
Structure-Functional Analysis of Human Cytochrome P450 2C8 Using Directed Evolution


Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Construction of P450 2C8 Random Mutant Libraries
2.3. Screening of CYP2C8 Libraries
2.4. Expression and Purification of Wild-Type and Mutant CYP2C8
2.5. Catalytic Activity Assay
2.6. Binding Spectral Titration of Wild-Type and Mutant CYP2C8s
2.7. Molecular Docking Modeling of CYP2C8
3. Results
3.1. Random Mutagenesis and Selection of CYP2C8 Mutants
3.2. Expression and Purification of Recombinant Wild-Type and Mutant CYP2C8s
3.3. Catalytic Activities of Selected CYP2C8 Mutants
3.4. Binding of Substrate to CYP2C8 Mutants
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Omura, T.; Sato, R. The Carbon Monoxide-Binding Pigment of Liver Microsomes. I. Evidence for Its Hemoprotein Nature. J. Biol. Chem. 1964, 239, 2370–2378. [Google Scholar] [CrossRef]
- Guengerich, F.P. Cytochrome p450 and chemical toxicology. Chem. Res. Toxicol. 2008, 21, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.R.; Zeldin, D.C.; Hoffman, S.M.; Maltais, L.J.; Wain, H.M.; Nebert, D.W. Comparison of cytochrome P450 (CYP) genes from the mouse and human genomes, including nomenclature recommendations for genes, pseudogenes and alternative-splice variants. Pharmacogenetics 2004, 14, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Guengerich, F.P. Human cytochrome P450 enzymes. In Cytochrome P450: Structure, Mechanism, and Biochemistry, 4th ed.; Ortiz de Montellano, P.R., Ed.; Springer: London, UK, 2015; pp. 523–785. [Google Scholar]
- Finta, C.; Zaphiropoulos, P.G. The human CYP2C locus: A prototype for intergenic and exon repetition splicing events. Genomics 2000, 63, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Achour, B.; Barber, J.; Rostami-Hodjegan, A. Expression of hepatic drug-metabolizing cytochrome p450 enzymes and their intercorrelations: A meta-analysis. Drug Metab. Dispos. 2014, 42, 1349–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, A.; Korzekwa, K.R.; Grogan, J.; Gonzalez, F.J.; Harris, J.W. Selective biotransformation of taxol to 6 alpha-hydroxytaxol by human cytochrome P450 2C8. Cancer Res. 1994, 54, 5543–5546. [Google Scholar] [PubMed]
- Daikh, B.E.; Lasker, J.M.; Raucy, J.L.; Koop, D.R. Regio- and stereoselective epoxidation of arachidonic acid by human cytochromes P450 2C8 and 2C9. J. Pharmacol. Exp. Ther. 1994, 271, 1427–1433. [Google Scholar] [PubMed]
- Rifkind, A.B.; Lee, C.; Chang, T.K.; Waxman, D.J. Arachidonic acid metabolism by human cytochrome P450s 2C8, 2C9, 2E1, and 1A2: Regioselective oxygenation and evidence for a role for CYP2C enzymes in arachidonic acid epoxygenation in human liver microsomes. Arch. Biochem. Biophys. 1995, 320, 380–389. [Google Scholar] [CrossRef]
- Zeldin, D.C.; DuBois, R.N.; Falck, J.R.; Capdevila, J.H. Molecular cloning, expression and characterization of an endogenous human cytochrome P450 arachidonic acid epoxygenase isoform. Arch. Biochem. Biophys. 1995, 322, 76–86. [Google Scholar] [CrossRef]
- Reynald, R.L.; Sansen, S.; Stout, C.D.; Johnson, E.F. Structural characterization of human cytochrome P450 2C19: Active site differences between P450s 2C8, 2C9, and 2C19. J. Biol. Chem. 2012, 287, 44581–44591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoch, G.A.; Yano, J.K.; Sansen, S.; Dansette, P.M.; Stout, C.D.; Johnson, E.F. Determinants of cytochrome P450 2C8 substrate binding: Structures of complexes with montelukast, troglitazone, felodipine, and 9-cis-retinoic acid. J. Biol. Chem. 2008, 283, 17227–17237. [Google Scholar] [CrossRef] [Green Version]
- Giver, L.; Gershenson, A.; Freskgard, P.O.; Arnold, F.H. Directed evolution of a thermostable esterase. Proc. Natl Acad. Sci. USA 1998, 95, 12809–12813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuchner, O.; Arnold, F.H. Directed evolution of enzyme catalysts. Trends Biotechnol. 1997, 15, 523–530. [Google Scholar] [CrossRef]
- Cadwell, R.C.; Joyce, G.F. Randomization of genes by PCR mutagenesis. PCR Methods Appl. 1992, 2, 28–33. [Google Scholar] [CrossRef] [Green Version]
- Park, H.G.; Lim, Y.R.; Han, S.; Jeong, D.; Kim, D. Enhanced Purification of Recombinant Rat NADPH-P450 Reductase by Using a Hexahistidine-Tag. J. Microbiol. Biotechnol. 2017, 27, 983–989. [Google Scholar] [CrossRef] [Green Version]
- Cho, M.A.; Yoon, J.G.; Kim, V.; Kim, H.; Lee, R.; Lee, M.G.; Kim, D. Functional Characterization of Pharmcogenetic Variants of Human Cytochrome P450 2C9 in Korean Populations. Biomol. Ther. 2019, 27, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.H.; Miller, G.P.; Guengerich, F.P. Rate-determining steps in phenacetin oxidations by human cytochrome P450 1A2 and selected mutants. Biochemistry 2000, 39, 11319–11329. [Google Scholar] [CrossRef]
- Lee, Y.; Park, H.G.; Kim, V.; Cho, M.A.; Kim, H.; Ho, T.H.; Cho, K.S.; Lee, I.S.; Kim, D. Inhibitory effect of alpha-terpinyl acetate on cytochrome P450 2B6 enzymatic activity. Chem. Biol. Interact. 2018, 289, 90–97. [Google Scholar] [CrossRef]
- Kim, D.; Wu, Z.L.; Guengerich, F.P. Analysis of coumarin 7-hydroxylation activity of cytochrome P450 2A6 using random mutagenesis. J. Biol. Chem. 2005, 280, 40319–40327. [Google Scholar] [CrossRef] [Green Version]
- Moodie, E.E.; Richardson, T.S.; Stephens, D.A. Demystifying optimal dynamic treatment regimes. Biometrics 2007, 63, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Schoch, G.A.; Yano, J.K.; Wester, M.R.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. Structure of human microsomal cytochrome P450 2C8. Evidence for a peripheral fatty acid binding site. J. Biol. Chem. 2004, 279, 9497–9503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, E.F.; Stout, C.D. Structural diversity of eukaryotic membrane cytochrome p450s. J. Biol. Chem. 2013, 288, 17082–17090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estrada, D.F.; Laurence, J.S.; Scott, E.E. Substrate-modulated cytochrome P450 17A1 and cytochrome b5 interactions revealed by NMR. J. Biol. Chem. 2013, 288, 17008–17018. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Kim, V.; McCarty, K.D.; Guengerich, F.P. Tight binding of cytochrome b5 to cytochrome P450 17A1 is a critical feature of stimulation of C21 steroid lyase activity and androgen synthesis. J. Biol. Chem. 2021, 296, 100571. [Google Scholar] [CrossRef]
- Guengerich, F.P. Rate-limiting steps in cytochrome P450 catalysis. Biol. Chem. 2002, 383, 1553–1564. [Google Scholar] [CrossRef]
- Kim, D.; Guengerich, F.P. Selection of human cytochrome P450 1A2 mutants with enhanced catalytic activity for heterocyclic amine N-hydroxylation. Biochemistry 2004, 43, 981–988. [Google Scholar] [CrossRef]
- Lee, H.; Kim, J.H.; Han, S.; Lim, Y.R.; Park, H.G.; Chun, Y.J.; Park, S.W.; Kim, D. Directed-evolution analysis of human cytochrome P450 2A6 for enhanced enzymatic catalysis. J. Toxicol. Environ. Health A 2014, 77, 1409–1418. [Google Scholar] [CrossRef]
- Arnold, F.H. Directed Evolution: Bringing New Chemistry to Life. Angew. Chem. Int. Ed. Engl. 2018, 57, 4143–4148. [Google Scholar] [CrossRef] [Green Version]
- Wittmann, B.J.; Yue, Y.; Arnold, F.H. Informed training set design enables efficient machine learning-assisted directed protein evolution. Cell Syst. 2021. [Google Scholar] [CrossRef]
- Peng, L.; Liao, B.; Zhu, W.; Li, Z.; Li, K. Predicting Drug-Target Interactions With Multi-Information Fusion. IEEE J. Biomed. Health Inform. 2017, 21, 561–572. [Google Scholar] [CrossRef]
- Zhou, L.; Wang, J.; Liu, G.; Lu, Q.; Dong, R.; Tian, G.; Yang, J.; Peng, L. Probing antiviral drugs against SARS-CoV-2 through virus-drug association prediction based on the KATZ method. Genomics 2020, 112, 4427–4434. [Google Scholar] [CrossRef]
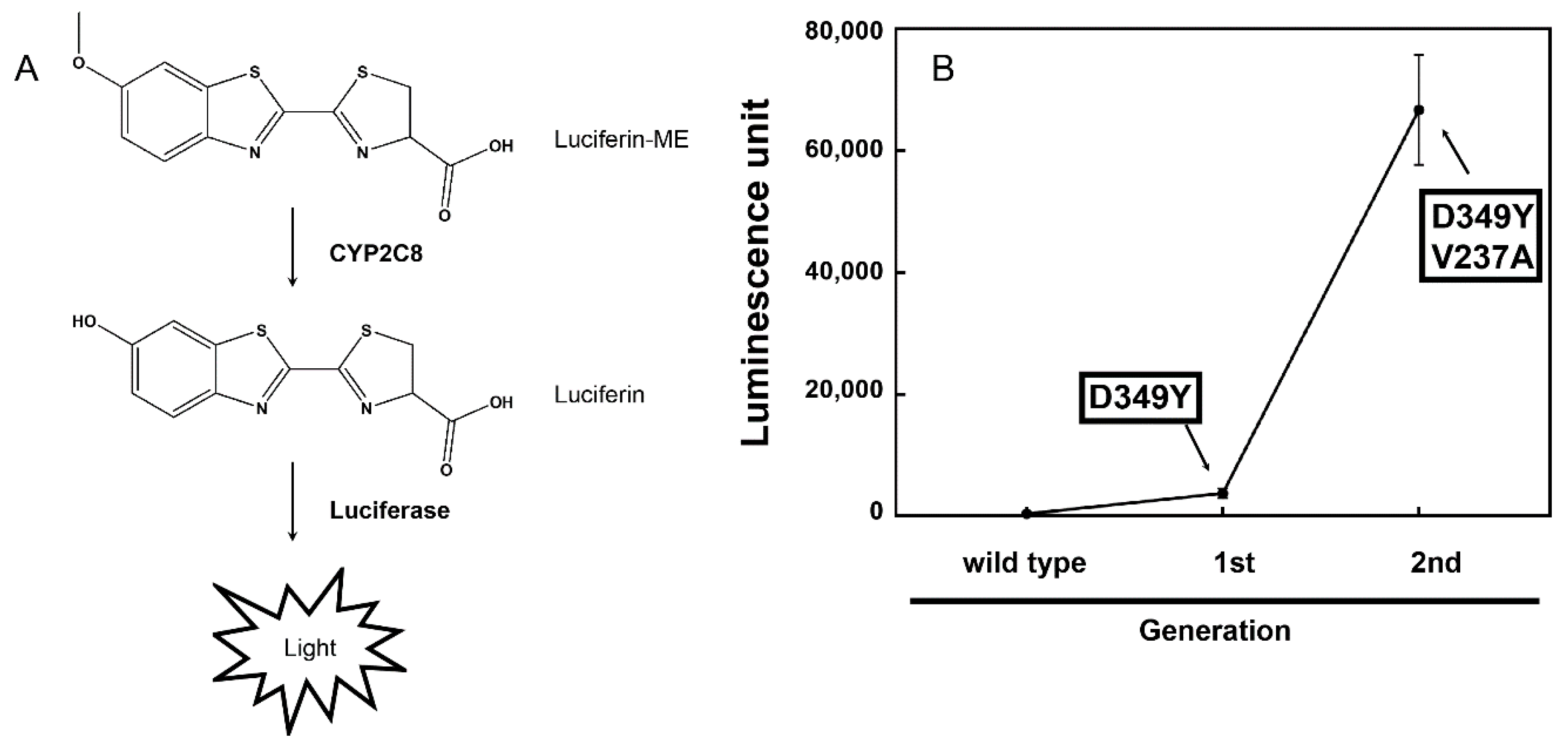
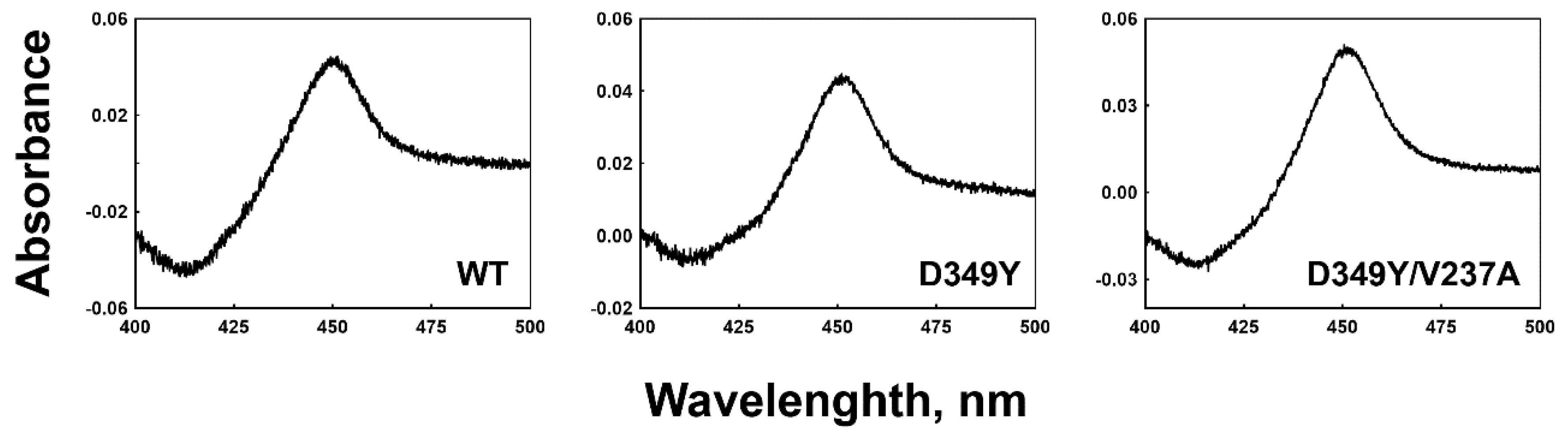
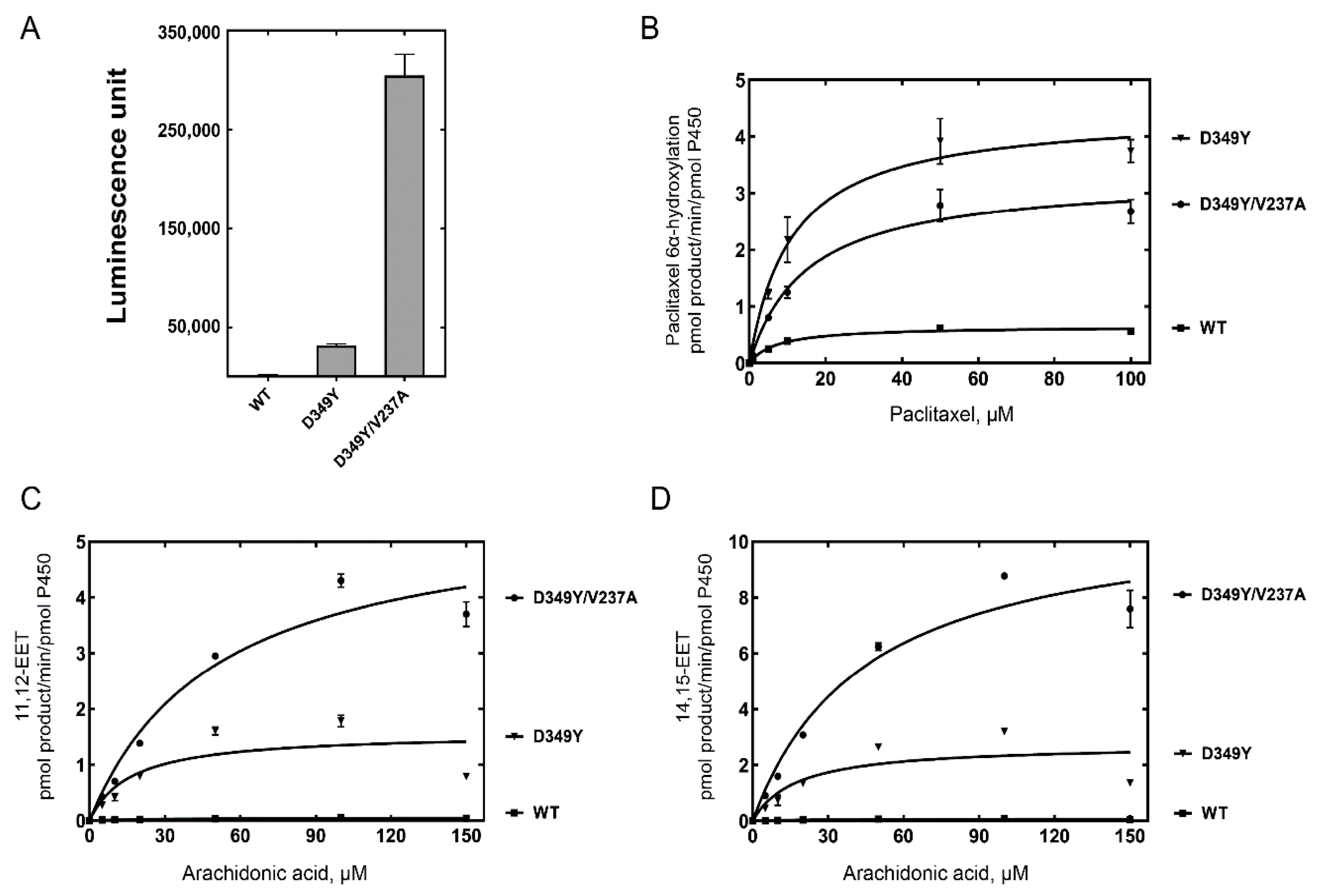



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CYP2C8 Mutants | 6α-Hydroxylation of Paclitaxel | ||
| kcat (min−1) | KM (μM) | kcat/KM | |
| WT | 0.65 ± 0.03 | 7.4 ± 1.1 | 0.09 ± 0.01 |
| D349Y | 4.4 ± 0.2 | 11.1 ± 2.0 | 0.40 ± 0.04 |
| D349Y/V237A | 3.3 ± 0.2 | 15.0 ± 2.8 | 0.22 ± 0.04 |
| ω-9 Epoxidation of Arachidonic Acid | |||
| kcat (min−1) | KM (μM) | kcat/KM | |
| WT | 0.055 ± 0.005 | 36.5 ± 8.5 | 0.0015 ± 0.0004 |
| D349Y | 1.6 ± 0.3 | 16.5 ± 10.2 | 0.10 ± 0.06 |
| D349Y/V237A | 5.6 ± 0.6 | 50.6 ± 12.8 | 0.11 ± 0.03 |
| ω-6 Epoxidation of Arachidonic Acid | |||
| kcat (min−1) | KM (μM) | kcat/KM | |
| WT | 0.069 ± 0.013 | 19.4 ± 12.0 | 0.004 ± 0.002 |
| D349Y | 2.7 ± 0.5 | 17.2 ± 10.7 | 0.16 ± 0.10 |
| D349Y/V237A | 11.1 ± 1.0 | 45.1 ± 10.7 | 0.25 ± 0.06 |
| CYP2C8 Mutants | Kd (μM) | |
|---|---|---|
| Paclitaxel | Arachidonic Acid | |
| WT | 20.3 ± 5.1 | 40.5 ± 6.0 |
| D349Y | 26.7 ± 5.6 | 44.5 ± 10.0 |
| D349Y/V237A | 5.7 ± 1.3 | 10.2 ± 1.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, R.; Kim, V.; Chun, Y.; Kim, D. Structure-Functional Analysis of Human Cytochrome P450 2C8 Using Directed Evolution. Pharmaceutics 2021, 13, 1429. https://doi.org/10.3390/pharmaceutics13091429
Lee R, Kim V, Chun Y, Kim D. Structure-Functional Analysis of Human Cytochrome P450 2C8 Using Directed Evolution. Pharmaceutics. 2021; 13(9):1429. https://doi.org/10.3390/pharmaceutics13091429
Chicago/Turabian StyleLee, Rowoon, Vitchan Kim, Youngjin Chun, and Donghak Kim. 2021. "Structure-Functional Analysis of Human Cytochrome P450 2C8 Using Directed Evolution" Pharmaceutics 13, no. 9: 1429. https://doi.org/10.3390/pharmaceutics13091429
APA StyleLee, R., Kim, V., Chun, Y., & Kim, D. (2021). Structure-Functional Analysis of Human Cytochrome P450 2C8 Using Directed Evolution. Pharmaceutics, 13(9), 1429. https://doi.org/10.3390/pharmaceutics13091429