
Mechanistic PBPK Modelling to Predict the Advantage of the Salt Form of a Drug When Dosed with Acid Reducing Agents


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Modelling of In Vitro Aqueous and Biorelevant Solubility Experiments
2.3. PBPK Model Development for the Free Form of the Drug
2.4. Drug Free Form PBPK Model Verification: Simulations with the Acid Reducing Agent—Famotidine
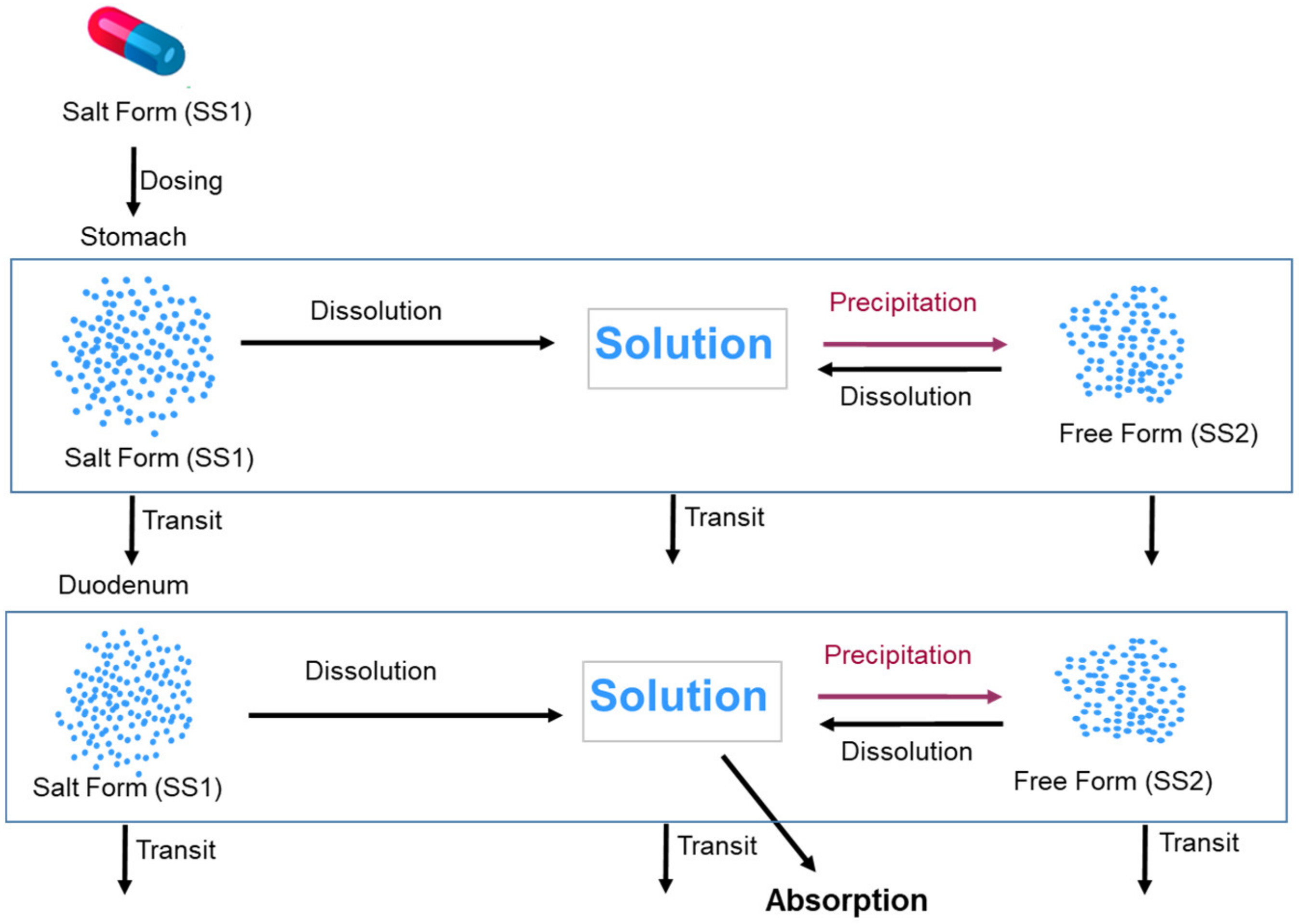
2.5. PBPK Model Development and Simulations of the Salt Formulation
3. Results
3.1. Modelling of In Vitro Aqueous and Biorelevant Solubility Experiments
3.2. Simulation and Prediction of Systemic Plasma Concentration Profiles of Free Form of Drug
3.3. Prediction of Systemic Plasma Concentration Profiles of Salt Form of Drug
4. Discussion
4.1. Prediction of the Effect of ARAs on the Absorption of the Free Form of the Model Compound
4.2. Prediction of the Effect of ARAs on the Absorption of the Salt Form of the Model Compound
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Buckley, S.T.; Frank, K.J.; Fricker, G.; Brandl, M. Biopharmaceutical classification of poorly soluble drugs with respect to “enabling formulations”. Eur. J. Pharm. Sci. 2013, 50, 8–16. [Google Scholar] [CrossRef] [Green Version]
- Mitra, A.; Kesisoglou, F. Impaired drug absorption due to high stomach pH: A review of strategies for mitigation of such effect to enable pharmaceutical product development. Mol. Pharm. 2013, 10, 3970–3979. [Google Scholar] [CrossRef]
- Mitra, A.; Kesisoglou, F.; Beauchamp, M.; Zhu, W.; Chiti, F.; Wu, Y. Using absorption simulation and gastric pH modulated dog model for formulation development to overcome achlorhydria effect. Mol. Pharm. 2011, 8, 2216–2223. [Google Scholar] [CrossRef]
- Dickinson, P.A.; Rmaileh, R.A.; Ashworth, L.; Barker, R.A.; Burke, W.M.; Patterson, C.M.; Stainforth, N.; Yasin, M. An investigation into the utility of a multi-compartmental, dynamic, system of the upper gastrointestinal tract to support formulation development and establish bioequivalence of poorly soluble drugs. AAPS J. 2012, 14, 196–205. [Google Scholar] [CrossRef] [Green Version]
- Bloomer, J.C.; Ambery, C.; Miller, B.E.; Connolly, P.; Garden, H.; Henley, N.; Hodnett, N.; Keel, S.; Kreindler, J.L.; Lloyd, R.S. Identification and characterisation of a salt form of Danirixin with reduced pharmacokinetic variability in patient populations. Eur. J. Pharm. Biopharm. 2017, 117, 224–231. [Google Scholar] [CrossRef]
- Seiler, D.; Doser, K.; Salem, I. Relative bioavailability of prasugrel free base in comparison to prasugrel hydrochloride in the presence and in the absence of a proton pump inhibitor. Arzneim. Forsch. 2011, 61, 247–251. [Google Scholar] [CrossRef]
- Kesisoglou, F.; Vertzoni, M.; Reppas, C. Physiologically Based Absorption Modeling of Salts of Weak Bases Based on Data in Hypochlorhydric and Achlorhydric Biorelevant Media. AAPS PharmSciTech. 2018, 19, 2851–2858. [Google Scholar] [CrossRef] [PubMed]
- Gesenberg, C.; Mathias, N.R.; Xu, Y.; Crison, J.; Savant, I.; Saari, A.; Good, D.J.; Hemenway, J.N.; Narang, A.S.; Schartman, R.R. Utilization of In Vitro, In Vivo and In Silico Tools to Evaluate the pH-Dependent Absorption of a BCS Class II Compound and Identify a pH-Effect Mitigating Strategy. Pharm. Res. 2019, 36, 164. [Google Scholar] [CrossRef] [PubMed]
- Jamei, M.; Turner, D.; Yang, J.; Neuhoff, S.; Polak, S.; Rostami-Hodjegan, A.; Tucker, G. Population-based mechanistic prediction of oral drug absorption. AAPS J. 2009, 11, 225–237. [Google Scholar] [CrossRef] [Green Version]
- Pathak, S.M.; Ruff, A.; Kostewicz, E.S.; Patel, N.; Turner, D.B.; Jamei, M. Model-based analysis of biopharmaceutic experiments to improve mechanistic oral absorption modeling: An integrated in vitro in vivo extrapolation perspective using ketoconazole as a model drug. Mol. Pharm. 2017, 14, 4305–4320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shebley, M.; Sandhu, P.; Emami Riedmaier, A.; Jamei, M.; Narayanan, R.; Patel, A.; Peters, S.A.; Reddy, V.P.; Zheng, M.; de Zwart, L. Physiologically based pharmacokinetic model qualification and reporting procedures for regulatory submissions: A consortium perspective. Clin. Pharm. Ther. 2018, 104, 88–110. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Flanagan, D.R. General solution for diffusion-controlled dissolution of spherical particles. 1. Theory. J. Pharm. Sci. 1999, 88, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Pade, D.; Jamei, M.; Rostami-Hodjegan, A.; Turner, D. Application of the MechPeff model to predict passive effective intestinal permeability in the different regions of the rodent small intestine and colon. Biopharm. Drug Dispos. 2017, 38, 94–114. [Google Scholar] [CrossRef]
- Lindahl, A.; Sandström, R.; Ungell, A.L.; Abrahamsson, B.; Knutson, T.W.; Knutson, L.; Lennernäs, H. Jejunal permeability and hepatic extraction of fluvastatin in humans. Clin. Pharm. Ther. 1996, 60, 493–503. [Google Scholar] [CrossRef]
- Litou, C.; Vertzoni, M.; Goumas, C.; Vasdekis, V.; Xu, W.; Kesisoglou, F.; Reppas, C. Characteristics of the human upper gastrointestinal contents in the fasted state under hypo-and A-chlorhydric gastric conditions under conditions of typical drug–drug interaction studies. Pharm. Res. 2016, 33, 1399–1412. [Google Scholar] [CrossRef] [PubMed]
- Uekusa, T.; Sugano, K. Precipitation behavior of pioglitazone on the particle surface of hydrochloride salt in biorelevant media. J. Pharm. Biomed. Anal. 2018, 161, 45–50. [Google Scholar] [CrossRef]
- Oki, J.; Watanabe, D.; Uekusa, T.; Sugano, K. Mechanism of supersaturation suppression in dissolution process of acidic drug salt. Mol. Pharm. 2019, 16, 1669–1677. [Google Scholar] [CrossRef]
- Ozturk, S.S.; Palsson, B.O.; Dressman, J.B. Dissolution of ionizable drugs in buffered and unbuffered solutions. Pharm. Res. 1988, 5, 272–282. [Google Scholar] [CrossRef]
- Ozturk, S.S.; Dressman, J.B.; Palsson, B.O. On the use of the quasi-equilibrium assumption for drug dissolution. Pharm. Res. 1990, 7, 425–429. [Google Scholar] [CrossRef] [Green Version]
- Koziolek, M.; Grimm, M.; Becker, D.; Iordanov, V.; Zou, H.; Shimizu, J.; Wanke, C.; Garbacz, G.; Weitschies, W. Investigation of pH and Temperature Profiles in the GI Tract of Fasted Human Subjects Using the Intellicap(®) System. J. Pharm. Sci. 2015, 104, 2855–2863. [Google Scholar] [CrossRef]
- Humphries, T. Famotidine: A notable lack of drug interactions. Scand. J. Gastroenterol. 1987, 22, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Ohbuchi, M.; Noguchi, K.; Kawamura, A.; Usui, T. Different effects of proton pump inhibitors and famotidine on the clopidogrel metabolic activation by recombinant CYP2B6, CYP2C19 and CYP3A4. Xenobiotica 2012, 42, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Bourdet, D.L.; Pritchard, J.B.; Thakker, D.R. Differential substrate and inhibitory activities of ranitidine and famotidine toward human organic cation transporter 1 (hOCT1; SLC22A1), hOCT2 (SLC22A2), and hOCT3 (SLC22A3). J. Pharm. Exp. Ther. 2005, 315, 1288–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Medium | Solubility (mg/mL) | Medium | Solubility (mg/mL) |
|---|---|---|---|
| Aqueous | Aqueous | ||
| pH 1.5 | 6.5 | pH 8.5 | 0.021 |
| pH 3.9 | 3.235 | pH 9.1 | 0.03 |
| pH 5.4 | 0.136 | pH 10.6 | 0.442 |
| pH 6.4 | 0.021 | Biorelevant | |
| pH 6.6 | 0.019 | FaSSIF (pH 6.5) | 0.043 |
| pH 7.5 | 0.015 | FeSSIF V1 (pH 5) | 0.99 |
| pH 7.9 | 0.015 | ||
| Parameters | Value | Reference/Comments |
|---|---|---|
| Physicochemical Properties | ||
| Molecular weight (g/mol) | 500 | 500–600 # Gesenberg et al. [8] |
| Compound Type | Ampholyte | Modelled as diprotic base |
| logPo:w | 2.288 | Back calculated from logD at pH 6.5 Gesenberg et al. [8] |
| pKa | 6.25 (b), 2.1 (b), 9.8 (a) | Fitted in SIVA and based on Gesenberg et al. [8] |
| Absorption Parameters | ||
| Formulation | Immediate Release | |
| Intrinsic solubility (mg/mL) | 0.0142 | Fitted in SIVA |
| Solubility factor | 457.8 | Fitted in SIVA |
| logKm:w,unionsed | 4.34 | Fitted in SIVA |
| logKm:w,ionised | 3.97 | Fitted in SIVA |
| Precipitation Model | Model 2 | Simcyp default precipitation model |
| CSR | 10 | Simcyp default parameter |
| PRC (1/h) | 4 | Simcyp default parameter |
| Particle size distribution | Monodispersed | |
| Particle radius (microns) | 1.958 | Gesenberg et al. [8] |
| Particle dissolution model | Wang-Flanagan | |
| Particle handling model | Particle Population Balance (PPB) | |
| Particle heff model | Hintz-Johnson | |
| Fluid Volumes Model | Simcyp advanced FVD (aFVD) | See main text |
| Gut Physiology | Simcyp “New Anatomy” | See main text |
| Permeability | Predicted | |
| Permeability model | MechPeff | |
| Ptrans,0 (10−6 cm/s) | 214.656 | Predicted from logPo:w |
| Peff,man (10−4 cm/s) (Duodenum) | 6.03 * | Predicted |
| Peff,man (10−4 cm/s) (Jejunum I) | 8.27 * | Predicted |
| Peff,man (10−4 cm/s) (Jejunum II) | 5.70 * | Predicted |
| Peff,man (10−4 cm/s) (Ileum I) | 1.76 * | Predicted |
| Peff,man (10−4 cm/s) (Ileum II) | 1.76 * | Predicted |
| Peff,man (10−4 cm/s) (Ileum III) | 1.74 * | Predicted |
| Peff,man (10−4 cm/s) (Ileum IV) | 1.67 * | Predicted |
| Peff,man (10−4 cm/s) (Colon) | 0.58 * | Predicted |
| Distribution and Elimination Parameters | ||
| fu | 0.23 | Simcyp Predicted |
| B/P | 1.144 | Simcyp predicted |
| Distribution Model | Minimal PBPK | |
| Vss (L/Kg) | 0.78 | Fitted to observed PO data (w/o famotidine) |
| Elimination CLiv (L/h) | 10 | Fitted to observed PO data (w/o famotidine) |
| Percentage CLH by 3A (%) | 100 | |
| fugut | 1 | |
| Trial Design | ||
| No of trials in each simulation | 10 | |
| No. of subjects in each trial | 10 | |
| Min. (Max.) age of subjects | 20 (50) | |
| Proportion of females | 0.5 | |
| Simulation duration (h) | 24 | |
| Fluid volume taken with dose (mL) | 250 | Between subject CV on fluid volume intake was 0% |
| Dose (mg) | 150 | |
| Parameters | Value | Reference/Comments |
|---|---|---|
| Solid State 1 (SS1) Parameters | ||
| Form | Salt | |
| Percentage in dose (%) | 100 | Refers to the % of SS1 in the dosage form prior to dosing. |
| Salt Solubility at pHmax (mg/mL) | 6.5 | Free base highest solubility (pH 1.5) |
| Ksp (mM2) | 117.6 | Back calculated from solubility at pHmax |
| Intrinsic solubility, So (mg/mL) | 0.0142 | Free base intrinsic solubility |
| Drug:Counterion Stoichiometry | 1:1 | |
| Concentration of counterion in drink (mg/mL) | 0 | |
| Counterion | Sulphate | |
| Counterion type | Strong acid | |
| Counterion pKa1 | −3 | |
| Counterion pKa2 | 1.92 | |
| Particle surface solubility | Mechanistic Surface pH model | |
| Precipitation to SS1 | Deselected | See main text for further explanation |
| Solid State 2 (SS2) Parameters | ||
| Form | Free | |
| Percentage in dose (%) | 0 | |
| Intrinsic solubility, So (mg/mL) | 0.0142 | Fitted in SIVA |
| Solubility factor (SF) | 457.8 | Fitted in SIVA |
| Precipitation to SS2 | Permitted | |
| Precipitation Model | Model 2 | |
| CSR | 10 | Default |
| PRC (1/h) | 4 | Default |
| Dosing Condition | Cmax (mg/L) | AUC0–t (mg/L∙h) | Tmax (h) | |||||
|---|---|---|---|---|---|---|---|---|
| Pred | Obs | Pred/Obs | Pred | Obs | Pred/Obs | Pred | Obs | |
| Without Famotidine | 1.22 | 1.23 | 0.99 | 9.80 | 8.15 | 1.20 | 1.3 | 3.0 |
| With Famotidine | 0.35 | 0.24 | 1.45 | 3.48 | 2.33 | 1.49 | 3.6 | 3.0 |
| Dosing Condition | Cmax (mg/L) | AUC0–t (mg/L·h) | Tmax (h) |
|---|---|---|---|
| Without Famotidine | 1.26 ± 0.51 | 9.8 ± 4.04 | 1.49 ± 0.58 |
| With Famotidine | 0.69 ± 0.31 | 6.19 ± 2.92 | 2.17 ± 1.15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chirumamilla, S.K.; Banala, V.T.; Jamei, M.; Turner, D.B. Mechanistic PBPK Modelling to Predict the Advantage of the Salt Form of a Drug When Dosed with Acid Reducing Agents. Pharmaceutics 2021, 13, 1169. https://doi.org/10.3390/pharmaceutics13081169
Chirumamilla SK, Banala VT, Jamei M, Turner DB. Mechanistic PBPK Modelling to Predict the Advantage of the Salt Form of a Drug When Dosed with Acid Reducing Agents. Pharmaceutics. 2021; 13(8):1169. https://doi.org/10.3390/pharmaceutics13081169
Chicago/Turabian StyleChirumamilla, Siri Kalyan, Venkatesh Teja Banala, Masoud Jamei, and David B. Turner. 2021. "Mechanistic PBPK Modelling to Predict the Advantage of the Salt Form of a Drug When Dosed with Acid Reducing Agents" Pharmaceutics 13, no. 8: 1169. https://doi.org/10.3390/pharmaceutics13081169
APA StyleChirumamilla, S. K., Banala, V. T., Jamei, M., & Turner, D. B. (2021). Mechanistic PBPK Modelling to Predict the Advantage of the Salt Form of a Drug When Dosed with Acid Reducing Agents. Pharmaceutics, 13(8), 1169. https://doi.org/10.3390/pharmaceutics13081169