemical characterization of chitosan by means of TGA coupled on-line with FTIR and GCMS: Thermal degradation and water adsorption capacity. Polym. Degrad. Stab. 2015, 112, 1–9. [Google Scholar] [CrossRef]
- Lemma, S.M.; Bossard, F.; Rinaudo, M. Preparation of Pure and Stable Chitosan Nanofibers by Electrospinning in the Presence of Poly (ethylene oxide). Int. J. Mol. Sci. 2016, 17, 1790. [Google Scholar] [CrossRef] [Green Version]
- Dhivya, S.; Padma, V.V.; Santhini, E. Wound dressings—A review. BioMedicine 2015, 5, 22. [Google Scholar] [CrossRef]
- Garciam, C.E.; Bossard, F.; Rinaudo, M. Electrospun Biomaterials from Chitosan Blends Applied as Scaffold for Tissue Regeneration. Polymers 2021, 13, 1037. [Google Scholar] [CrossRef]
- Ostrowska-Czubenko, J.; Gierszewska, M.; Pieróg, M. pH-responsive hydrogel membranes based on modified chitosan: Water transport and kinetics of swelling. J. Polym. Res. 2015, 22, 153. [Google Scholar] [CrossRef] [Green Version]
- Ono, S.; Imai, R.; Ida, Y.; Shibata, D.; Komiya, T.; Matsumura, H. Increased wound pH as an indicator of local wound infection in second degree burns. Burns 2015, 41, 820–824. [Google Scholar] [CrossRef]
- Hasmann, A.; Wehrschuetz-Sigl, E.; Kanzler, G.; Gewessler, U.; Hulla, E.; Schneider, K.P.; Binder, B.; Schintler, M.; Guebitz, G.M. Novel peptidoglycan-based diagnostic devices for detection of wound infection. Diagn. Microbiol. Infect. Dis. 2011, 71, 12–23. [Google Scholar] [CrossRef]
- Hirano, S.; Yagi, Y. The effects of N-substitution of chitosan and the physical form of the products on the rate of hydrolysis by chitinase from Streptomyces griseus. Carbohydr. Res. 1980, 83, 103–108. [Google Scholar] [CrossRef]
- Jones, E.M.; Cochrane, C.A.; Percival, S.L. The Effect of pH on the Extracellular Matrix and Biofilms. Adv. Wound Care 2015, 4, 431–439. [Google Scholar] [CrossRef]
- Rippke, F.; Berardesca, E.; Weber, T.M. pH and Microbial Infections. Curr. Probl. Dermatol. 2018, 54, 87–94. [Google Scholar] [CrossRef]
- Goy, R.C.; de Britto, D.; Assis, O.B.G. A review of the antimicrobial activity of chitosan. Polímeros 2009, 19, 241–247. [Google Scholar] [CrossRef]
- Marin, L.; Stoica, I.; Mares, M.; Dinu, V.; Simionescu, B.C.; Barboiu, M. Antifungal vanillin-imino-chitosan biodynameric films. J. Mater. Chem. B 2013, 1, 3353–3358. [Google Scholar] [CrossRef] [PubMed]
- Borys, K.M.; Wieczorek, D.; Pecura, K.; Lipok, J.; Adamczyk-Woźniak, A. Antifungal activity and tautomeric cyclization equilibria of formylphenylboronic acids. Bioorg. Chem. 2019, 91, 103081. [Google Scholar] [CrossRef]
- Alebachew, T.; Yismaw, G.; Derabe, A.; Sisay, Z. Staphylococcus aureus burn wound infection among patients attending yekatit 12 hospital burn unit, addis ababa, ethiopia. Ethiop. J. Health Sci. 2012, 22, 209–213. [Google Scholar] [PubMed]
- Church, D.; Elsayed, S.; Reid, O.; Winston, B.; Lindsay, R. Burn Wound Infections. Clin. Microbiol. Rev. 2006, 19, 403–434. [Google Scholar] [CrossRef] [Green Version]
- ISO. Biological Evaluation of Medical Devices—Part 5: Tests for In Vitro Cytotoxicity; International Organization for Standardization: Geneva, Switzerland, 2009. [Google Scholar]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anisiei, A.; Rosca, I.; Sandu, A.-I.; Bele, A.; Cheng, X.; Marin, L. Imination of Microporous Chitosan Fibers—A Route to Biomaterials with “On Demand” Antimicrobial Activity and Biodegradation for Wound Dressings. Pharmaceutics 2022, 14, 117. https://doi.org/10.3390/pharmaceutics14010117
Anisiei A, Rosca I, Sandu A-I, Bele A, Cheng X, Marin L. Imination of Microporous Chitosan Fibers—A Route to Biomaterials with “On Demand” Antimicrobial Activity and Biodegradation for Wound Dressings. Pharmaceutics. 2022; 14(1):117. https://doi.org/10.3390/pharmaceutics14010117
Chicago/Turabian StyleAnisiei, Alexandru, Irina Rosca, Andreea-Isabela Sandu, Adrian Bele, Xinjian Cheng, and Luminita Marin. 2022. "Imination of Microporous Chitosan Fibers—A Route to Biomaterials with “On Demand” Antimicrobial Activity and Biodegradation for Wound Dressings" Pharmaceutics 14, no. 1: 117. https://doi.org/10.3390/pharmaceutics14010117
APA StyleAnisiei, A., Rosca, I., Sandu, A.-I., Bele, A., Cheng, X., & Marin, L. (2022). Imination of Microporous Chitosan Fibers—A Route to Biomaterials with “On Demand” Antimicrobial Activity and Biodegradation for Wound Dressings. Pharmaceutics, 14(1), 117. https://doi.org/10.3390/pharmaceutics14010117