
Glioblastoma Multiforme—A Look at the Past and a Glance at the Future



Abstract
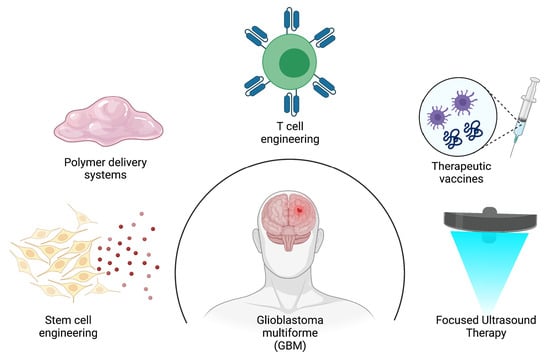
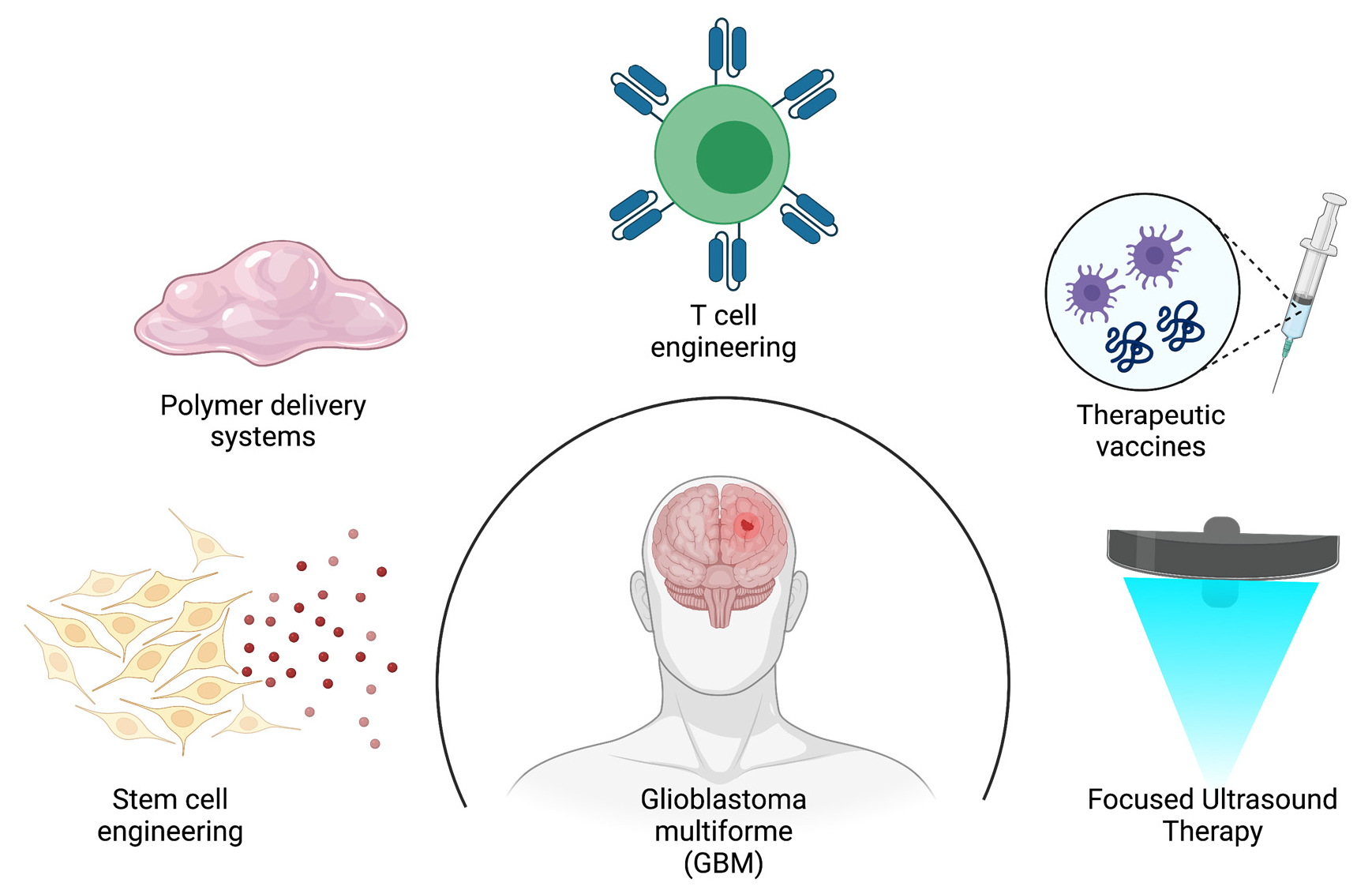
1. Introduction
2. Current Treatment for Glioblastoma Multiforme (GBM)—Where We Are Now?
2.1. Surgical Resection
2.2. Radiotherapy
2.3. Chemotherapy
3. Immune Checkpoint Inhibitors in GBM
4. Clinical Need to Target Tumor Infiltration
5. Clinical Need for Drug Delivery Systems
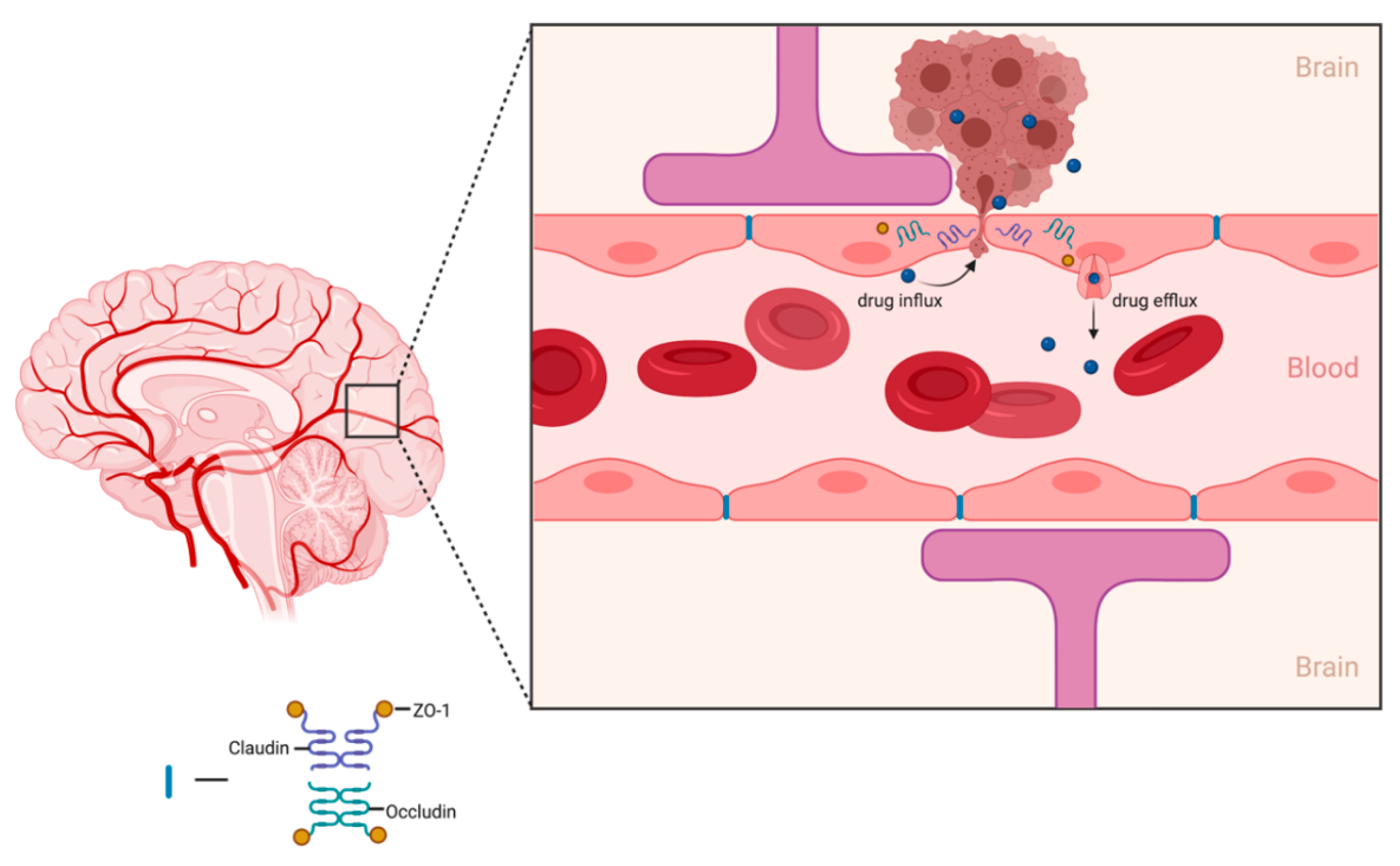
6. Clinical Need to Increase Delivery of Therapeutics across Blood–Brain Barrier (BBB)
7. A Glance at the Future
7.1. Anticancer Stem Cell Therapy for GBM
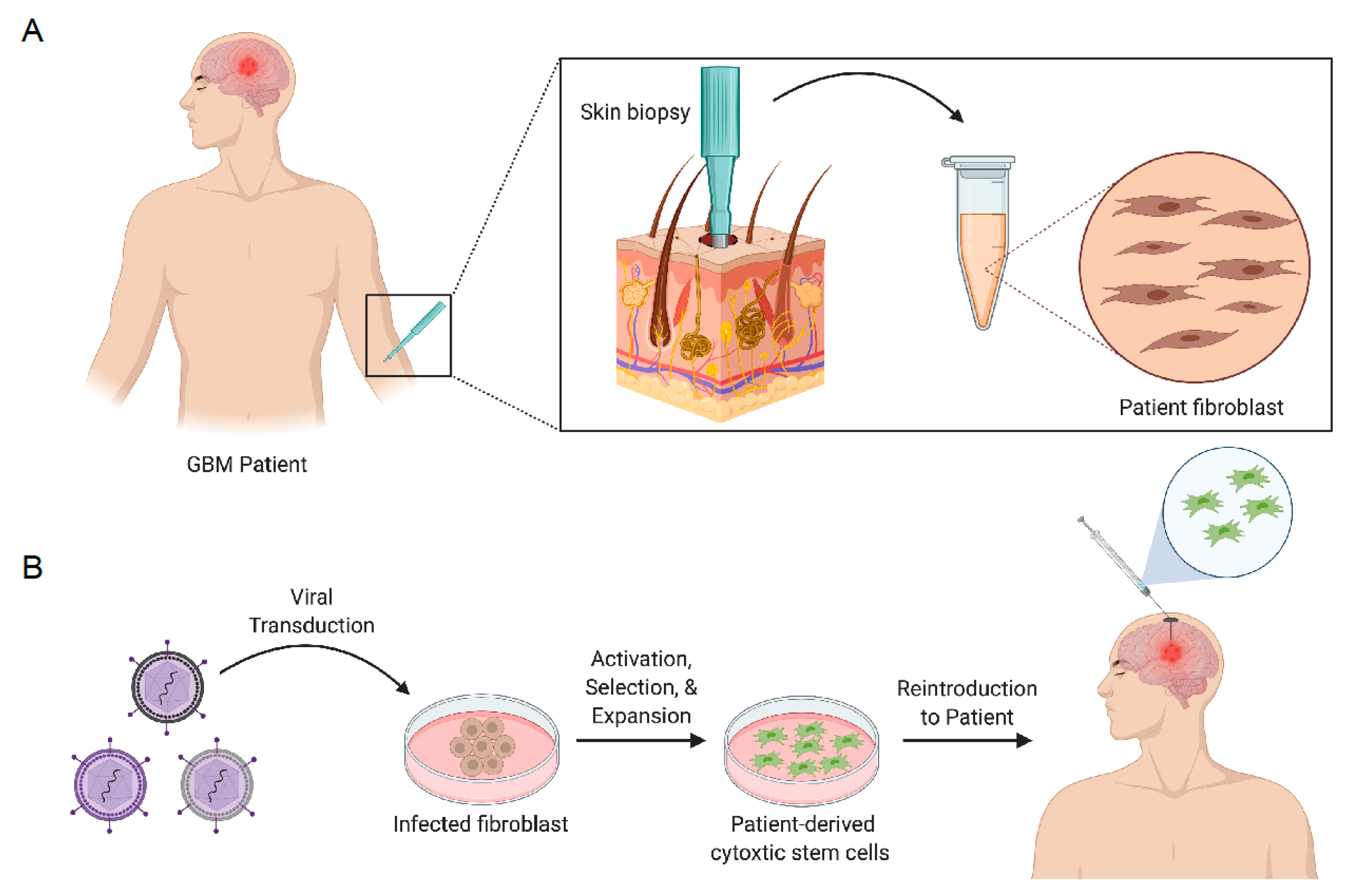
7.1.1. Engineering Stem Cells to Secrete Anticancer Proteins
7.1.2. Engineering Stem Cells to Induce Cancer Cell Suicide
7.1.3. Engineering Stem Cells with Oncolytic Virus
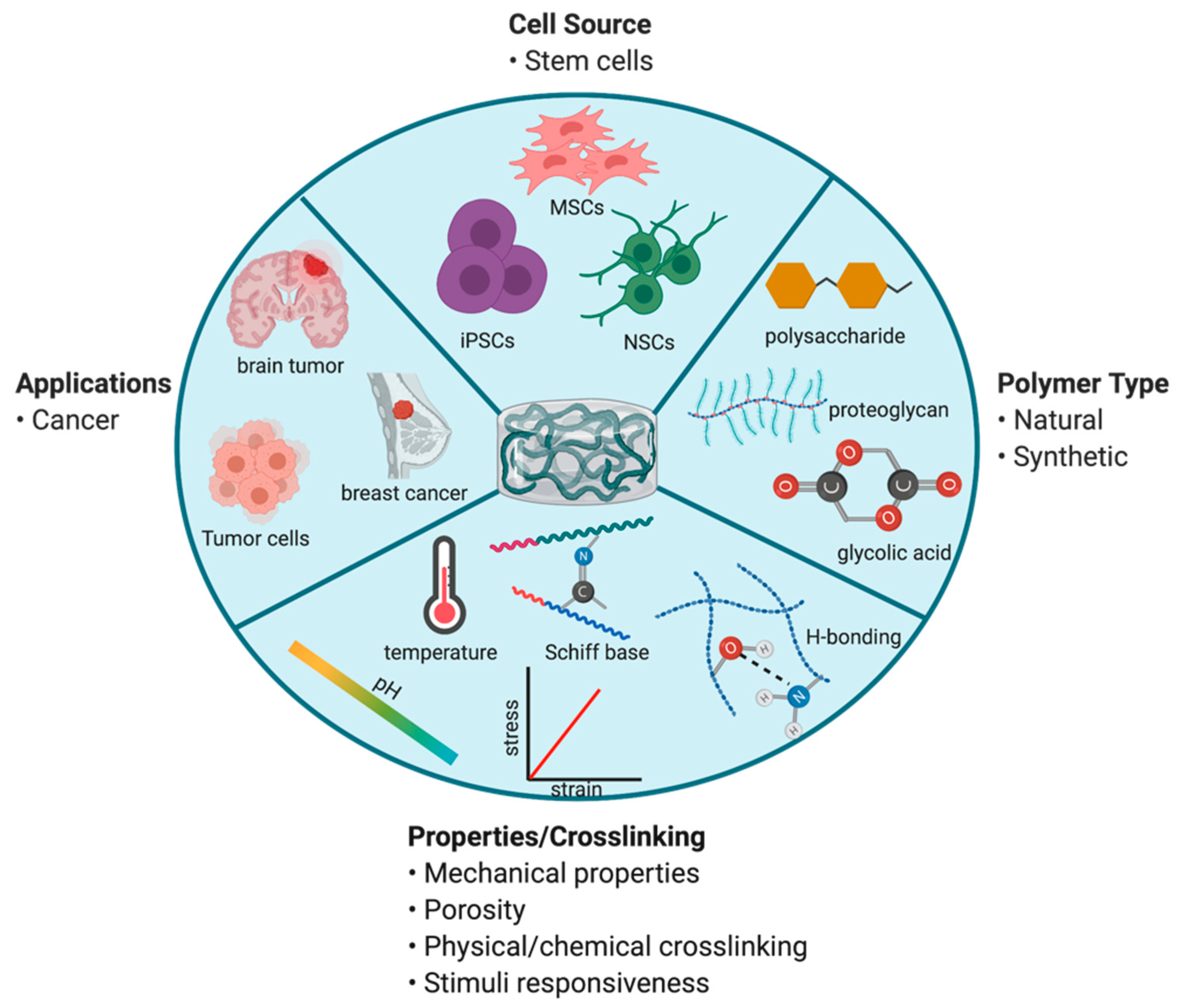
7.2. Polymer-Based Scaffolds for Tumoricidal Stem Cells (SCs)
7.3. Immunotherapeutic Strategies for Improving GBM Therapy
7.3.1. Engineering T Cells to Recognize GBM-Associated Antigens and Induce Tumor Cell Death
7.3.2. Engineering Vaccines to Stimulate Specific Immune Responses against GBM
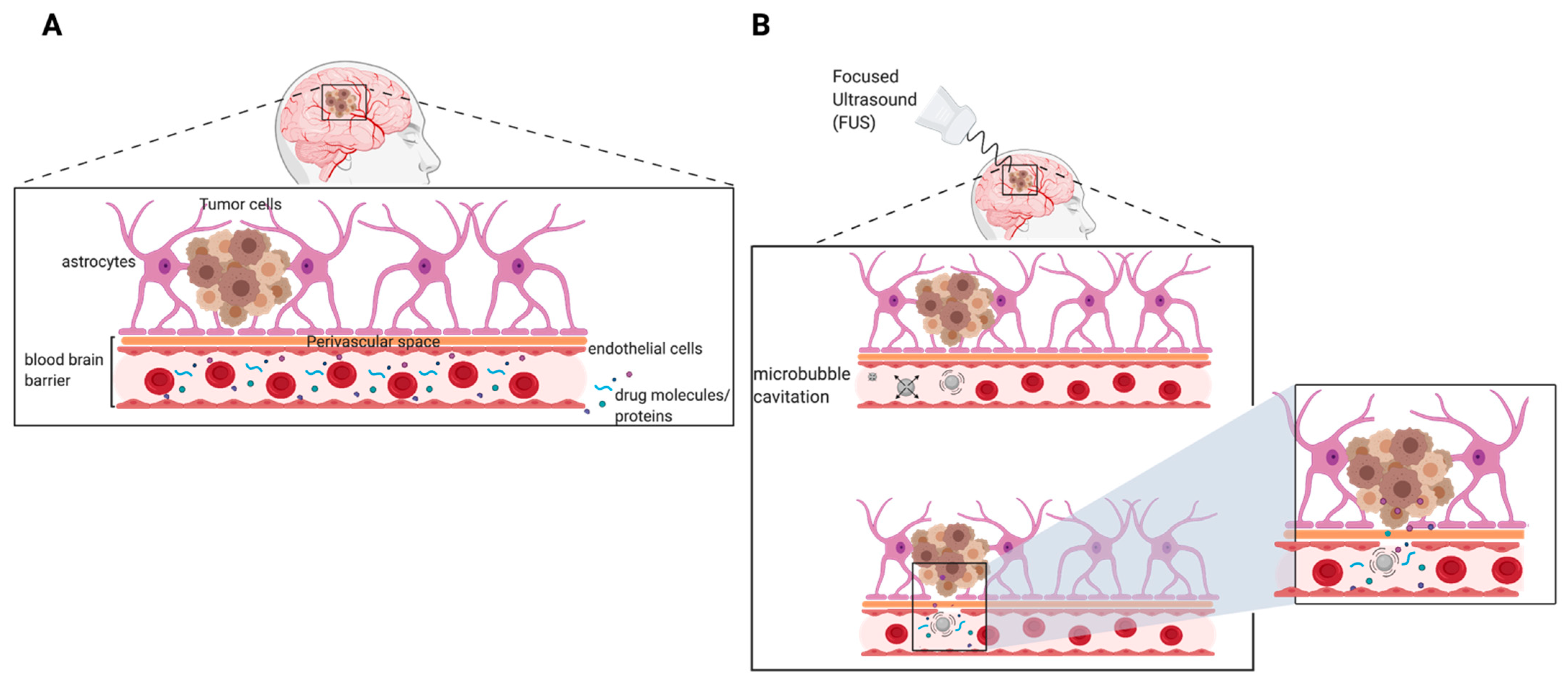
7.4. Focused Ultrasound-Mediated Therapy to Improve Delivery of Therapeutics for GBM

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Unit | Definition |
|---|---|---|
| Frequency | MHz, Hz | Number of cycles or oscillations per second |
| Pressure | MPa | Pressure caused by a sound wave minus the ambient pressure in a medium resulting from the sound wave |
| Pulse repetition frequency | Hz | Number of emitted pulses that occur per second |
| Burst duration | ms | The length of time designated for repeat pulses at a constant frequency |
| Total exposure time/total time (TT) | s | The total amount of time the transducer is emitting ultrasonic energy in an area |
| Preclinical | |||
|---|---|---|---|
| Animal Species/Therapeutic Agent | US Parameters | Key Findings | Ref. |
| Species: New Zealand white rabbits | Intensity: 16–690 W/cm2 Pressure: 0.7–4.7 MPa Burst duration: 10 or 100 ms PRF: 1 Hz TT: 20 s | Low acoustic power levels were able to consistently enhance BBB permeability following administration of an US contrast agent No neuronal damage was observed at pressure amplitudes 0.7 and 1.0 MPa. Opening of the BBB was independent of burst duration and acoustic power. | [96] |
| Species: Orthotopic xenograft model Drug: Doxorubicin, ado-trastuzumab emtansine (T-DM1) | Frequency: 1 MHz Peak negative pressure (PNP): 480 kPa Burst duration: 10 ms every 1 s TT: 2 min | Extravasation of doxorubicin and T-DM1 was significantly increased using FUS in combination with microbubble contrast agent in comparison to non-FUS group via multiphoton microscopy (7-fold and 2-fold higher). Drug penetration was significantly increased in both treatment groups (>100 vs. <20 μm and 42 ± 7 vs. 12 ± 4 μm for doxorubicin and T-DM1). | [105] |
| Species: Fischer 344 rats Drug: TMZ | Power: 3 W PNP: 0.6 MPa Burst duration: 10 ms PRF: 1 Hz TT: 60 s | Accumulation of TMZ in CSF/plasma increased following FUS treatment (22.7% to 38.6%). Reduction in 7 day tumor progression ratio was observed following FUS treatment (24.03 to 5.06) Median survival was extended from 20 to 23 following FUS treatment. | [106] |
| Species: Sprague-Dawley rats Drug: liposomal doxorubicin | Pressure: 1.2 MPa Burst duration: 10 ms PRF: 1 Hz TT: 60–120 s | Reduction in tumor growth was observed in the FUS + DOX treated group in comparison to DOX alone (indicated by tumor volume doubling time 3.7 ± 0.5 days vs. 2.7 ± 0.4 days). A significant increase (>24%) in median survival was observed in FUS + DOX treated group in comparison to non-treated group (p = 0.0007). | [107] |
| Species: Nu/Nu mice Drug: BVZ | Frequency: 400 kHz PNP: 0.4–0.8 MPa Burst duration: 10 ms PRF: 1 Hz TT: 60 s | Penetration of BVZ into the CNS was statistically enhanced in the FUS + BVZ in comparison to BVZ alone (5.73-fold increase at 0.4 MPa and 56.7-fold increase at 0.8 MPa). Median survival time was significantly increased in FUS + BVZ treated group in comparison to BVZ alone (135% vs. 48%; p = 0.0002). | [108] |
| NCT Number/Study Completion Date | Status/Location | FUS Device + Drug | Primary Outcome Measures |
|---|---|---|---|
| NCT03616860 Study Completion Date: December 2024 | Recruiting Location: Canada | Device: ExAblate Neuro Model 4000 Type 2 Drug: TMZ | Device and procedure related adverse events (safety) |
| NCT03551249 Study Completion Date: December 2024 | Recruiting Location: US | Device: ExAblate Neuro Model 4000 Type 2 Drug: TMZ | Device and procedure related adverse events (safety) |
| NCT04440358 Study Completion Date: April 2023 | Recruiting Location: Canada | Device: ExAblate Neuro Model 4000 Type 2 Drug: Carboplatin | Adverse events (safety) Contrast intensity on MR imaging |
| NCT04417088 Study Completion Date: November 2023 | Recruiting Location: US | Device: ExAblate Neuro Model 4000 Type 2 Drug: Carboplatin | Adverse events (safety) Contrast intensity on MR imaging |
| NCT03712293 Study Completion Date: December 2021 | Recruiting Location: Korea | Device: ExAblate Neuro Model 4000 Type 2 Drug: TMZ | Adverse events (safety) |
| NCT04446416 Study Completion Date: December 2022 | Recruiting Location: Taiwan | Device: NaviFUS System Drug: BVZ | Adverse events (safety) PFS at 6 months |
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Goodenberger, M.; Jenkins, R.B. Genetics of adult glioma. Cancer Genet. 2012, 205, 613–621. [Google Scholar] [CrossRef]
- Jäkel, S.; Dimou, L. Glial Cells and Their Function in the Adult Brain: A Journey through the History of Their Ablation. Front. Cell. Neurosci. 2017, 11, 24. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Armstrong, T.; Gilbert, M.R. Biology and management of ependymomas. Neuro-Oncology 2016, 18, 902–913. [Google Scholar] [CrossRef] [PubMed]
- Bradl, M.; Lassmann, H. Oligodendrocytes: Biology and pathology. Acta Neuropathol. 2009, 119, 37–53. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Gittleman, H.; Farah, P.; Ondracek, A.; Chen, Y.; Wolinsky, Y.; Stroup, N.E.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2006–2010. Neuro-Oncology 2013, 15 (Suppl. 2), ii1–ii56. [Google Scholar] [CrossRef]
- Wrensch, M.; Minn, Y.; Chew, T.; Bondy, M.; Berger, M.S. Epidemiology of primary brain tumors: Current concepts and review of the literature. Neuro-Oncology 2002, 4, 278–299. [Google Scholar] [CrossRef]
- Prager, B.C.; Bhargava, S.; Mahadev, V.; Hubert, C.G.; Rich, J.N. Glioblastoma Stem Cells: Driving Resilience through Chaos. Trends Cancer 2020, 6, 223–235. [Google Scholar] [CrossRef]
- Olar, A.; Aldape, K.D. Using the molecular classification of glioblastoma to inform personalized treatment. J. Pathol. 2014, 232, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Bleeker, F.E.; Molenaar, R.J.; Leenstra, S. Recent advances in the molecular understanding of glioblastoma. J. Neuro Oncol. 2012, 108, 11–27. [Google Scholar] [CrossRef]
- Young, R.M.; Jamshidi, A.; Davis, G.; Sherman, J.H. Current trends in the surgical management and treatment of adult glioblastoma. Ann. Transl. Med. 2015, 3, 121. [Google Scholar] [CrossRef]
- Brown, T.J.; Brennan, M.C.; Li, M.; Church, E.W.; Brandmeir, N.J.; Rakszawski, K.L.; Patel, A.S.; Rizk, E.B.; Suki, D.; Sawaya, R.; et al. Association of the Extent of Resection With Survival in Glioblastoma: A Systematic Review and Meta-analysis. JAMA Oncol. 2016, 2, 1460–1469. [Google Scholar] [CrossRef]
- Han, Q.; Liang, H.; Cheng, P.; Yang, H.; Zhao, P. Gross Total vs. Subtotal Resection on Survival Outcomes in Elderly Patients With High-Grade Glioma: A Systematic Review and Meta-Analysis. Front. Oncol. 2020, 10, 151. [Google Scholar] [CrossRef]
- Tunthanathip, T.; Madteng, S. Factors associated with the extent of resection of glioblastoma. Precis. Cancer Med. 2020, 3, 12. [Google Scholar] [CrossRef]
- Mann, J.; Ramakrishna, R.; Magge, R.; Wernicke, A.G. Advances in radiotherapy for glioblastoma. Front. Neurol. 2017, 8, 748. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, A.R.; Kirkpatrick, J.; Fiveash, J.B.; Shih, H.; Koay, E.J.; Lutz, S.; Petit, J.; Chao, S.T.; Brown, P.D.; Vogelbaum, M.; et al. Radiation therapy for glioblastoma: Executive summary of an American Society for Radiation Oncology Evidence-Based Clinical Practice Guideline. Pract Radiat Oncol. 2016, 6, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Nabors, L.; Portnow, J.; Ammirati, M.; Baehring, J.; Brem, H.; Butowski, N.; Fenstermaker, R.A.; Forsyth, P.; Hattangadi-Gluth, J.; Holdhoff, M.; et al. NCCN guidelines insights: Central nervous system cancers, version 1. J. Natl. Compr. Cancer Netw. 2017, 15, 1331–1345. [Google Scholar] [CrossRef]
- Roa, W.; Brasher, P.M.A.; Bauman, G.; Anthes, M.; Bruera, E.; Chan, A.; Fisher, B.; Fulton, D.; Gulavita, S.; Hao, C.; et al. Abbreviated course of radiation therapy in older patients with glioblastoma multiforme: A prospective randomized clinical trial. J. Clin. Oncol. 2004, 22, 1583–1588. [Google Scholar] [CrossRef]
- Roa, W.; Kepka, L.; Kumar, N.; Sinaika, V.; Matiello, J.; Lomidze, D.; Hentati, D.; de Castro, D.G.; Dyttus-Cebulok, K.; Drodge, S.; et al. International Atomic Energy Agency Randomized Phase III Study of Radiation Therapy in Elderly and/or Frail Patients With Newly Diagnosed Glioblastoma Multiforme. J. Clin. Oncol. 2015, 33, 4145–4150. [Google Scholar] [CrossRef]
- Taylor, A.; Powell, M.E.B. Intensity-modulated radiotherapy—What is it? Cancer Imaging 2004, 4, 68–73. [Google Scholar] [CrossRef]
- MacDonald, S.M.; Ahmad, S.; Kachris, S.; Vogds, B.J.; DeRouen, M.; Gittleman, A.E.; DeWyngaert, K.; Vlachaki, M.T. Intensity modulated radiation therapy versus three-dimensional conformal radiation therapy for the treatment of high grade glioma: A dosimetric comparison. J. Appl. Clin. Med. Phys. 2007, 8, 47–60. [Google Scholar] [CrossRef]
- Ding, M.; Newman, F.; Chen, C.; Stuhr, K.; Gaspar, L.E. Dosimetric comparison between 3DCRT and IMRT using different multileaf collimators in the treatment of brain tumors. Med. Dosim. 2009, 34, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A Randomized Trial of Bevacizumab for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef]
- Giladi, M.; Munster, M.; Schneiderman, R.S.; Voloshin, T.; Porat, Y.; Blat, R.; Zielinska-Chomej, K.; Hååg, P.; Bomzon, Z.; Kirson, E.D.; et al. Tumor treating fields (TTFields) delay DNA damage repair following radiation treatment of glioma cells. Radiat. Oncol. 2017, 12, 206. [Google Scholar] [CrossRef]
- Kim, E.S.; Kim, J.E.; Patel, M.A.; Mangraviti, A.; Ruzevick, J.; Lim, M. Immune checkpoint modulators: An emerging antiglioma armamentarium. J. Immunol. Res. 2016, 2016, 4683607. [Google Scholar] [CrossRef]
- Preusser, M.; Lim, M.; Hafler, D.A.; Reardon, D.A.; Sampson, J.H. Prospects of immune checkpoint modulators in the treatment of glioblastoma. Nat. Rev. Neurol. 2015, 11, 504–514. [Google Scholar] [CrossRef]
- Gil-Gil, M.J.; Mesia, C.; Rey, M.; Bruna, J. Bevacizumab for the treatment of glioblastoma. Clin. Med. Insights Oncol. 2013, 7, 123–135. [Google Scholar] [CrossRef]
- Bent, M.V.D.; Gorlia, T.; Bendszus, M.; Sahm, F.; Domont, J.; Idbaih, A.; Platten, M.; Weller, M.; Golfoinopoulos, V.; Wick, W.; et al. EH1.3 EORTC 26101 phase III trial exploring the combination of bevacizumab and lomustine versus lomustine in patients with first progression of a glioblastoma. Neuro-Oncology 2016, 18, iv1–iv2. [Google Scholar] [CrossRef][Green Version]
- Brandes, A.A.; Bartolotti, M.; Tosoni, A.; Poggi, R.; Franceschi, E. Practical management of bevacizumab-related toxicities in glioblastoma. Oncologist 2015, 20, 166–175. [Google Scholar] [CrossRef]
- Weber, E.L.; Goebel, E.A. Cerebral edema associated with Gliadel wafers: Two case studies. Neuro-Oncology 2005, 7, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Gallego, J.M.; Barcia, J.A.; Barcia-Mariño, C. Fatal outcome related to carmustine implants in glioblastoma multiforme. Acta Neurochir. 2007, 149, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Graeber, M.B.; Scheithauer, B.W.; Kreutzberg, G.W. Microglia in brain tumors. Glia 2002, 40, 252–259. [Google Scholar] [CrossRef]
- Reardon, D.A.; Freeman, G.; Wu, C.; Chiocca, E.A.; Wucherpfennig, K.W.; Wen, P.Y.; Fritsch, E.F.; Curry, W.T.; Sampson, J.; Dranoff, G. Immunotherapy advances for glioblastoma. Neuro-Oncology 2014, 16, 1441–1458. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bähr, O.; et al. Effect of nivolumab vs bevacizumab in patients with recurrent glioblastoma: The checkmate 143 phase 3 randomized clinical trial. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Manjila, S.; Hdeib, A.M.; Radhakrishnan, A.; Nock, C.J.; Cohen, M.L.; Sloan, A.E. Extracranial metastasis of gliobastoma: Three illustrative cases and current review of the molecular pathology and management strategies. Mol. Clin. Oncol. 2015, 3, 479–486. [Google Scholar] [CrossRef]
- Rosen, J.; Blau, T.; Grau, S.J.; Barbe, M.T.; Fink, G.R.; Galldiks, N. Extracranial metastases of a cerebral glioblastoma: A case report and review of the literature. Case Rep. Oncol. 2018, 11, 591–600. [Google Scholar] [CrossRef]
- Rossi, J.; Giaccherini, L.; Cavallieri, F.; Napoli, M.; Moratti, C.; Froio, E.; Serra, S.; Fraternali, A.; Ghadirpour, R.; Cozzi, S.; et al. Extracranial metastases in secondary glioblastoma multiforme: A case report. BMC Neurol. 2020, 20, 382. [Google Scholar] [CrossRef]
- Mehrotra, A.; Das, K.K.; Jamdar, J.; Jaiswal, A.K.; Behari, S.; Singh, G.; Sardhara, J.; Pal, L.; Srivastava, A.K.; Sahu, R.N. Multiple glioblastomas: Are they different from their solitary counterparts? Asian J. Neurosurg. 2015, 10, 266–271. [Google Scholar] [CrossRef]
- Mallick, S.; Benson, R.; Hakim, A.; Rath, G.K. Management of glioblastoma after recurrence: A changing paradigm. J. Egypt. Natl. Cancer Inst. 2016, 28, 199–210. [Google Scholar] [CrossRef]
- Bota, D.A.; Desjardins, A.; Quinn, J.A.; Affronti, M.L.; Friedman, H.S. Interstitial chemotherapy with biodegradable BCNU (Gliadel) wafers in the treatment of malignant gliomas. Ther. Clin. Risk Manag. 2007, 3, 707–715. [Google Scholar] [PubMed]
- Haque, R.M.; Amundson, E.; Dorsi, M.; Brem, H. Interstitial Chemotherapy and Polymer-Drug Delivery. In Handbook of Brain Tumor Chemotherapy; Elsevier: Amsterdam, The Netherlands, 2006; pp. 274–294. [Google Scholar]
- Attenello, F.; Raza, S.M.; Dimeco, F.; Olivi, A. Chemotherapy for brain tumors with polymer drug delivery. Handb. Clin. Neurol. 2012, 104, 339–353. [Google Scholar] [PubMed]
- Mikitsh, J.L.; Chacko, A.-M. Pathways for small molecule delivery to the central nervous system across the blood-brain barrier. Perspect. Med. Chem. 2014, 6, 11–24. [Google Scholar] [CrossRef]
- Pardridge, W.M. The blood-brain barrier: Bottleneck in brain drug development. NeuroRx 2005, 2, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Bart, J.; Groen, H.J.; Hendrikse, N.H.; van der Graaf, W.T.; Vaalburg, W.; de Vries, E.G. The blood-brain barrier and oncology: New insights into function and modulation. Cancer Treat. Rev. 2000, 26, 449–462. [Google Scholar] [CrossRef]
- Nagy, J.A.; Chang, S.-H.; Shih, S.-C.; Dvorak, A.M.; Dvorak, H.F. Heterogeneity of the tumor vasculature. Semin. Thromb. Hemost. 2010, 36, 321–331. [Google Scholar] [CrossRef]
- van Tellingen, O.; Yetkin-Arik, B.; de Gooijer, M.C.; Wesseling, P.; Wurdinger, T.; de Vries, H.E. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist. Updat. 2015, 19, 1–12. [Google Scholar] [CrossRef]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar]
- Yhee, J.Y.; Son, S.; Son, S.; Joo, M.K.; Kwon, I.C. The EPR effect in cancer therapy. In Cancer Targeted Drug Delivery; Bae, Y.H., Mrsny, R.J., Park, K., Eds.; Springer: New York, NY, USA, 2013; pp. 621–632. [Google Scholar]
- Stylianopoulos, T.; Jain, R.K. Design considerations for nanotherapeutics in oncology. Nanomedicine 2015, 11, 1893–1907. [Google Scholar] [CrossRef]
- Shi, Y.; van der Meel, R.; Chen, X.; Lammers, T. The EPR effect and beyond: Strategies to improve tumor targeting and cancer nanomedicine treatment efficacy. Theranostics 2020, 10, 7921–7924. [Google Scholar] [CrossRef]
- Golombek, S.K.; May, J.-N.; Theek, B.; Appold, L.; Drude, N.; Kiessling, F.; Lammers, T. Tumor targeting via EPR: Strategies to enhance patient responses. Adv. Drug Deliv. Rev. 2018, 130, 17–38. [Google Scholar] [CrossRef] [PubMed]
- Aboody, K.S.; Brown, A.; Rainov, N.G.; Bower, K.A.; Liu, S.; Yang, W.; Small, J.E.; Herrlinger, U.; Ourednik, V.; Black, P.M.; et al. Neural stem cells display extensive tropism for pathology in adult brain: Evidence from intracranial gliomas. Proc. Natl. Acad. Sci. USA 2000, 97, 12846–12851. [Google Scholar] [CrossRef] [PubMed]
- Lourenco, S.; Teixeira, V.H.; Kalber, T.; Jose, R.J.; Floto, R.A.; Janes, S.M. Macrophage migration inhibitory factor-CXCR4 is the dominant chemotactic axis in human mesenchymal stem cell recruitment to tumors. J. Immunol. 2015, 194, 3463–3474. [Google Scholar] [CrossRef]
- Guo, Y.; Hangoc, G.; Bian, H.; Pelus, L.M.; Broxmeyer, H.E. SDF-1/CXCL12 enhances survival and chemotaxis of murine embryonic stem cells and production of primitive and definitive hematopoietic progenitor cells. Stem Cells 2005, 23, 1324–1332. [Google Scholar] [CrossRef]
- Serfozo, P.; Schlarman, M.S.; Pierret, C.; Maria, B.L.; Kirk, M.D. Selective migration of neuralized embryonic stem cells to stem cell factor and media conditioned by glioma cell lines. Cancer Cell Int. 2006, 6, 1. [Google Scholar] [CrossRef][Green Version]
- Yamazoe, T.; Koizumi, S.; Yamasaki, T.; Amano, S.; Tokuyama, T.; Namba, H. Potent tumor tropism of induced pluripotent stem cells and induced pluripotent stem cell-derived neural stem cells in the mouse intracerebral glioma model. Int. J. Oncol. 2015, 46, 147–152. [Google Scholar] [CrossRef]
- Kosztowski, T.; Zaidi, H.A.; Quiñones-Hinojosa, A. Applications of neural and mesenchymal stem cells in the treatment of gliomas. Expert Rev. Anticancer Ther. 2009, 9, 597–612. [Google Scholar] [CrossRef]
- Balyasnikova, I.V.; Ferguson, S.D.; Sengupta, S.; Han, Y.; Lesniak, M.S. Mesenchymal stem cells modified with a single-chain antibody against EGFRvIII successfully inhibit the growth of human xenograft malignant glioma. PLoS ONE 2010, 5, e9750. [Google Scholar] [CrossRef]
- Shah, K.; Bureau, E.; Kim, D.-E.; Yang, K.; Tang, Y.; Weissleder, R.; Breakefield, X.O. Glioma therapy and real-time imaging of neural precursor cell migration and tumor regression. Ann. Neurol. 2004, 57, 34–41. [Google Scholar] [CrossRef]
- Sasportas, L.S.; Kasmieh, R.; Wakimoto, H.; Hingtgen, S.; van de Water, J.; Mohapatra, G.; Figueiredo, J.L.; Martuza, R.L.; Weissleder, R.; Shah, K. Assessment of therapeutic efficacy and fate of engineered human mesenchymal stem cells for cancer therapy. Proc. Natl. Acad. Sci. USA 2009, 106, 4822–4827. [Google Scholar] [CrossRef]
- Balyasnikova, I.V.; Ferguson, S.D.; Han, Y.; Liu, F.; Lesniak, M.S. Therapeutic effect of neural stem cells expressing TRAIL and bortezomib in mice with glioma xenografts. Cancer Lett. 2011, 310, 148–159. [Google Scholar] [CrossRef]
- Kim, S.M.; Woo, J.S.; Jeong, C.H.; Ryu, C.H.; Lim, J.Y.; Jeun, S.-S. Effective combination therapy for malignant glioma with TRAIL-secreting mesenchymal stem cells and lipoxygenase inhibitor MK886. Cancer Res. 2012, 72, 4807–4817. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.-J.; Lu, J.-T.; Tu, H.-J.; Huang, K.-M.; Fu, R.; Cao, G.; Huang, M.; Cheng, L.-H.; Dai, L.-J.; Zhang, L. TRAIL-engineered bone marrow-derived mesenchymal stem cells: TRAIL expression and cytotoxic effects on C6 glioma cells. Anticancer Res. 2014, 34, 729–734. [Google Scholar] [PubMed]
- Bago, J.R.; Alfonso-Pecchio, A.; Okolie, O.; Dumitru, R.; Rinkenbaugh, A.; Baldwin, A.S.; Miller, C.; Magness, S.T.; Hingtgen, A.R.A.S.B.C.R.M.S.D. Therapeutically engineered induced neural stem cells are tumour-homing and inhibit progression of glioblastoma. Nat. Commun. 2016, 7, 10593. [Google Scholar] [CrossRef]
- Buckley, A.; Hagler, S.B.; Lettry, V.; Bago, J.R.; Maingi, S.M.; Khagi, S.; Ewend, M.G.; Miller, C.; Hingtgen, S.D. Generation and Profiling of Tumor-Homing Induced Neural Stem Cells from the Skin of Cancer Patients. Mol. Ther. 2020, 28, 1614–1627. [Google Scholar] [CrossRef] [PubMed]
- Bomba, H.N.; Sheets, K.T.; Valdivia, A.; Khagi, S.; Ruterbories, L.; Mariani, C.L.; Borst, L.B.; Tokarz, D.A.; Hingtgen, S.D. Personalized-induced neural stem cell therapy: Generation, transplant, and safety in a large animal model. Bioeng. Transl. Med. 2020, 6, e10171. [Google Scholar] [PubMed]
- Portnow, J.; Synold, T.; Badie, B.; Tirughana, R.; Lacey, S.F.; D’Apuzzo, M.; Metz, M.Z.; Najbauer, J.; Bedell, V.; Vo, T.; et al. Neural Stem Cell–Based Anticancer Gene Therapy: A First-in-Human Study in Recurrent High-Grade Glioma Patients. Clin. Cancer Res. 2017, 23, 2951–2960. [Google Scholar] [CrossRef]
- Li, S.; Tokuyama, T.; Yamamoto, J.; Koide, M.; Yokota, N.; Namba, H. Bystander effect-mediated gene therapy of gliomas using genetically engineered neural stem cells. Cancer Gene Ther. 2005, 12, 600–607. [Google Scholar] [CrossRef]
- Matuskova, M.; Hlubinova, K.; Pastorakova, A.; Hunakova, L.; Altanerova, V.; Altaner, C.; Kucerova, L. HSV-tk expressing mesenchymal stem cells exert bystander effect on human glioblastoma cells. Cancer Lett. 2010, 290, 58–67. [Google Scholar] [CrossRef]
- Uhl, M.; Weiler, M.; Wick, W.; Jacobs, A.H.; Weller, M.; Herrlinger, U. Migratory neural stem cells for improved thymidine kinase-based gene therapy of malignant gliomas. Biochem. Biophys. Res. Commun. 2005, 328, 125–129. [Google Scholar] [CrossRef]
- Markert, J.M.; Razdan, S.N.; Kuo, H.-C.; Cantor, A.; Knoll, A.; Karrasch, M.; Nabors, L.; Markiewicz, M.; Agee, B.S.; Coleman, J.M.; et al. A Phase 1 Trial of Oncolytic HSV-1, G207, Given in Combination With Radiation for Recurrent GBM Demonstrates Safety and Radiographic Responses. Mol. Ther. 2014, 22, 1048–1055. [Google Scholar] [CrossRef]
- Forsyth, P.; Roldán, G.; George, D.; Wallace, C.; Palmer, C.; Morris, D.; Cairncross, G.; Matthews, M.V.; Markert, J.; Gillespie, Y.; et al. A Phase I Trial of Intratumoral Administration of Reovirus in Patients With Histologically Confirmed Recurrent Malignant Gliomas. Mol. Ther. 2008, 16, 627–632. [Google Scholar] [CrossRef]
- Ahmed, A.U.; Thaci, B.; Alexiades, N.G.; Han, Y.; Qian, S.; Liu, F.; Balyasnikova, I.V.; Ulasov, I.; Aboody, K.S.; Lesniak, M.S. Neural Stem Cell-based Cell Carriers Enhance Therapeutic Efficacy of an Oncolytic Adenovirus in an Orthotopic Mouse Model of Human Glioblastoma. Mol. Ther. 2011, 19, 1714–1726. [Google Scholar] [CrossRef]
- Ahmed, A.U.; Thaci, B.; Tobias, A.L.; Auffinger, B.; Zhang, L.; Cheng, Y.; Kim, C.K.; Yunis, C.; Han, Y.; Alexiades, N.G.; et al. A Preclinical Evaluation of Neural Stem Cell–Based Cell Carrier for Targeted Antiglioma Oncolytic Virotherapy. J. Natl. Cancer Inst. 2013, 105, 968–977. [Google Scholar] [CrossRef]
- Tyler, M.A.; Ulasov, I.V.; Sonabend, A.M.; Nandi, S.; Han, Y.; Marler, S.; Roth, J.; Lesniak, M.S. Neural stem cells target intracranial glioma to deliver an oncolytic adenovirus in vivo. Gene Ther. 2008, 16, 262–278. [Google Scholar] [CrossRef]
- Thaci, B.; Ahmed, A.U.; Ulasov, I.; Tobias, A.L.; Han, Y.; Aboody, K.S.; Lesniak, M.S. Pharmacokinetic study of neural stem cell-based cell carrier for oncolytic virotherapy: Targeted delivery of the therapeutic payload in an orthotopic brain tumor model. Cancer Gene Ther. 2012, 19, 431–442. [Google Scholar] [CrossRef]
- Brem, H.; Gabikian, P. Biodegradable polymer implants to treat brain tumors. J. Control. Release 2001, 74, 63–67. [Google Scholar] [CrossRef]
- Westphal, M.; Hilt, D.C.; Bortey, E.; Delavault, P.; Olivares, R.; Warnke, P.C.; Whittle, I.R.; Jääskeläinen, J.; Ram, Z. A phase 3 trial of local chemotherapy with biodegradable carmustine (BCNU) wafers (Gliadel wafers) in patients with primary malignant glioma. Neuro-Oncology 2003, 5, 79–88. [Google Scholar] [CrossRef]
- Ashby, L.S.; Smith, K.A.; Stea, B. Gliadel wafer implantation combined with standard radiotherapy and concurrent followed by adjuvant temozolomide for treatment of newly diagnosed high-grade glioma: A systematic literature review. World J. Surg. Oncol. 2016, 14, 225. [Google Scholar] [CrossRef]
- Li, X.; Sun, Q.; Li, Q.; Kawazoe, N.; Chen, G. Functional hydrogels with tunable structures and properties for tissue engineering applications. Front. Chem. 2018, 6, 499. [Google Scholar] [CrossRef]
- Vasile, C.; Pamfil, D.; Stoleru, E.; Baican, M. New developments in medical applications of hybrid hydrogels containing natural polymers. Molecules 2020, 25, 1539. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Klaikherd, A.; Thayumanavan, S. Temperature sensitivity trends and multi-stimuli sensitive behavior in amphiphilic oligomers. J. Am. Chem. Soc. 2011, 133, 13496–13503. [Google Scholar] [CrossRef]
- Kauer, T.M.; Figueiredo, J.-L.; Hingtgen, S.; Shah, K. Encapsulated therapeutic stem cells implanted in the tumor resection cavity induce cell death in gliomas. Nat. Neurosci. 2011, 15, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Bagó, J.R.; Pegna, G.J.; Okolie, O.; Mohiti-Asli, M.; Loboa, E.G.; Hingtgen, S.D. Electrospun nanofibrous scaffolds increase the efficacy of stem cell-mediated therapy of surgically resected glioblastoma. Biomaterials 2016, 90, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Bagó, J.R.; Pegna, G.J.; Okolie, O.; Hingtgen, S.D. Fibrin matrices enhance the transplant and efficacy of cytotoxic stem cell therapy for post-surgical cancer. Biomaterials 2016, 84, 42–53. [Google Scholar] [CrossRef]
- Sheets, K.T.; Ewend, M.G.; Mohiti-Asli, M.; Tuin, S.A.; Loboa, E.G.; Aboody, K.S.; Hingtgen, S.D. Developing Implantable Scaffolds to Enhance Neural Stem Cell Therapy for Post-Operative Glioblastoma. Mol. Ther. 2020, 28, 1056–1067. [Google Scholar] [CrossRef] [PubMed]
- Marei, H.E.; Althani, A.; Caceci, T.; Arriga, R.; Sconocchia, T.; Ottaviani, A.; Lanzilli, G.; Roselli, M.; Caratelli, S.; Cenciarelli, C.; et al. Recent perspective on CAR and Fcγ-CR T cell immunotherapy for cancers: Preclinical evidence versus clinical outcomes. Biochem. Pharmacol. 2019, 166, 335–346. [Google Scholar] [CrossRef]
- Bagley, S.J.; Desai, A.S.; Linette, G.P.; June, C.H.; O’Rourke, D.M. CAR T-cell therapy for glioblastoma: Recent clinical advances and future challenges. Neuro-Oncology 2018, 20, 1429–1438. [Google Scholar] [CrossRef]
- Caratelli, S.; Sconocchia, T.; Arriga, R.; Coppola, A.; Lanzilli, G.; Lauro, D.; Venditti, A.; Del Principe, M.I.; Buccisano, F.; Maurillo, L.; et al. FCγ Chimeric Receptor-Engineered T Cells: Methodology, Advantages, Limitations, and Clinical Relevance. Front. Immunol. 2017, 8, 457. [Google Scholar] [CrossRef]
- Falzone, L.; Salomone, S.; Libra, M. Evolution of Cancer Pharmacological Treatments at the Turn of the Third Millennium. Front. Pharmacol. 2018, 9, 1300. [Google Scholar] [CrossRef]
- Yao, Y.; Luo, F.; Tang, C.; Chen, D.; Qin, Z.; Hua, W.; Xu, M.; Zhong, P.; Yu, S.; Chen, D.; et al. Molecular subgroups and B7-H4 expression levels predict responses to dendritic cell vaccines in glioblastoma: An exploratory randomized phase II clinical trial. Cancer Immunol. Immunother. 2018, 67, 1777–1788. [Google Scholar] [CrossRef]
- Müller, S.; Agnihotri, S.; Shoger, K.E.; Myers, M.I.; Smith, N.; Chaparala, S.; Villanueva, C.R.; Chattopadhyay, A.; Lee, A.V.; Butterfield, L.H.; et al. Peptide vaccine immunotherapy biomarkers and response patterns in pediatric gliomas. JCI Insight 2018, 3, e98791. [Google Scholar] [CrossRef]
- Kovacs, Z.I.; Kim, S.; Jikaria, N.; Qureshi, F.; Milo, B.; Lewis, B.K.; Bresler, M.; Burks, S.R.; Frank, J.A. Disrupting the blood–brain barrier by focused ultrasound induces sterile inflammation. Proc. Natl. Acad. Sci. USA 2017, 114, E75–E84. [Google Scholar] [CrossRef]
- Hynynen, K.; McDannold, N.; Vykhodtseva, N.; Jolesz, F.A. Noninvasive MR Imaging–guided Focal Opening of the Blood-Brain Barrier in Rabbits. Radiology 2001, 220, 640–646. [Google Scholar] [CrossRef]
- Sheikov, N.; McDannold, N.; Sharma, S.; Hynynen, K. Effect of Focused Ultrasound Applied With an Ultrasound Contrast Agent on the Tight Junctional Integrity of the Brain Microvascular Endothelium. Ultrasound Med. Biol. 2008, 34, 1093–1104. [Google Scholar] [CrossRef]
- McDannold, N.; Arvanitis, C.D.; Vykhodtseva, N.; Livingstone, M.S. Temporary Disruption of the Blood–Brain Barrier by Use of Ultrasound and Microbubbles: Safety and Efficacy Evaluation in Rhesus Macaques. Cancer Res. 2012, 72, 3652–3663. [Google Scholar] [CrossRef]
- Carpentier, A.; Canney, M.; Vignot, A.; Reina, V.; Beccaria, K.; Horodyckid, C.; Karachi, C.; Leclercq, D.; Lafon, C.; Chapelon, J.-Y.; et al. Clinical trial of blood-brain barrier disruption by pulsed ultrasound. Sci. Transl. Med. 2016, 8, 343re2. [Google Scholar] [CrossRef]
- Mainprize, T.; Lipsman, N.; Huang, Y.; Meng, Y.; Bethune, A.; Ironside, S.; Heyn, C.; Alkins, R.; Trudeau, M.; Sahgal, A.; et al. Blood-Brain Barrier Opening in Primary Brain Tumors with Non-invasive MR-Guided Focused Ultrasound: A Clinical Safety and Feasibility Study. Sci. Rep. 2019, 9, 321. [Google Scholar] [CrossRef]
- Idbaih, A.; Canney, M.; Belin, L.; Desseaux, C.; Vignot, A.; Bouchoux, G.; Asquier, N.; Law-Ye, B.; Leclercq, D.; Bissery, A.; et al. Safety and Feasibility of Repeated and Transient Blood–Brain Barrier Disruption by Pulsed Ultrasound in Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2019, 25, 3793–3801. [Google Scholar] [CrossRef]
- Aryal, M.; Fischer, K.; Gentile, C.; Gitto, S.; Zhang, Y.-Z.; McDannold, N. Effects on P-Glycoprotein Expression after Blood-Brain Barrier Disruption Using Focused Ultrasound and Microbubbles. PLoS ONE 2017, 12, e0166061. [Google Scholar] [CrossRef]
- Lee, J.; Chang, W.S.; Shin, J.; Seo, Y.; Kong, C.; Song, B.-W.; Na, Y.C.; Kim, B.S.; Chang, J.W. Corrigendum to “Non-invasively enhanced intracranial transplantation of mesenchymal stem cells using focused ultrasound mediated by overexpression of cell-adhesion molecules” [Stem Cell Res. 43 (2020) 101726]. Stem Cell Res. 2021, 51, 102179. [Google Scholar] [CrossRef]
- Blackmore, J.; Shrivastava, S.; Sallet, J.; Butler, C.; Cleveland, R.O. Ultrasound Neuromodulation: A Review of Results, Mechanisms and Safety. Ultrasound Med. Biol. 2019, 45, 1509–1536. [Google Scholar] [CrossRef]
- Arvanitis, C.D.; Askoxylakis, V.; Guo, Y.; Datta, M.; Kloepper, J.; Ferraro, G.B.; Bernabeu, M.O.; Fukumura, D.; McDannold, N.; Jain, R.K. Mechanisms of enhanced drug delivery in brain metastases with focused ultrasound-induced blood–tumor barrier disruption. Proc. Natl. Acad. Sci. USA 2018, 115, E8717–E8726. [Google Scholar] [CrossRef]
- Wei, K.-C.; Chu, P.-C.; Wang, H.-Y.J.; Huang, C.-Y.; Chen, P.-Y.; Tsai, H.-C.; Lu, Y.J.; Lee, P.Y.; Tseng, I.C.; Feng, L.Y.; et al. Focused ultrasound-induced blood-brain barrier opening to enhance temozolomide delivery for glioblastoma treatment: A preclinical study. PLoS ONE 2013, 8, e58995. [Google Scholar] [CrossRef]
- Treat, L.H.; McDannold, N.; Zhang, Y.; Vykhodtseva, N.; Hynynen, K. Improved Anti-Tumor Effect of Liposomal Doxorubicin After Targeted Blood-Brain Barrier Disruption by MRI-Guided Focused Ultrasound in Rat Glioma. Ultrasound Med. Biol. 2012, 38, 1716–1725. [Google Scholar] [CrossRef]
- Liu, H.-L.; Hsu, P.; Lin, C.-Y.; Huang, C.-W.; Chai, W.-Y.; Chu, P.-C.; Huang, C.-Y.; Chen, P.-Y.; Yang, L.-Y.; Kuo, J.; et al. Focused Ultrasound Enhances Central Nervous System Delivery of Bevacizumab for Malignant Glioma Treatment. Radiology 2016, 281, 99–108. [Google Scholar] [CrossRef] [PubMed]




| NCT Number | Official Title | Primary Endpoint(s) | Endpoint Status |
|---|---|---|---|
| NCT02667587 | A Randomized Phase 3 Single Blind Study of Temozolomide Plus Radiation Therapy Combined with Nivolumab or Placebo in Newly Diagnosed Adult Subjects with MGMT-Methylated (Tumor-O6-methylguanine DNA Methyltransferase) Glioblastoma (CheckMate-548) | Progression-free survival (PFS) Overall survival (OS) | PFS endpoint not met OS in progress |
| NCT02337686 | Pharmacodynamic Study of Pembrolizumab in Patients with Recurrent Glioblastoma | PFS | Endpoint in progress |
| NCT03174197 | Phase I/II Study to Evaluate the Safety and Clinical Efficacy of Atezolizumab (aPDL1) in Combination with Temozolomide and Radiation in Patients with Newly Diagnosed Glioblastoma | Dose-limiting toxicities (DLT; Phase I) Overall survival (Phase II) Incidence of adverse events | DLT endpoint met OS endpoint not met Study in progress |
| NCT03047473 | Avelumab in Patients with Newly Diagnosed Glioblastoma Multiforme | Safety and tolerability | Endpoint in progress |
| Chemotherapeutic | Structure (MW) |
|---|---|
| Temozolomide |  (194.1508 g/mol) b |
| Lomustine a |  (233.695 g/mol) b |
| Carmustine a |  (214.05 g/mol) b |
| Carboplatin a |  (371.254 g/mol) b |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
King, J.L.; Benhabbour, S.R. Glioblastoma Multiforme—A Look at the Past and a Glance at the Future. Pharmaceutics 2021, 13, 1053. https://doi.org/10.3390/pharmaceutics13071053
King JL, Benhabbour SR. Glioblastoma Multiforme—A Look at the Past and a Glance at the Future. Pharmaceutics. 2021; 13(7):1053. https://doi.org/10.3390/pharmaceutics13071053
Chicago/Turabian StyleKing, Jasmine L., and Soumya Rahima Benhabbour. 2021. "Glioblastoma Multiforme—A Look at the Past and a Glance at the Future" Pharmaceutics 13, no. 7: 1053. https://doi.org/10.3390/pharmaceutics13071053
APA StyleKing, J. L., & Benhabbour, S. R. (2021). Glioblastoma Multiforme—A Look at the Past and a Glance at the Future. Pharmaceutics, 13(7), 1053. https://doi.org/10.3390/pharmaceutics13071053