
An Effective and Safe Enkephalin Analog for Antinociception

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
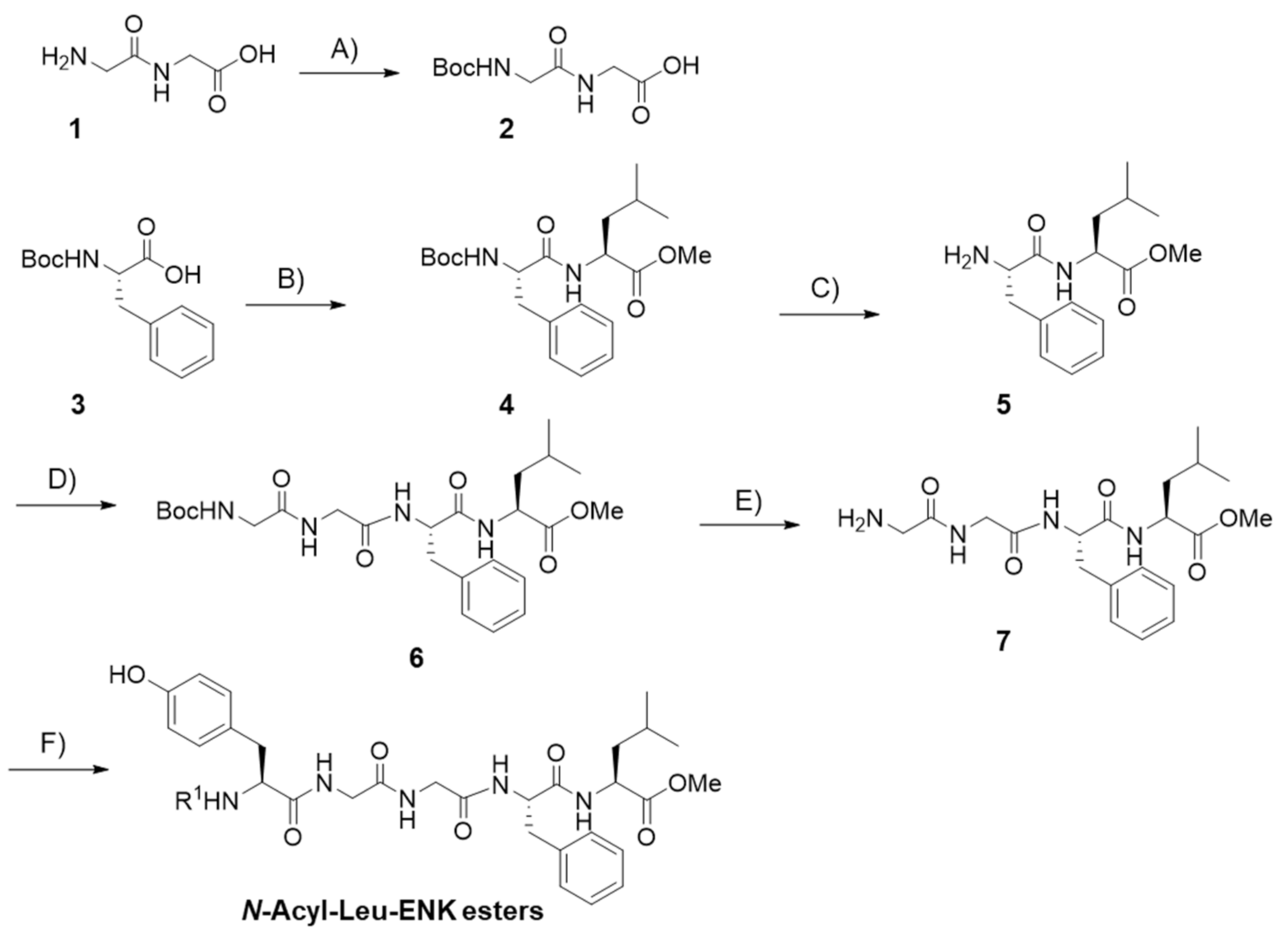
2.1. Peptide Synthesis
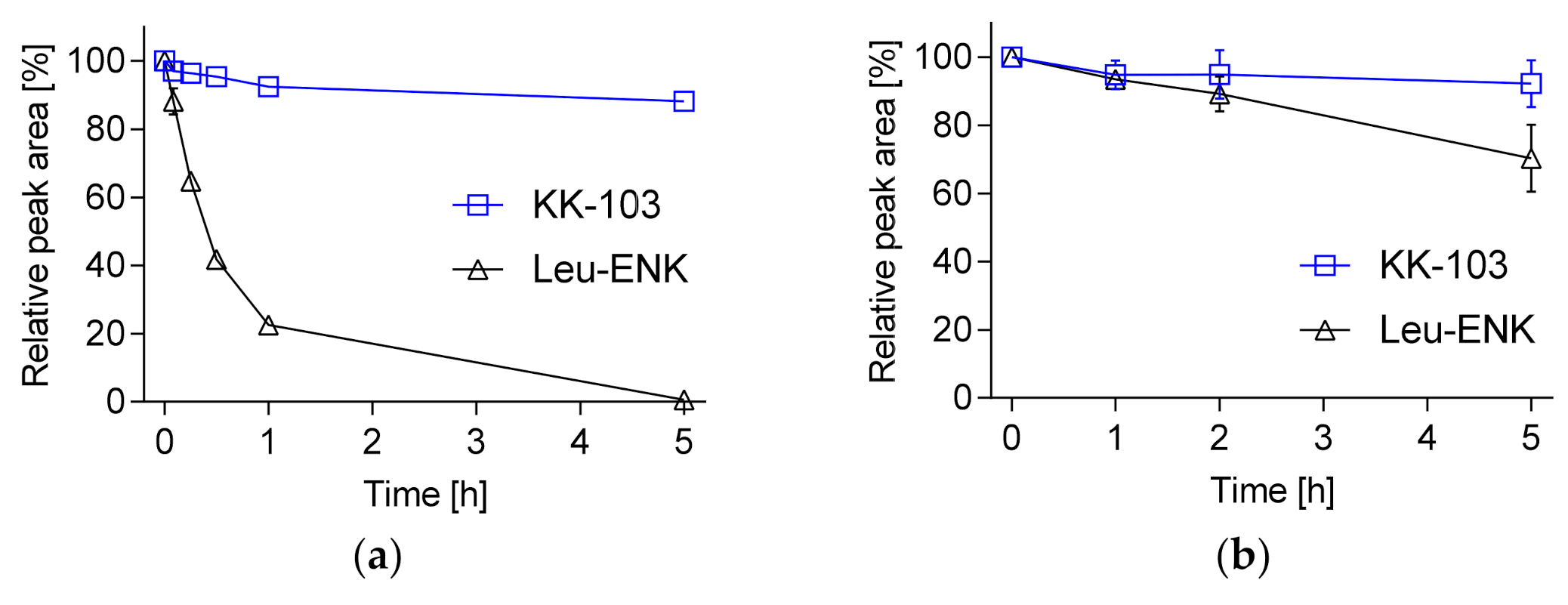
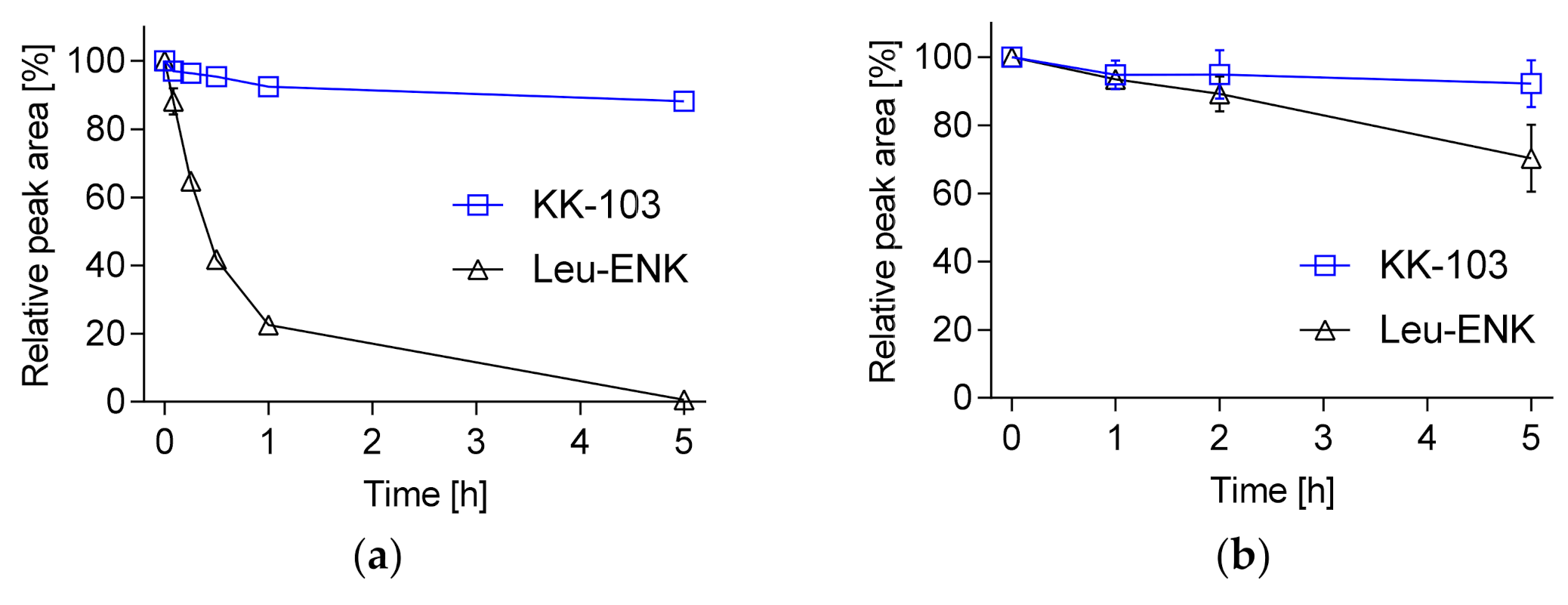
2.2. Peptide Stability
2.3. UPLC
2.4. Binding to DOR
2.5. Animals
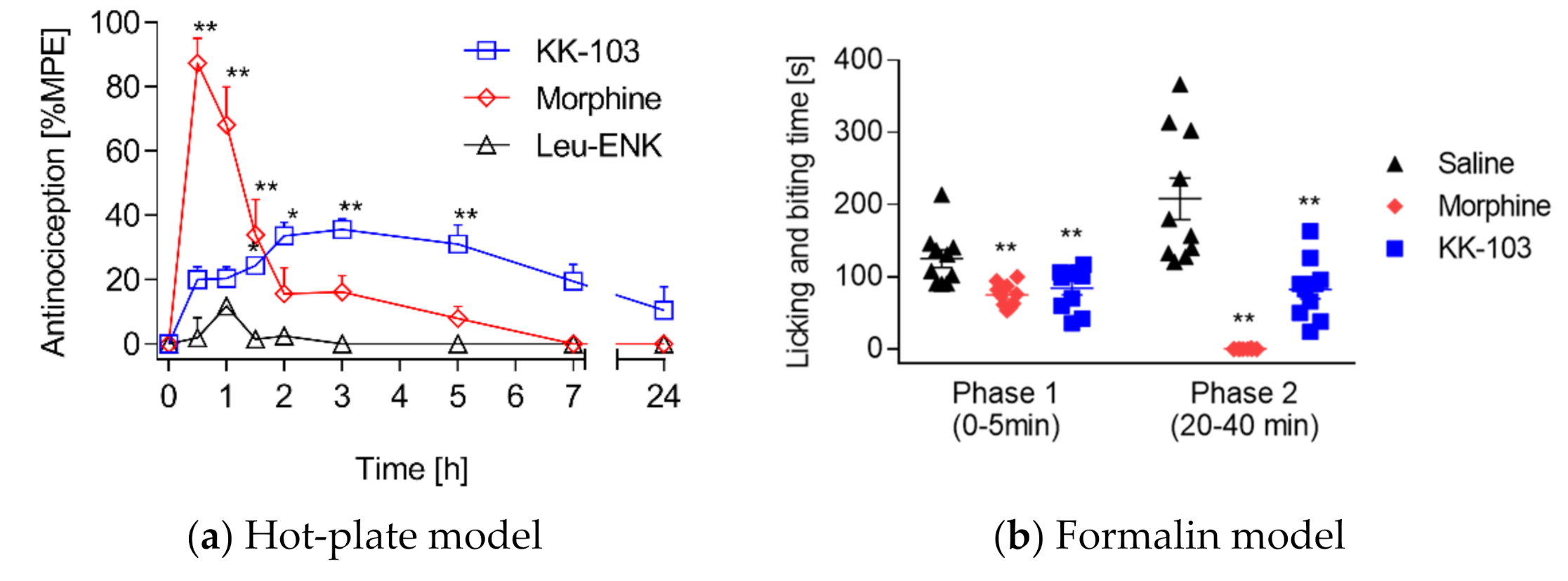
2.6. Hot-Plate Test
2.7. Formalin Foot Assay
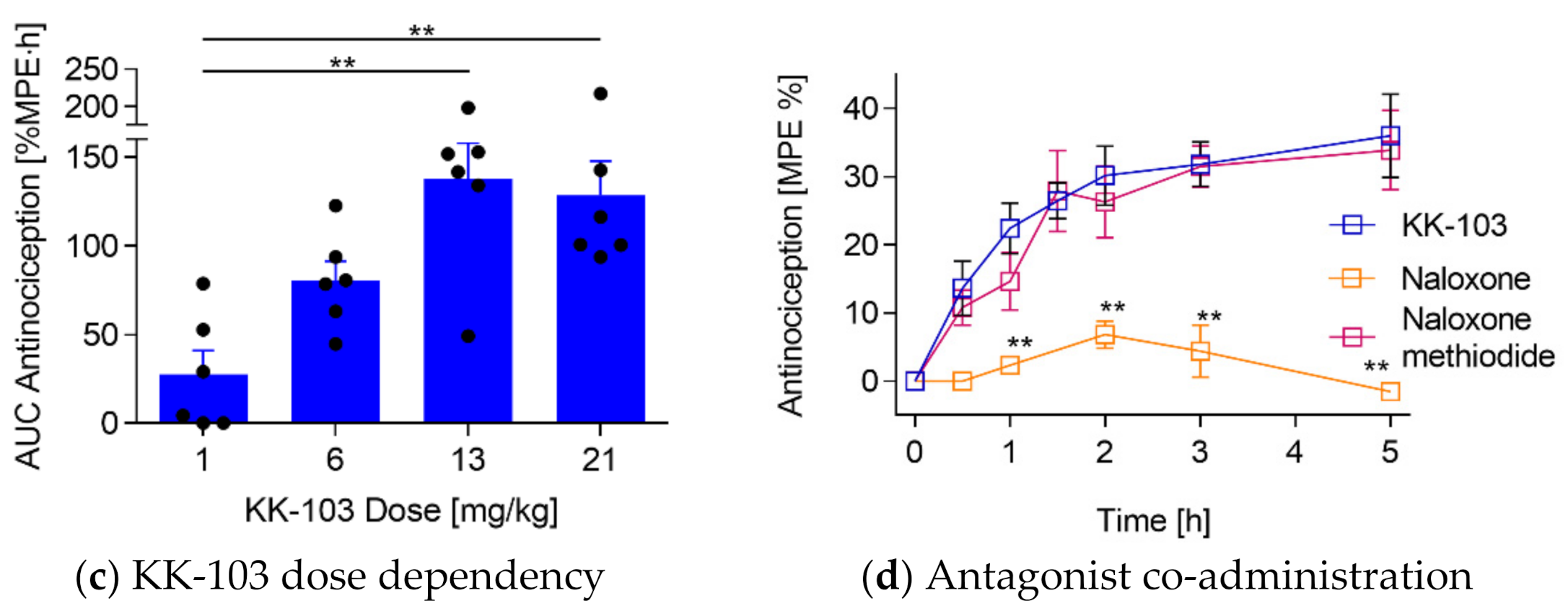
2.8. Antagonist Co-Injection
2.9. Safety Test in Mice
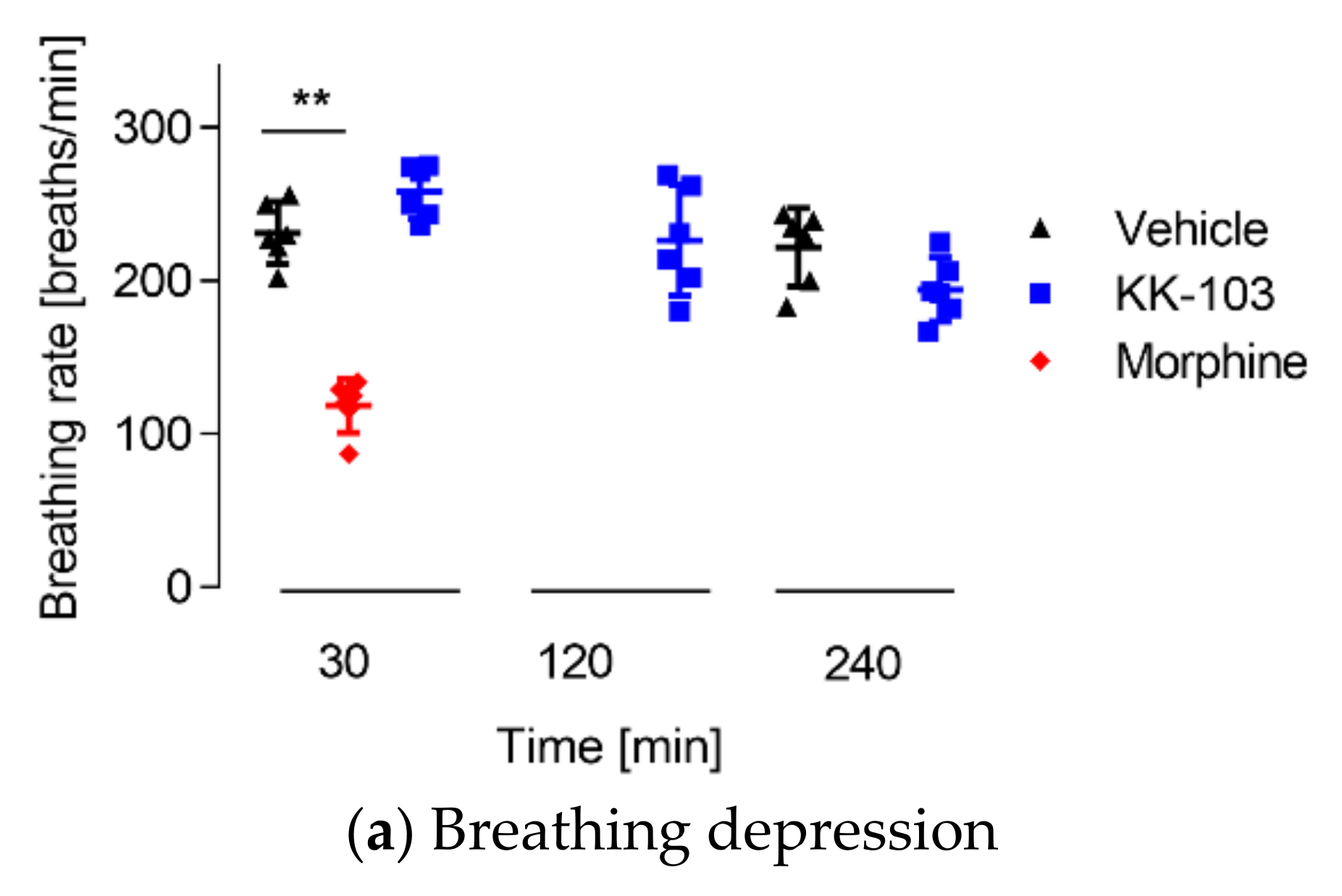
2.10. Respiratory Depression Assay
2.11. Dependence Study
2.12. Tolerance Study
2.13. Statistical Analysis
3. Results and Discussion
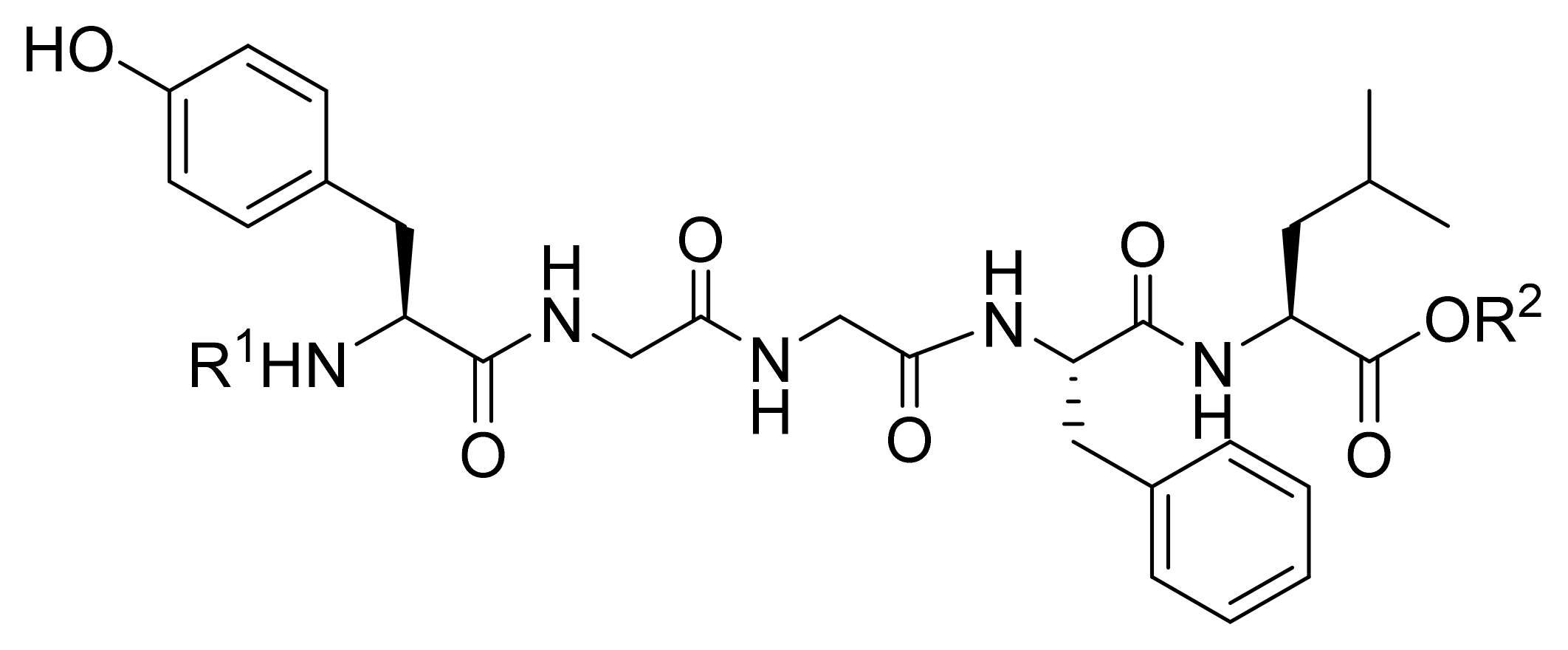
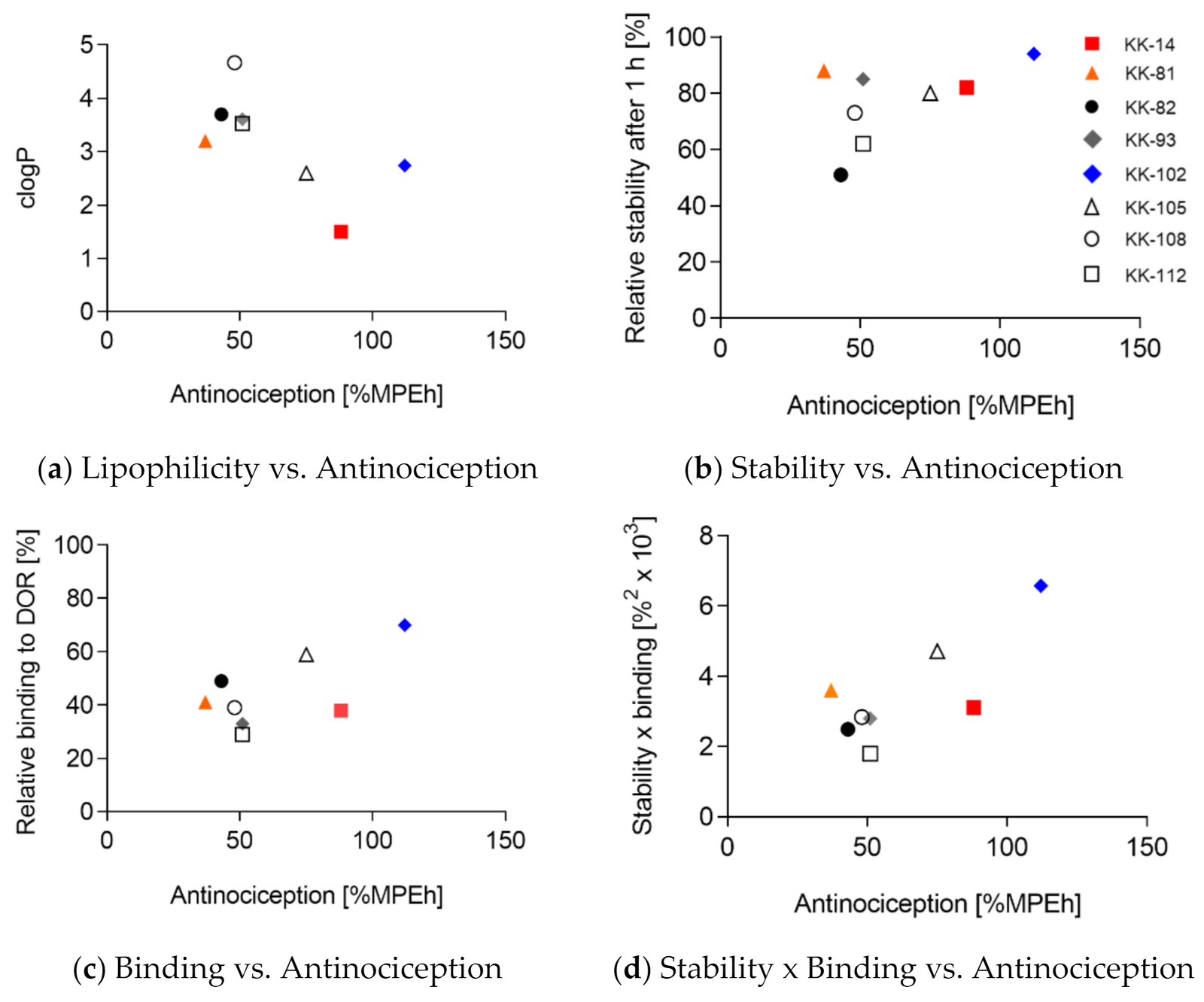
3.1. Design of Leu-ENK Analogs
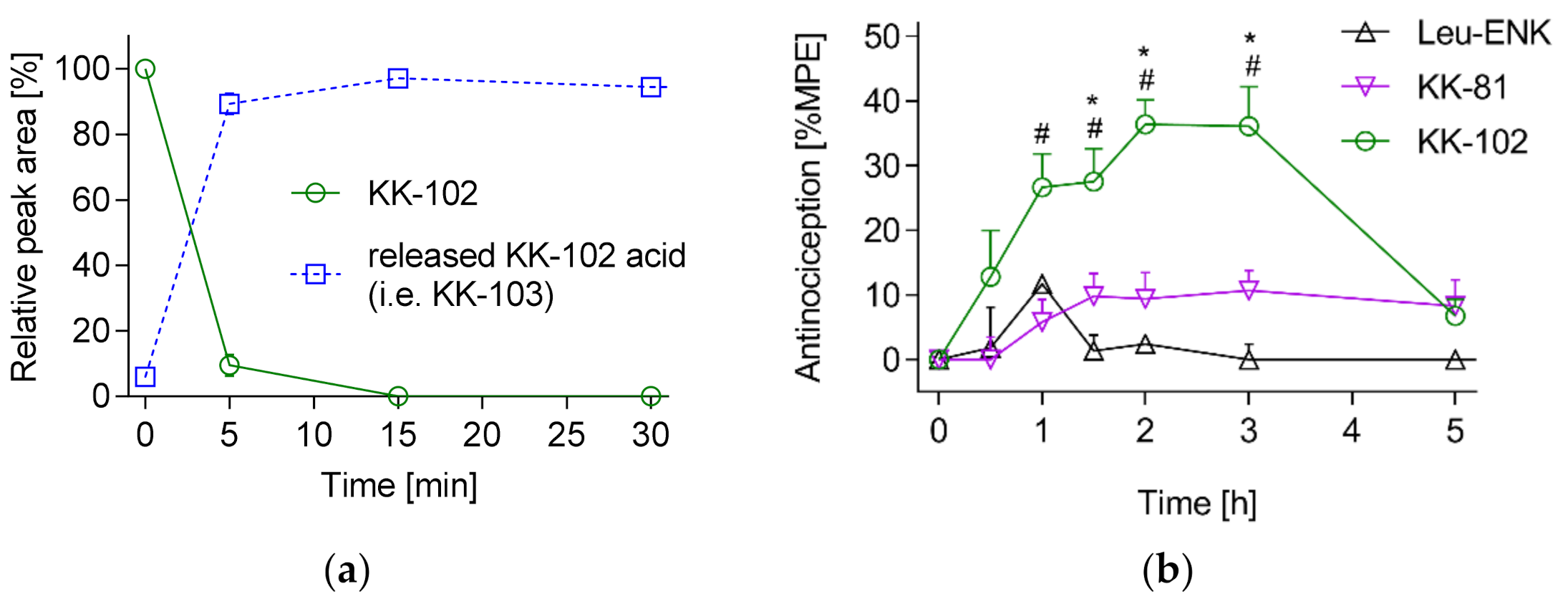
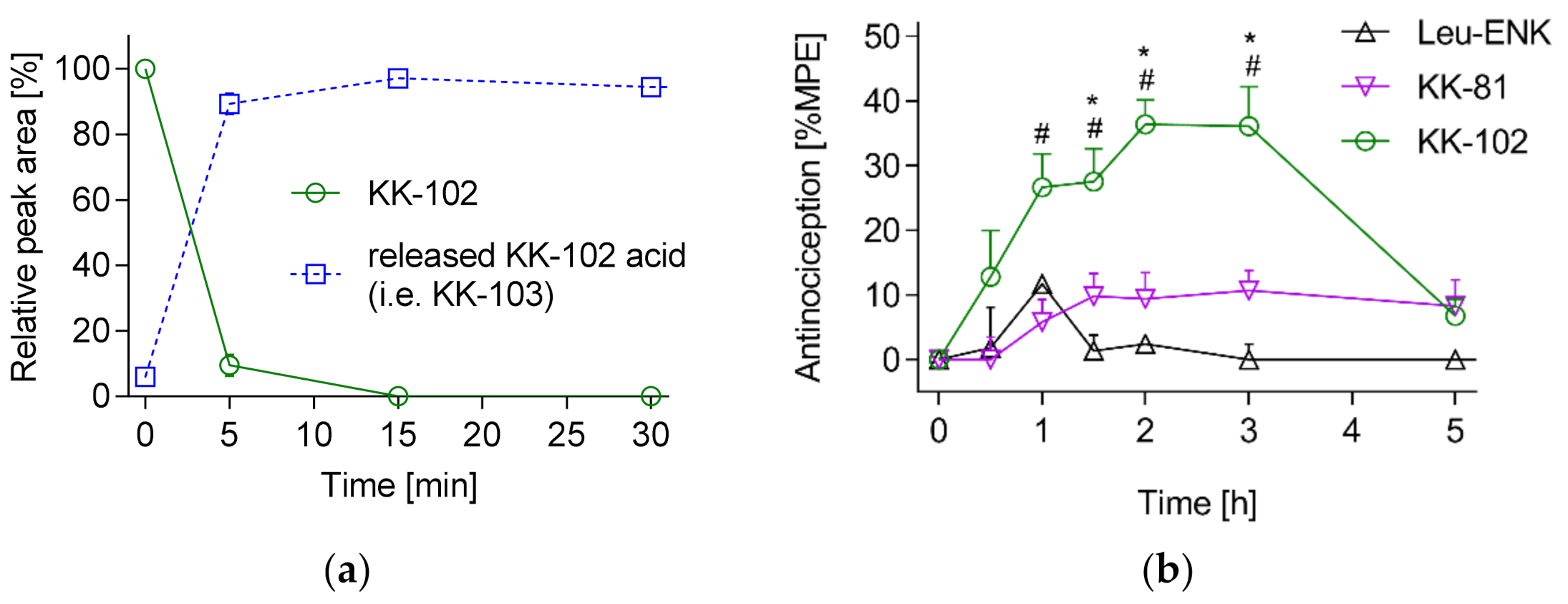
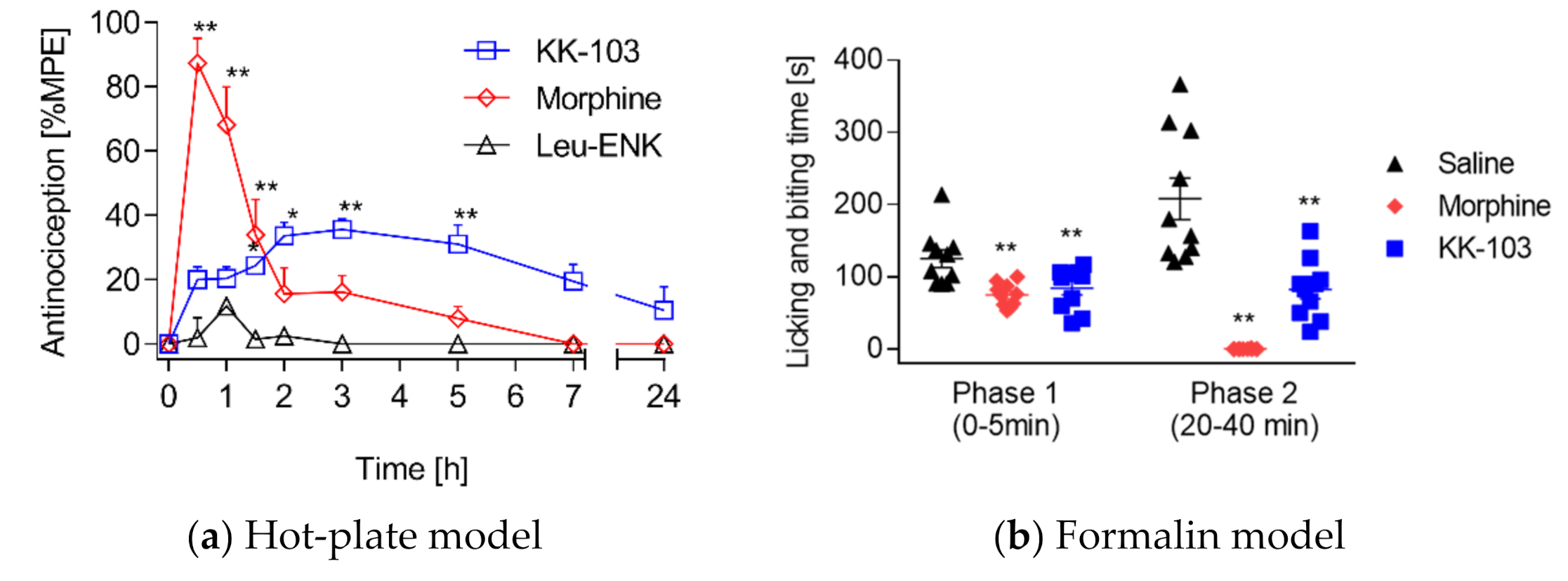
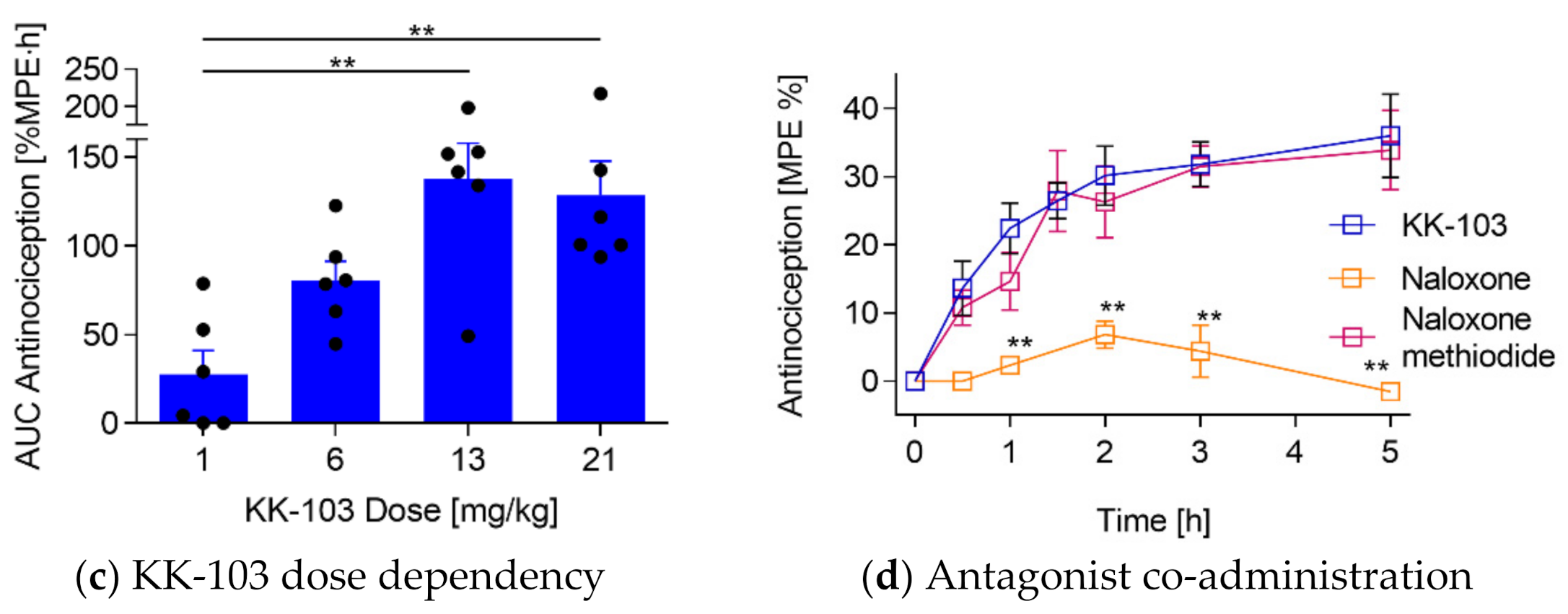
3.2. Analgesic Effect of KK-103 Compared to Morphine
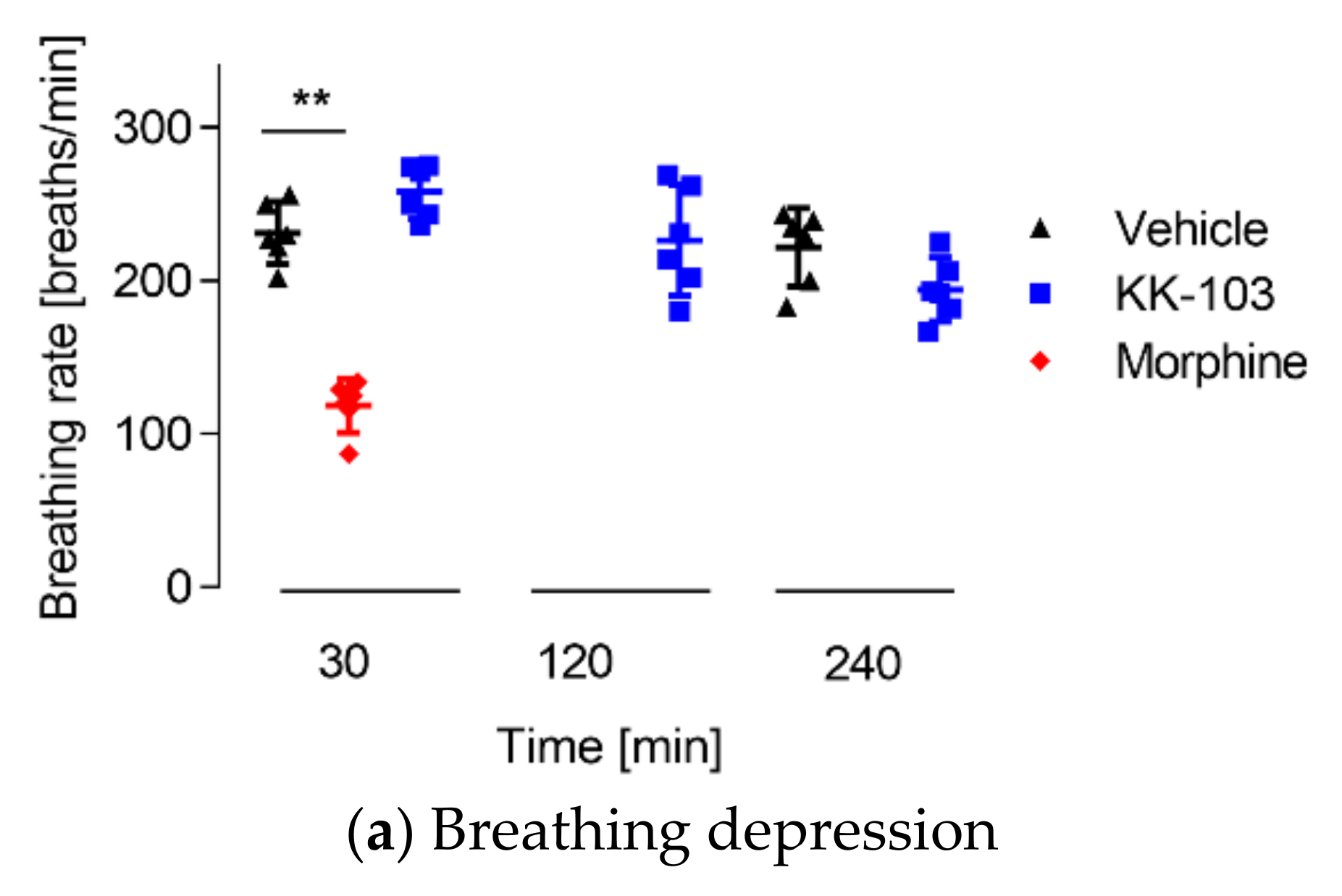
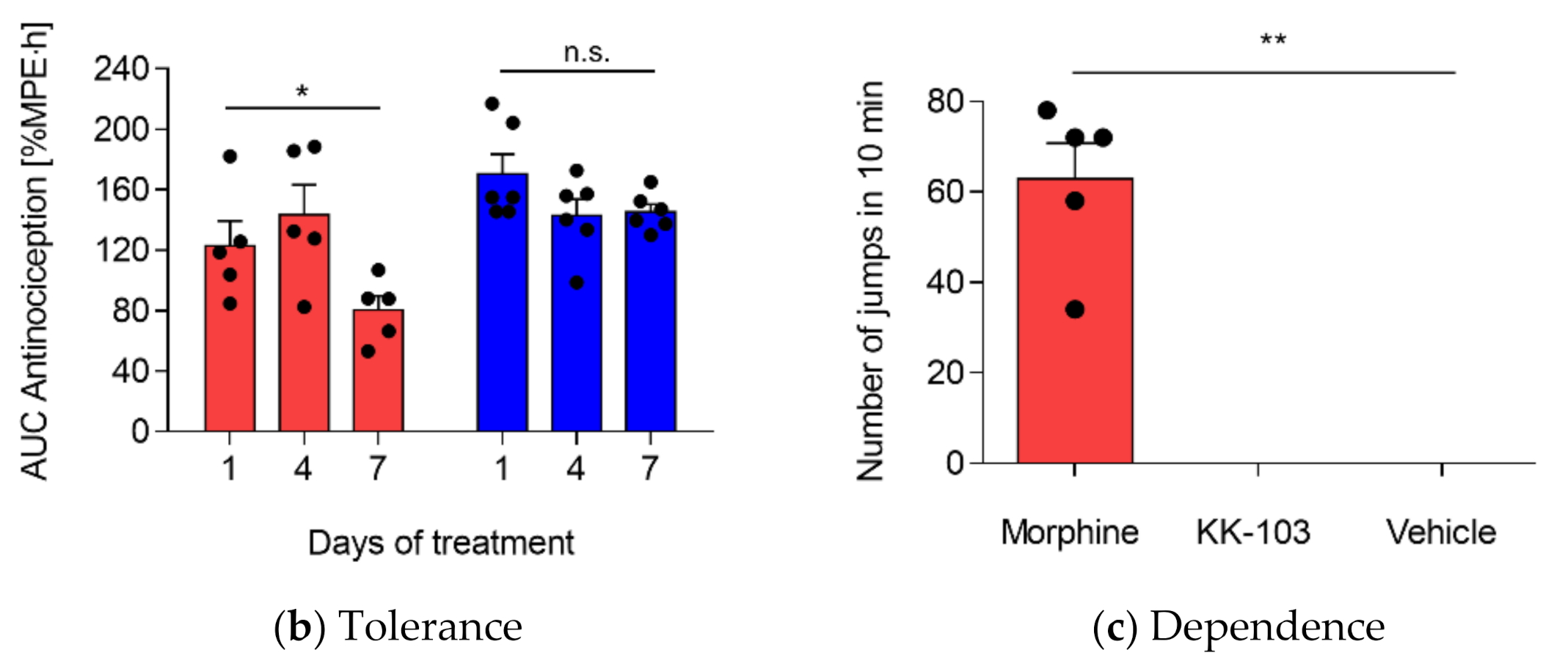
3.3. Safety Profiles of KK-103 Compared to Morphine
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- GSK Global Pain Index 2017 Research Report. Available online: https://www.gsk.com/media/3814/global-pain-index-2017-report.pdf (accessed on 22 June 2021).
- IASP Unrelieved Pain is a Major Global Healthcare Problem. Available online: https://s3.amazonaws.com/rdcms-iasp/files/production/public/Content/ContentFolders/GlobalYearAgainstPain2/20042005RighttoPainRelief/factsheet.pdf (accessed on 22 June 2021).
- Gaskin, D.J.; Richard, P. The economic costs of pain in the United States. J. Pain 2012, 13, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Kieffer, B.L. Opioids: First lessons from knockout mice. Trends Pharmacol. Sci. 1999, 20, 19–26. [Google Scholar] [CrossRef]
- Bodnar, R.J.; Klein, G.E. Endogenous opiates and behavior: 2004. Peptides 2005, 26, 2629–2711. [Google Scholar] [CrossRef] [PubMed]
- Wilson, N.; Kariisa, M.; Seth, P.; Smith, H.; Davis, N.L. Drug and Opioid-Involved Overdose Deaths—United States, 2017–2018. MMWR Morb. Mortal. Wkly. Rep. 2020, 69, 290–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanderah, T.W. Delta and kappa opioid receptors as suitable drug targets for pain. Clin. J. Pain 2010, 26, S10–S15. [Google Scholar] [CrossRef]
- Roscetti, G.; Possenti, R.; Bassano, E.; Roda, L.G. Mechanisms of leu-enkephalin hydrolysis in human plasma. Neurochem. Res. 1985, 10, 1393–1404. [Google Scholar] [CrossRef]
- Bolacchi, F.; Marini, M.; Urbani, A.; Giorgio Roda, L. Enzymes and inhibitors in leu-enkephalin in metabolism in human plasma. Neurochem. Res. 1995, 20, 991–999. [Google Scholar] [CrossRef]
- Gentilucci, L.; De Marco, R.; Cerisoli, L. Chemical Modifications Designed to Improve Peptide Stability: Incorporation of Non-Natural Amino Acids, Pseudo-Peptide Bonds, and Cyclization. Curr. Pharm. Des. 2010, 16, 3185–3203. [Google Scholar] [CrossRef]
- Galeazzi, R.; Martelli, G.; Marcucci, E.; Orena, M.; Rinaldi, S.; Lattanzi, R.; Negri, L. Analogues of both Leu- and Met-enkephalin containing a constrained dipeptide isostere prepared from a Baylis-Hillman adduct. Amino Acids 2010, 38, 1057–1065. [Google Scholar] [CrossRef]
- Shinada, T.; Ishida, T.; Hayashi, K.I.; Yoshida, Y.; Shigeri, Y.; Ohfune, Y. Synthesis of leucine-enkephalin analogs containing α-amino squaric acid. Tetrahedron Lett. 2007, 48, 7614–7617. [Google Scholar] [CrossRef]
- Martin, S.F.; Dwyer, M.P.; Hartmann, B.; Knight, K.S. Cyclopropane-derived peptidomimetics. Design, synthesis, and evaluation of novel enkephalin analogues. J. Org. Chem. 2000, 65, 1305–1318. [Google Scholar] [CrossRef]
- Proteau-Gagné, A.; Bournival, V.; Rochon, K.; Dory, Y.L.; Gendron, L. Exploring the backbone of enkephalins to adjust their pharmacological profile for the δ-opioid receptor. ACS Chem. Neurosci. 2010, 1, 757–769. [Google Scholar] [CrossRef] [Green Version]
- Rochon, K.; Proteau-Gagné, A.; Bourassa, P.; Nadon, J.F.; Coîté, J.; Bournival, V.; Gobeil, F.; Guérin, B.; Dory, Y.L.; Gendron, L. Preparation and evaluation at the delta opioid receptor of a series of linear Leu-enkephalin analogues obtained by systematic replacement of the amides. ACS Chem. Neurosci. 2013, 4, 1204–1216. [Google Scholar] [CrossRef] [Green Version]
- Benovitz, D.E.; Spatola, A.F. Enkephalin pseudopeptides: Resistance to in vitro proteolytic degradation afforded by amide bond replacements extends to remote sites. Peptides 1985, 6, 257–261. [Google Scholar] [CrossRef]
- Lord, J.A.H.; Waterfield, A.A.; Hughes, J.; Kosterlitz, H.W. Endogenous opioid peptides: Multiple agonists and receptors. Nature 1977, 267, 495–499. [Google Scholar] [CrossRef]
- Mosberg, H.I.; Hurst, R.; Hruby, V.J.; Gee, K.; Akiyama, K.; Yamamura, H.I.; Galligan, J.J.; Burks, T.F. Cyclic penicillamine containing enkephalin analogs display profound delta receptor selectivities. Life Sci. 1983, 33, 447–450. [Google Scholar] [CrossRef]
- Harris, H.M.; Eans, S.O.; Ganno, M.L.; Davis, J.C.; Dooley, C.T.; McLaughlin, J.P.; Nefzi, A. Antinociceptive activity of thiazole-containing cyclized DAMGO and Leu-(Met) enkephalin analogs. Org. Biomol. Chem. 2019, 17, 5305–5315. [Google Scholar] [CrossRef]
- Oldendorf, W.H. Lipid Solubility and Drug Penetration of the Blood Brain Barrier (38444). Proc. Soc. Exp. Biol. Med. 1974, 147, 813–816. [Google Scholar] [CrossRef]
- Cros, C.D.; Toth, I.; Blanchfield, J.T. Lipophilic derivatives of leu-enkephalinamide: In vitro permeability, stability and in vivo nasal delivery. Bioorg. Med. Chem. 2011, 19, 1528–1534. [Google Scholar] [CrossRef]
- Shechter, Y.; Heldman, E.; Sasson, K.; Bachar, T.; Popov, M.; Fridkin, M. Delivery of neuropeptides from the periphery to the brain: Studies with enkephalin. ACS Chem. Neurosci. 2010, 1, 399–406. [Google Scholar] [CrossRef] [Green Version]
- Summers, M.C.; Hayes, R.J. The interaction of N(α)-alkylenkephalins with opiate receptors. Tissue-dependent shifts in the opiate activity of methionine-enkephalin following N(α)-alkylation. J. Biol. Chem. 1981, 256, 4951–4956. [Google Scholar] [CrossRef]
- Uchiyama, T.; Kotani, A.; Tatsumi, H.; Kishida, T.; Okamoto, A.; Okada, N.; Murakami, M.; Fujita, T.; Fujiwara, Y.; Kiso, Y.; et al. Development of novel lipophilic derivatives of DADLE (Leucine enkephalin analogue): Intestinal permeability characteristics of DADLE derivatives in rats. Pharm. Res. 2000, 17, 1461–1467. [Google Scholar] [CrossRef] [PubMed]
- Beaudeau, J.L.; Blais, V.; Holleran, B.J.; Bergeron, A.; Pineyro, G.; Guérin, B.; Gendron, L.; Dory, Y.L. N-Guanidyl and C-Tetrazole Leu-Enkephalin Derivatives: Efficient Mu and Delta Opioid Receptor Agonists with Improved Pharmacological Properties. ACS Chem. Neurosci. 2019, 10, 1615–1626. [Google Scholar] [CrossRef] [PubMed]
- Böttger, R.; Knappe, D.; Hoffmann, R. PEGylated prodrugs of antidiabetic peptides amylin and GLP-1. J. Control. Release 2018, 292. [Google Scholar] [CrossRef]
- Simonin, F.; Befort, K.; Gaveriaux-Ruff, C.; Matthes, H.; Nappey, V.; Lannes, B.; Micheletti, G.; Kieffer, B. The human δ-opioid receptor: Genomic organization, cDNA cloning, functional expression, and distribution in human brain. Mol. Pharmacol. 1994, 46, 1015–1021. [Google Scholar]
- Jayawardene, D.S.; Dass, C. The effect of N-terminal acetylation and the inhibition activity of acetylated enkephalins on the aminopeptidase M-catalyzed hydrolysis of enkephalins. Peptides 1999, 20, 963–970. [Google Scholar] [CrossRef]
- Yao, J.-F.; Yang, H.; Zhao, Y.-Z.; Xue, M. Metabolism of Peptide Drugs and Strategies to Improve their Metabolic Stability. Curr. Drug Metab. 2018, 19, 892–901. [Google Scholar] [CrossRef]
- Morley, J.S. Structure-activity relationships of enkephalin-like peptides. Annu. Rev. Pharmacol. Toxicol. 1980, 20, 81–110. [Google Scholar] [CrossRef]
- Neurath, H. Evolution of proteolytic enzymes. Science 1984, 224, 350–357. [Google Scholar] [CrossRef]
- Werle, M.; Bernkop-Schnurch, A. Strategies to improve plasma half life time of peptide and protein drugs. Amino Acids 2006, 30, 351–367. [Google Scholar] [CrossRef]
- Böttger, R.; Hoffmann, R.; Knappe, D. Differential stability of therapeutic peptides with different proteolytic cleavage sites in blood, plasma and serum. PLoS ONE 2017, 12, e0178943. [Google Scholar] [CrossRef]
- Böttger, R.; Knappe, D.; Hoffmann, R. Readily adaptable release kinetics of prodrugs using protease-dependent reversible PEGylation. J. Control. Release 2016, 230, 88–94. [Google Scholar] [CrossRef]
- Dass, C.; Mahalakshmi, P. Phosphorylation of enkephalins enhances their proteolytic stability. Life Sci. 1996, 58, 1039–1045. [Google Scholar] [CrossRef]
- Rasmussen, G.J.; Bundgaard, H. Prodrugs of peptides. 10. Protection of di- and tripeptides against aminopeptidase by formation of bioreversible 4-imidazolidinone derivatives. Int. J. Pharm. 1991, 71, 45–53. [Google Scholar] [CrossRef]
- Feng, J.; Lepetre-Mouelhi, S.; Gautier, A.; Mura, S.; Cailleau, C.; Coudore, F.; Hamon, M.; Couvreur, P. A new painkiller nanomedicine to bypass the blood-brain barrier and the use of morphine. Sci. Adv. 2019, 5. [Google Scholar] [CrossRef] [Green Version]
- Mollica, A.; Pinnen, F.; Stefanucci, A.; Feliciani, F.; Campestre, C.; Mannina, L.; Sobolev, A.P.; Lucente, G.; Davis, P.; Lai, J.; et al. The cis-4-amino-l-proline residue as a scaffold for the synthesis of cyclic and linear endomorphin-2 analogues. J. Med. Chem. 2012, 55, 3027–3035. [Google Scholar] [CrossRef] [Green Version]
- Bender, A.M.; Griggs, N.W.; Gao, C.; Trask, T.J.; Traynor, J.R.; Mosberg, H.I. Rapid Synthesis of Boc-2′,6′-dimethyl- l -tyrosine and Derivatives and Incorporation into Opioid Peptidomimetics. ACS Med. Chem. Lett. 2015, 6, 1199–1203. [Google Scholar] [CrossRef] [Green Version]
- Burbach, J.P.H.; Loeber, J.G.; Verhoef, J.; De Kloet, E.R.; Van Ree, J.M.; De Wied, D. Schizophrenia and Degradation of Eendorphins in Cerebrospinal Fluid. Lancet 1979, 314, 480–481. [Google Scholar] [CrossRef]
- Schwartz, J.C.; Malfroy, B.; De La Baume, S. Biological inactivation of enkephalins and the role of enkephalin-dipeptidyl-carboxypeptidase (“enkephalinase”) as neuropeptidase. Life Sci. 1981, 29, 1715–1740. [Google Scholar] [CrossRef]
- Burbach, J.P.H. Neuropeptides and cerebrospinal fluid. Ann. Clin. Biochem. 1982, 19, 269–277. [Google Scholar] [CrossRef]
- Eddy, N.B.; Leimbach, D. Synthetic analgesics. II. Dithienylbutenyl- and dithienylbutylamines. J. Pharmacol. Exp. Ther. 1953, 107, 385–393. [Google Scholar]
- Pathan, H.; Williams, J. Basic opioid pharmacology: An update. Br. J. Pain 2012, 6, 11–16. [Google Scholar] [CrossRef] [Green Version]
- Pacifici, R.; Patrini, G.; Venier, I.; Parolaro, D.; Zuccaro, P.; Gori, E. Effect of morphine and methadone acute treatment on immunological activity in mice: Pharmacokinetic and pharmacodynamic correlates. J. Pharmacol. Exp. Ther. 1994, 269, 1112–1116. [Google Scholar]
- Le Bars, D.; Gozariu, M.; Cadden, S.W. Animal models of nociception. Pharmacol. Rev. 2001, 53, 597–652. [Google Scholar] [PubMed]
- Tjølsen, A.; Berge, O.G.; Hunskaar, S.; Rosland, J.H.; Hole, K. The formalin test: An evaluation of the method. Pain 1992, 51, 5–17. [Google Scholar] [CrossRef]
- Jourdan, D.; Ardid, D.; Bardin, L.; Neuzeret, D.; Lanphouthacoul, L.; Eschalier, A. A new automated method of pain scoring in the formalin test in rats. Pain 1997, 71, 265–270. [Google Scholar] [CrossRef]
- Hill, R.; Disney, A.; Conibear, A.; Sutcliffe, K.; Dewey, W.; Husbands, S.; Bailey, C.; Kelly, E.; Henderson, G. The novel μ-opioid receptor agonist PZM21 depresses respiration and induces tolerance to antinociception. Br. J. Pharmacol. 2018, 175, 2653–2661. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Traub, R.J.; Murphy, A.Z. Persistent pain model reveals sex difference in morphine potency. Am. J. Physiol. Integr. Comp. Physiol. 2006, 291, R300–R306. [Google Scholar] [CrossRef] [Green Version]
- Grant, G.J.; Vermeulen, K.; Zakowski, M.I.; Stenner, M.; Turndorf, H.; Langerman, L. Prolonged analgesia and decreased toxicity with liposomal morphine in a mouse model. Anesth. Analg. 1994, 79, 706–709. [Google Scholar] [CrossRef]
- Dahan, A.; Van Der Schrier, R.; Smith, T.; Aarts, L.; Van Velzen, M.; Niesters, M. Averting opioid-induced respiratory depression without affecting analgesia. Anesthesiology 2018, 128, 1027–1037. [Google Scholar] [CrossRef]
- Hill, R.; Lyndon, A.; Withey, S.; Roberts, J.; Kershaw, Y.; Maclachlan, J.; Lingford-Hughes, A.; Kelly, E.; Bailey, C.; Hickman, M.; et al. Ethanol reversal of tolerance to the respiratory depressant effects of morphine. Neuropsychopharmacology 2016, 41, 762–773. [Google Scholar] [CrossRef] [Green Version]
- Hill, R.; Santhakumar, R.; Dewey, W.; Kelly, E.; Henderson, G. Fentanyl depression of respiration: Comparison with heroin and morphine. Br. J. Pharmacol. 2020, 177, 254–266. [Google Scholar] [CrossRef]
- Kosten, T.R.; George, T.P. The neurobiology of opioid dependence: Implications for treatment. Sci. Pract. Perspect. 2002, 1, 13–20. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Coller, J.K.; Watkins, L.R.; Somogyi, A.A.; Hutchinson, M.R. Naloxone-precipitated morphine withdrawal behavior and brain IL-1β expression: Comparison of different mouse strains. Brain. Behav. Immun. 2011, 25, 1223–1232. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Yoshii, T.; Yanaura, S. Induction of Physical Dependence on Morphine in Mice by the Drug-Admixed Food Method. Jpn. J. Pharmacol. 1984, 34, 319–325. [Google Scholar] [CrossRef]
- Dumas, E.O.; Pollack, G.M. Opioid tolerance development: A pharmacokinetic/pharmacodynamic perspective. AAPS J. 2008, 10, 537–551. [Google Scholar] [CrossRef] [Green Version]
- Yuan, B.Y.; Liu, W.Z.; Wang, X.F.; Zhang, Y.Z.; Yang, D.J.; Wang, C.L. Endomorphin-1 analogs with oligoarginine-conjugation at C-terminus produce potent antinociception with reduced opioid tolerance in paw withdrawal test. Peptides 2018, 106, 96–101. [Google Scholar] [CrossRef]
- Le Naour, M.; Akgün, E.; Yekkirala, A.; Lunzer, M.M.; Powers, M.D.; Kalyuzhny, A.E.; Portoghese, P.S. Bivalent ligands that target μ opioid (MOP) and cannabinoid1 (CB 1) receptors are potent analgesics devoid of tolerance. J. Med. Chem. 2013, 56, 5505–5513. [Google Scholar] [CrossRef] [Green Version]
- Mansouri, M.T.; Khodayar, M.J.; Tabatabaee, A.; Ghorbanzadeh, B.; Naghizadeh, B. Modulation of morphine antinociceptive tolerance and physical dependence by co-administration of simvastatin. Pharmacol. Biochem. Behav. 2015, 137, 38–43. [Google Scholar] [CrossRef]
- Ren, X.; Noda, Y.; Mamiya, T.; Nagai, T.; Nabeshima, T. A neuroactive steroid, dehydroepiandrosterone sulfate, prevents the development of morphine dependence and tolerance via c-fos expression linked to the extracellular signal-regulated protein kinase. Behav. Brain Res. 2004, 152, 243–250. [Google Scholar] [CrossRef]
- Yamazaki, M.; Suzuki, T.; Narita, M.; Lipkowski, A.W. The opioid peptide analogue biphalin induces less physical dependence than morphine. Life Sci. 2001, 69, 1023–1028. [Google Scholar] [CrossRef]
- Sitbon, P.; Van Elstraete, A.; Hamdi, L.; Juarez-Perez, V.; Mazoit, J.X.; Benhamou, D.; Rougeot, C. STR-324, a Stable Analog of Opiorphin, Causes Analgesia in Postoperative Pain by Activating Endogenous Opioid Receptor-dependent Pathways. Anesthesiology 2016, 125, 1017–1029. [Google Scholar] [CrossRef]
- Roques, B.P.; Noble, F.; Daugé, V.; Fournié-Zaluski, M.C.; Beaumont, A. Neutral endopeptidase 24.11: Structure, inhibition, and experimental and clinical pharmacology. Pharmacol. Rev. 1993, 45, 87–146. [Google Scholar]
- Raffa, R.B.; Pergolizzi, J.V.; Taylor, R.; Ossipov, M.H. Indirect-acting strategy of opioid action instead of direct receptor activation: Dual-acting enkephalinase inhibitors (DENKIs). J. Clin. Pharm. Ther. 2018, 43, 443–449. [Google Scholar] [CrossRef] [Green Version]
- Popik, P.; Kamysz, E.; Kreczko, J.; Wróbel, M. Human opiorphin: The lack of physiological dependence, tolerance to antinociceptive effects and abuse liability in laboratory mice. Behav. Brain Res. 2010, 213, 88–93. [Google Scholar] [CrossRef]
- Mello, N.K.; Mendelson, J.H. Self-administration of an enkephalin analog by rhesus monkey. Pharmacol. Biochem. Behav. 1978. [Google Scholar] [CrossRef]
- Lowery, J.J.; Raymond, T.J.; Giuvelis, D.; Bidlack, J.M.; Polt, R.; Bilsky, E.J. In vivo characterization of MMP-2200, a mixed δ/μ opioid agonist, in mice. J. Pharmacol. Exp. Ther. 2011, 336, 767–778. [Google Scholar] [CrossRef] [Green Version]
- Hökfelt, T.; Bartfai, T.; Bloom, F. Neuropeptides: Opportunities for drug discovery. Lancet Neurol. 2003, 2, 463–472. [Google Scholar] [CrossRef]
- Godfrey, L.; Iannitelli, A.; Garrett, N.L.; Moger, J.; Imbert, I.; King, T.; Porreca, F.; Soundararajan, R.; Lalatsa, A.; Schätzlein, A.G.; et al. Nanoparticulate peptide delivery exclusively to the brain produces tolerance free analgesia. J. Control. Release 2018, 270, 135–144. [Google Scholar] [CrossRef]
- Horan, P.J.; Mattia, A.; Bilsky, E.J.; Weber, S.; Davis, T.P.; Yamamura, H.I.; Malatynska, E.; Appleyard, S.M.; Slaninova, J.; Misicka, A.; et al. Antinociceptive profile of biphalin, a dimeric enkephalin analog. J. Pharmacol. Exp. Ther. 1993, 265, 1446–1454. [Google Scholar]
- Noble, F.; Coric, P.; Fournié-Zaluski, M.C.; Roques, B.P. Lack of physical dependence in mice after repeated systemic administration of the mixed inhibitor prodrug of enkephalin-degrading enzymes, RB101. Eur. J. Pharmacol. 1992, 223, 91–96. [Google Scholar] [CrossRef]
- Boudinot, E.; Morin-Surun, M.-P.; Foutz, A.S.; Fournié-Zaluski, M.-C.; Roques, B.P.; Denavit-Saubié, M. Effects of the potent analgesic enkephalin-catabolizing enzyme inhibitors RB101 and kelatorphan on respiration. Pain 2001, 90, 7–13. [Google Scholar] [CrossRef]
- Heidbreder, C.; Gewiss, M.; Lallemand, S.; Roques, B.P.; de Witte, P. Inhibition of enkephalin metabolism and activation of mu- or delta-opioid receptors elicit opposite effects on reward and motility in the ventral mesencephalon. Neuropharmacology 1992, 31, 293–298. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
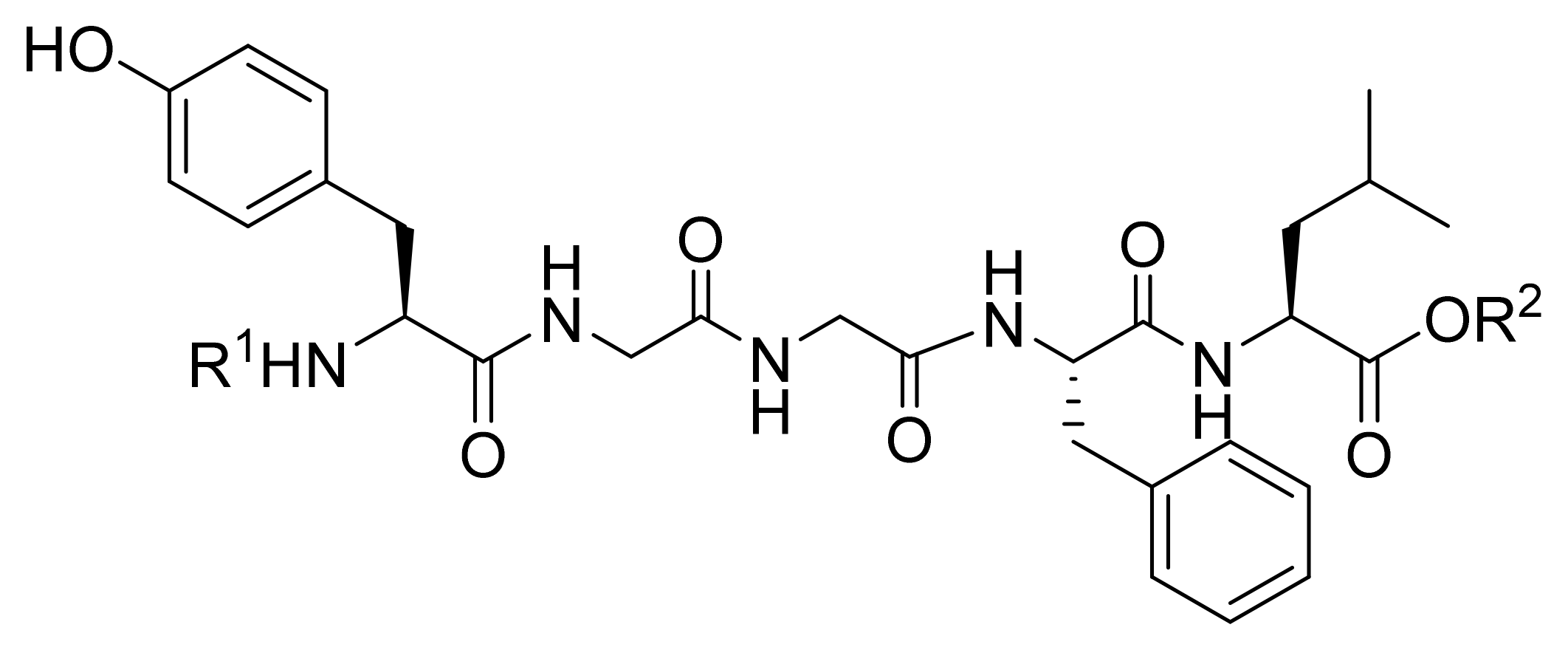
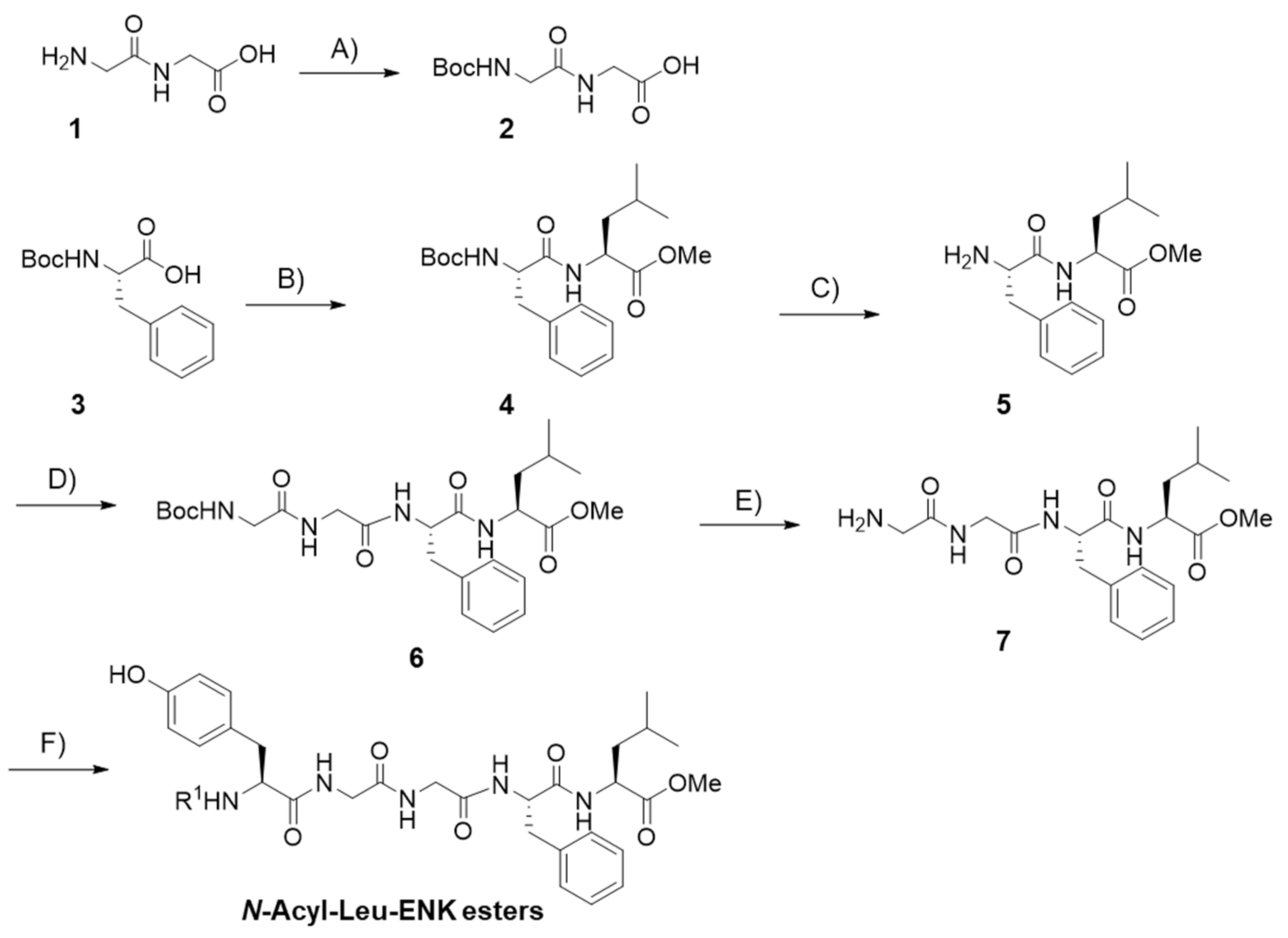
| Compound Code | N-Terminal Modification R 1,a | C-Terminal Modification R 2,a | clogP b | Recovered Peptide after 1 h in Mouse Plasma [%] | Relative Binding to DOR Compared to Leu-ENK [%] c | Antinociception (AUC0-5h, %MPE·h) d |
|---|---|---|---|---|---|---|
| Leu-ENK |  | H | −0.85 | 23 ± 2 | 100 ± 16 | 14 ± 6 |
| KK-14 e | 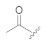 | CH3 | 1.50 | 82 ± 5 | 38 ± 4 | 89 ± 28 |
| KK-81 |  | CH3 | 3.20 | 88 ± 7 | 41 ± 25 | 37 ± 10 |
| KK-82 | 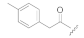 | CH3 | 3.70 | 51 ± 5 | 49 ± 20 | 43 ± 17 |
| KK-93 |  | CH3 | 3.61 | 85 ± 7 | 33 ± 11 | 51 ± 13 |
| KK-102 |  | CH3 | 2.74 | 94 ± 3 | 70 ± 11 | 112 ± 24 |
| KK-105 | 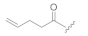 | CH3 | 2.60 | 80 ± 7 | 59 ± 22 | 75 ± 22 |
| KK-108 |  | CH3 | 4.67 | 73 ± 4 | 39 ± 27 | 48 ± 15 |
| KK-112 | 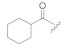 | CH3 | 3.53 | 62 ± 4 | 29 ± 21 | 51 ± 10 |
| KK-103 f | 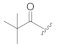 | H | 2.61 | 93 ± 1 | 68 ± 2 | 142 ± 15 |
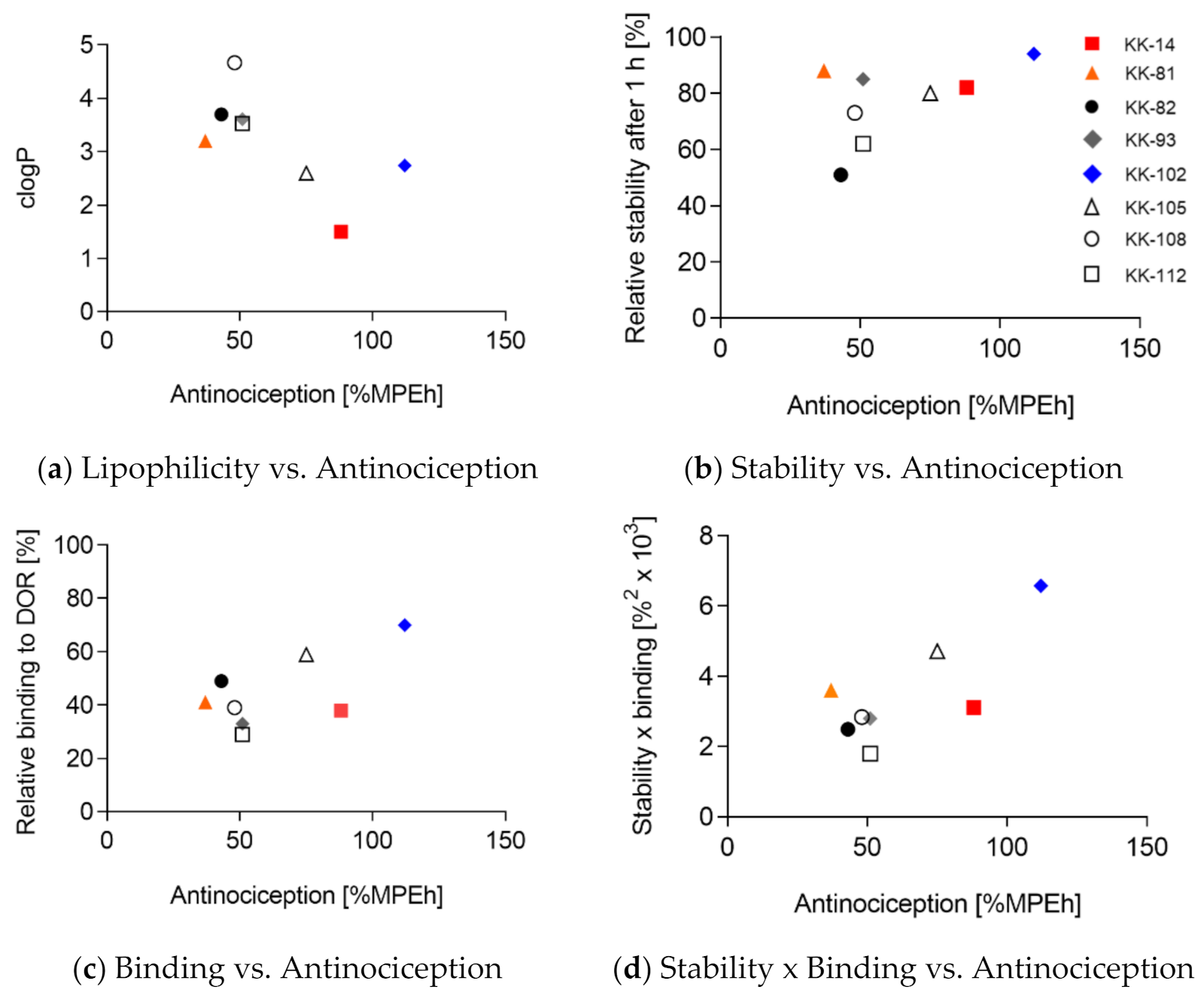
| R2 | p (Two-Tailed) | p Value Summary | |
|---|---|---|---|
| Lipophilicity vs. Antinociception | 0.4376 | 0.07 | n.s. |
| Stability vs. Antinociception | 0.2777 | 0.18 | n.s. |
| Binding vs. Antinociception | 0.4372 | 0.07 | n.s. |
| Stability × Binding vs. Antinociception | 0.5909 | 0.03 | * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Viswanadham, K.K.D.; Böttger, R.; Hohenwarter, L.; Nguyen, A.; Rouhollahi, E.; Smith, A.; Tsai, Y.-H.; Chang, Y.-Y.; Ortiz, C.L.; Yang, L.-W.; et al. An Effective and Safe Enkephalin Analog for Antinociception. Pharmaceutics 2021, 13, 927. https://doi.org/10.3390/pharmaceutics13070927
Viswanadham KKD, Böttger R, Hohenwarter L, Nguyen A, Rouhollahi E, Smith A, Tsai Y-H, Chang Y-Y, Ortiz CL, Yang L-W, et al. An Effective and Safe Enkephalin Analog for Antinociception. Pharmaceutics. 2021; 13(7):927. https://doi.org/10.3390/pharmaceutics13070927
Chicago/Turabian StyleViswanadham, K. K. DurgaRao, Roland Böttger, Lukas Hohenwarter, Anne Nguyen, Elham Rouhollahi, Alexander Smith, Yi-Hsuan Tsai, Yuan-Yu Chang, Christopher Llynard Ortiz, Lee-Wei Yang, and et al. 2021. "An Effective and Safe Enkephalin Analog for Antinociception" Pharmaceutics 13, no. 7: 927. https://doi.org/10.3390/pharmaceutics13070927
APA StyleViswanadham, K. K. D., Böttger, R., Hohenwarter, L., Nguyen, A., Rouhollahi, E., Smith, A., Tsai, Y.-H., Chang, Y.-Y., Ortiz, C. L., Yang, L.-W., Jimenez, L., Li, S., Hur, C., & Li, S.-D. (2021). An Effective and Safe Enkephalin Analog for Antinociception. Pharmaceutics, 13(7), 927. https://doi.org/10.3390/pharmaceutics13070927