
Model-Informed Optimization of a Pediatric Clinical Pharmacokinetic Trial of a New Spironolactone Liquid Formulation


 , , ,
, , , 

Abstract
1. Introduction
2. Material and Methods
2.1. Study Design and General Overview of the Data Analysis Steps
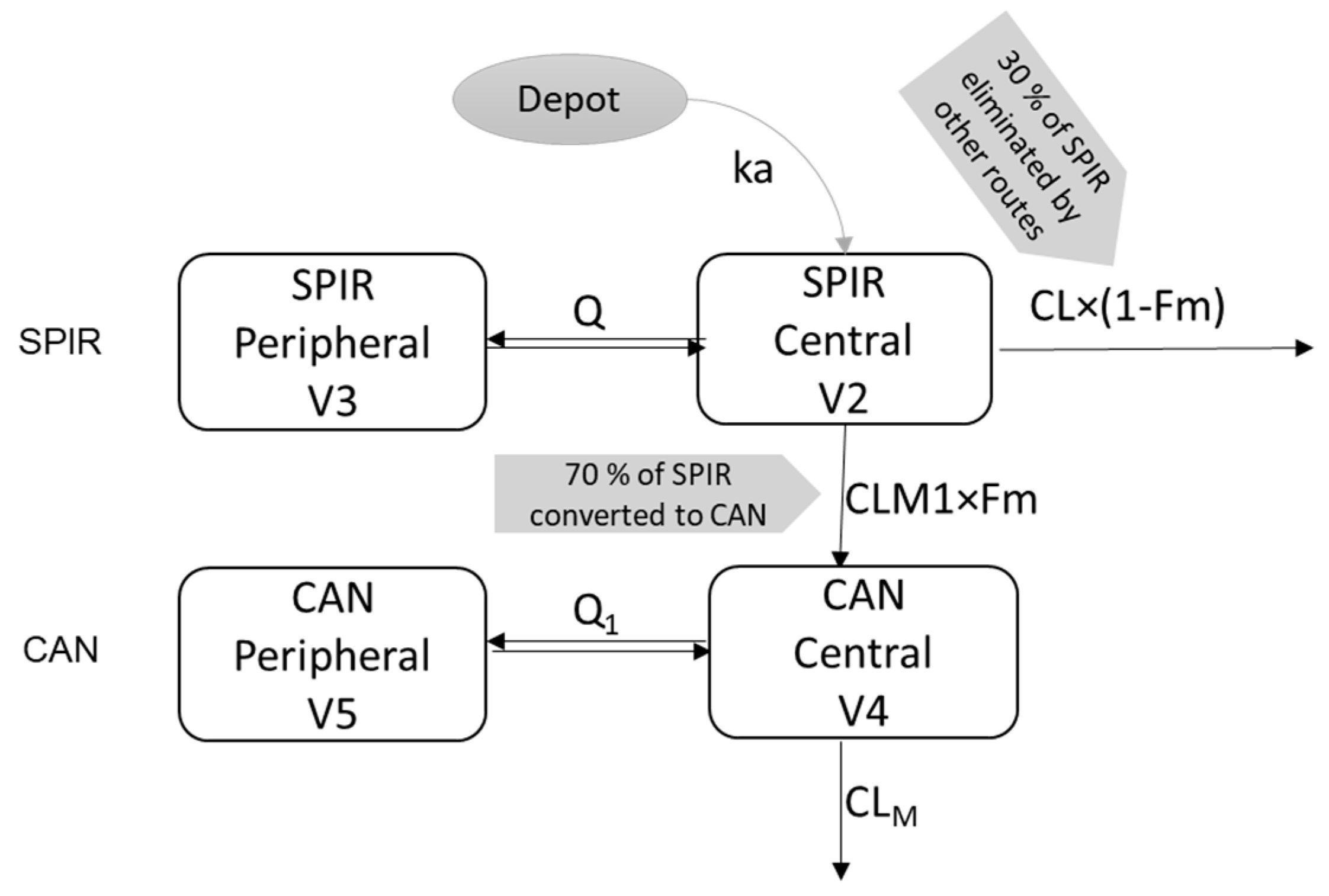
2.2. Population PK Analysis
2.3. Extrapolation of the Parent Drug-Metabolite Model to Pediatrics
2.4. Dose Selection
2.5. Sampling Scheme Selection
3. Results
3.1. Parent—Metabolite popPK Model in Adults
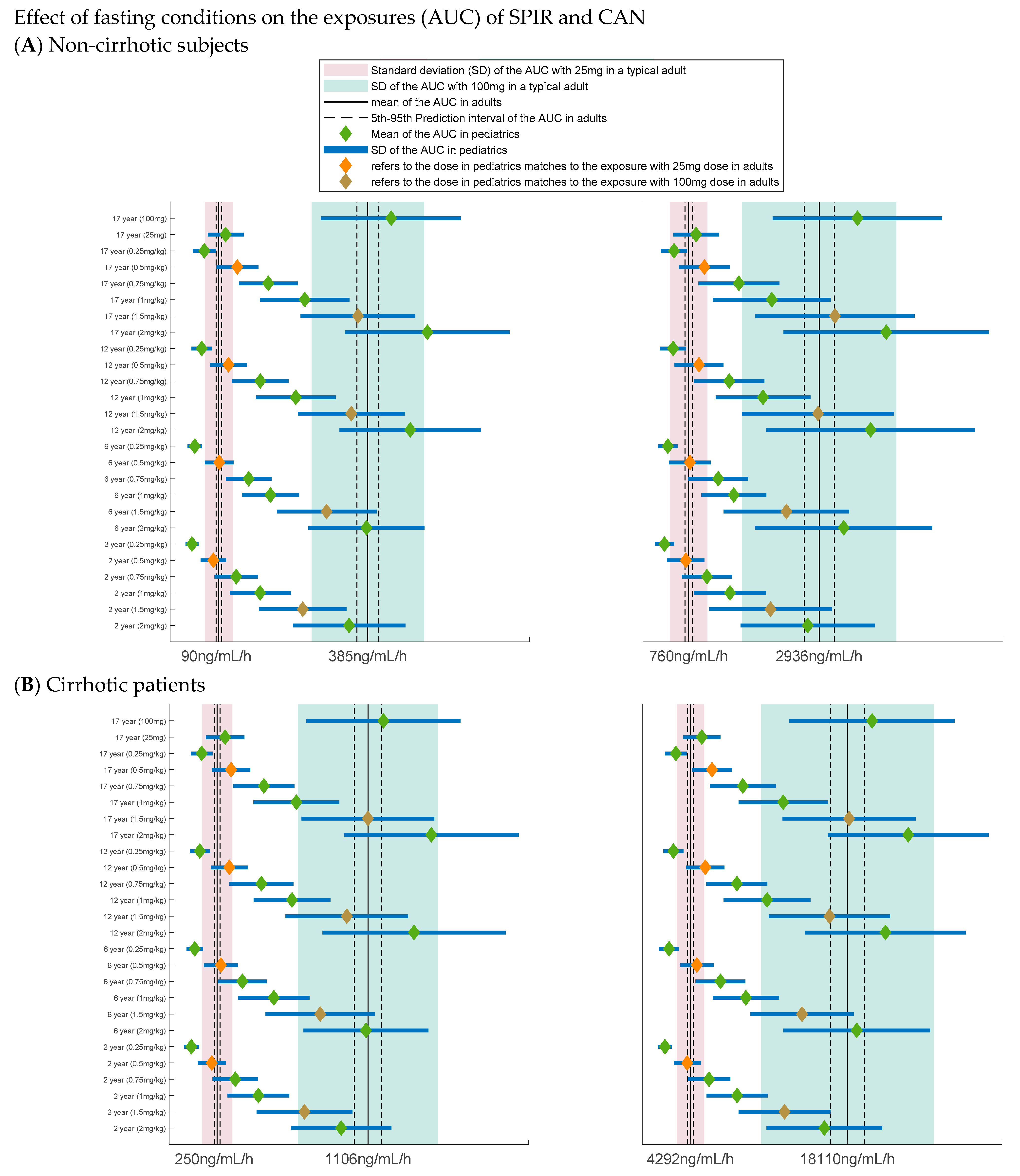
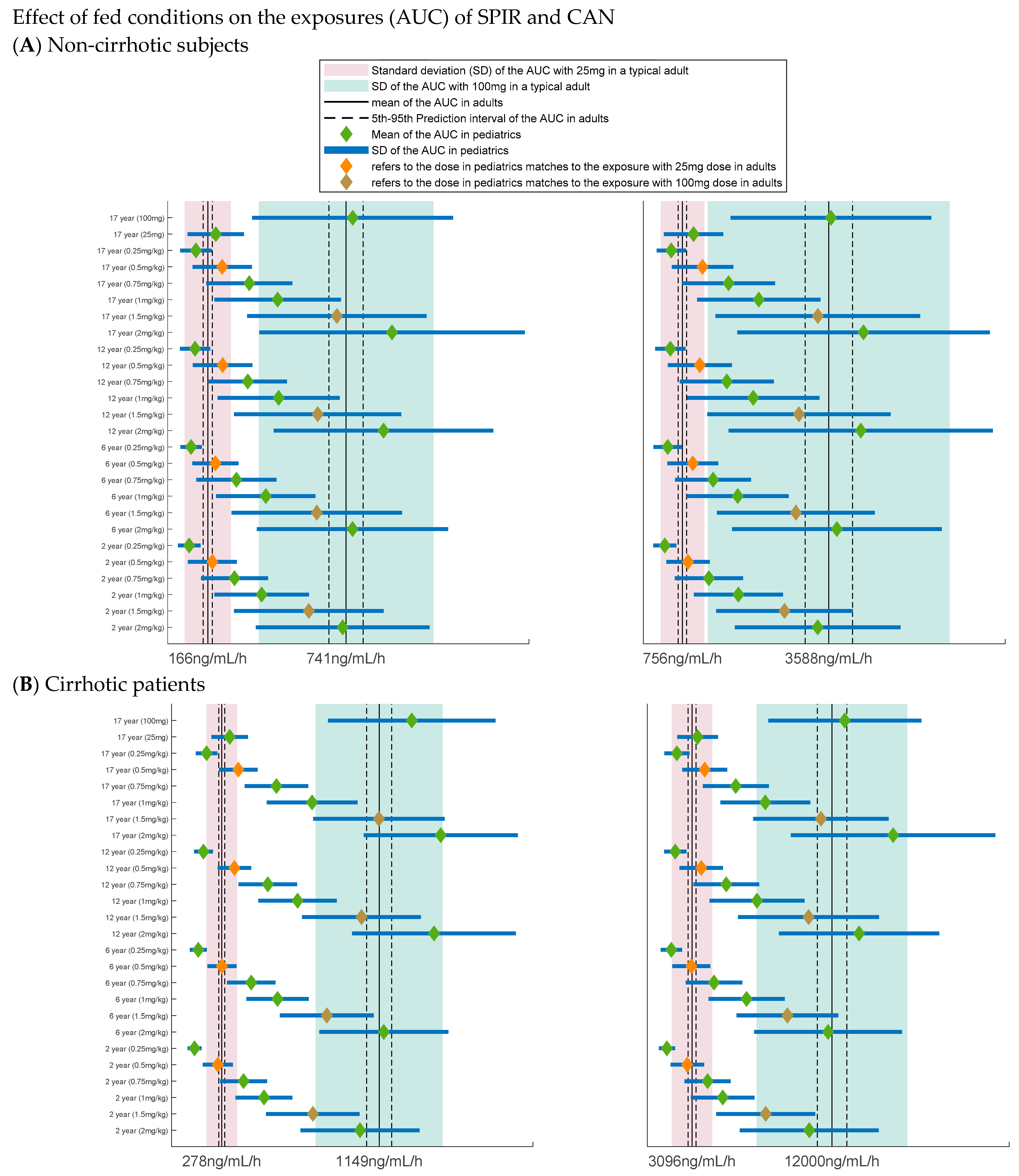
3.2. Extrapolation of the Parent Metabolite Model to Pediatrics
3.3. Application of the Model to Select the Dose and Design the Pediatric Clinical Trial
3.4. Dose Selection
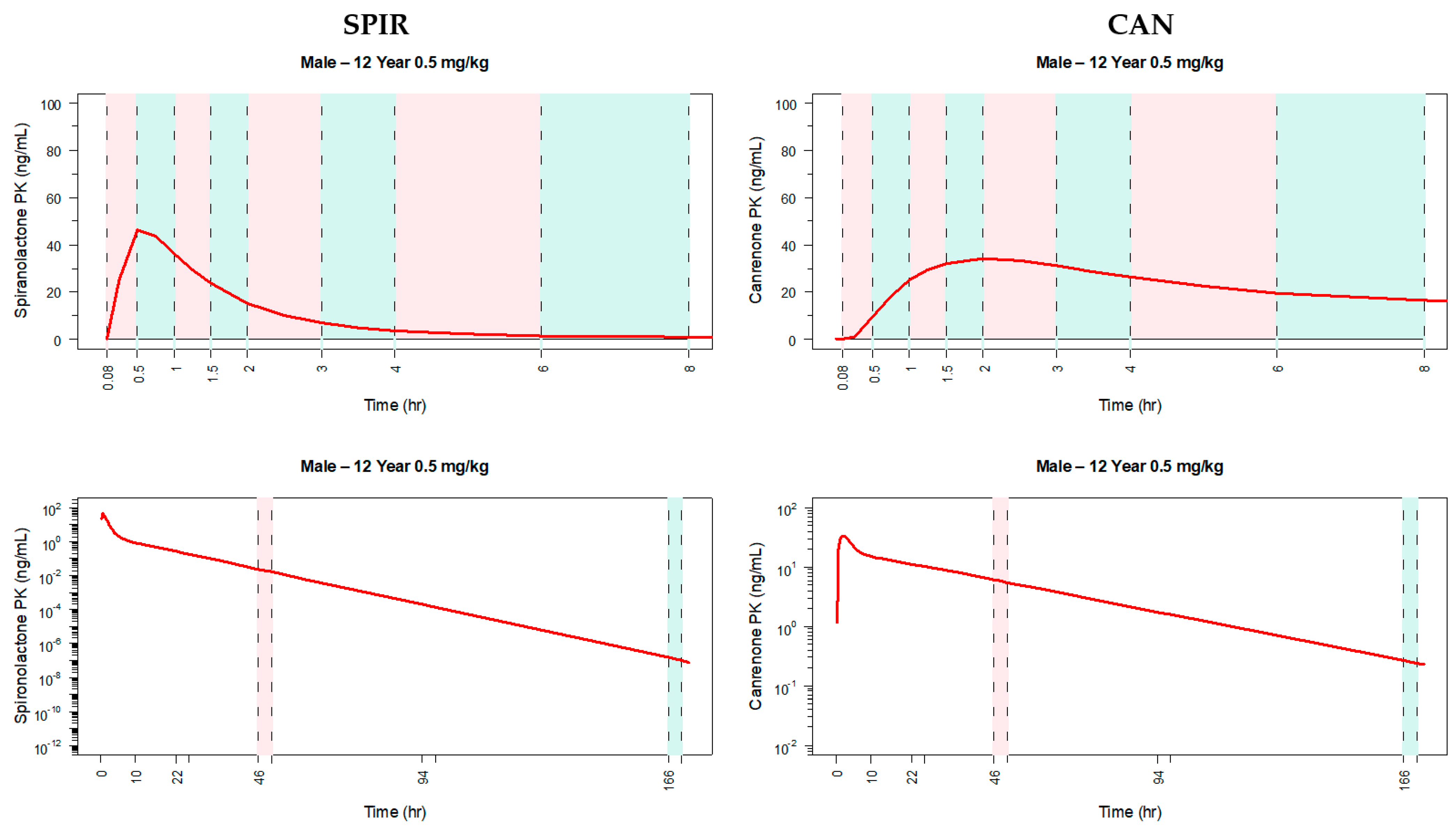
3.5. PK Sampling Scheme for a Pediatric PK Trial
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carone, L.; Oxberry, S.G.; Twycross, R.; Charlesworth, S.; Mihalyo, M.; Wilcock, A. Spironolactone. J. Pain Symptom Manag. 2017, 53, 288–292. [Google Scholar] [CrossRef]
- Aldactone Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/012151s075lbl.pdf (accessed on 20 February 2021).
- Buck, M.L. Clinical experience with spironolactone in pediatrics. Ann. Pharmacother. 2005, 39, 823–828. [Google Scholar] [CrossRef]
- World Health Organization. WHO Model Formulary for Children. 2010. Available online: https://www.who.int/selection_medicines/list/WMFc_2010.pdf (accessed on 20 February 2021).
- Spironolactone. British National Formulary (BNF) for Children; British Medical Journal Group, Pharmaceutical Press and Royal College of Paediatrics and Child Health: London, UK, 2017. [Google Scholar]
- Allen, L.V., Jr.; Erickson, M.A., 3rd. Stability of ketoconazole, metolazone, metronidazole, procainamide hydrochloride, and spironolactone in extemporaneously compounded oral liquids. Am. J. Health Syst. Pharm. 1996, 53, 2073–2078. [Google Scholar] [CrossRef] [PubMed]
- Committee for Human Medicinal Products. ICH E11(R1) Guideline on Clinical Investigation of Medicinal Products in the Paediatric Population; European Medicines Agency: London, UK, 2017. [Google Scholar]
- McLean, A.J.; Isbister, C.; Bobik, A.; Dudley, F.J. Reduction of first-pass hepatic clearance of propranolol by food. Clin. Pharmacol. Ther. 1981, 30, 31–34. [Google Scholar] [CrossRef] [PubMed]
- Welling, P.G. Effects of Food on Drug Absorption. Annu. Rev. Nutr. 1996, 16, 383–415. [Google Scholar] [CrossRef]
- Winstanley, P.A.; Orme, M.L. The effects of food on drug bioavailability. Br. J. Clin. Pharmacol. 1989, 28, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Charman, W.N.; Porter, C.J.; Mithani, S.; Dressman, J.B. Physiochemical and physiological mechanisms for the effects of food on drug absorption: The role of lipids and pH. J. Pharm. Sci. 1997, 86, 269–282. [Google Scholar] [CrossRef] [PubMed]
- CMP Pharma USA Clinical Study 084-15. An Open Label, Balanced, Randomized, Single Dose, Two Treatment (Fed vs. Fasting), Two-Period, Two-Way cross over, Oral Food Effect Study of Spironolactone Suspension 100 mg (20 mL of 25 mg/5 mL) of CMP Pharma, USA. Available online: https://clinicaltrials.gov/ct2/show/record/NCT01083290?cond=spironolactone&cntry=IN&draw=2 (accessed on 20 February 2021).
- CMP Pharma USA Clinical Study 063-15. An Open Label, Randomized, Two Treatment, Two Period, Two Sequence, Crossover, Single Dose, Oral Pharmacokinetic and Comparative Bioavailability Study of Spironolactone Suspension 25 mg/5 mL of CMP Pharma, USA. Available online: https://clinicaltrials.gov/ct2/show/record/NCT01083290?cond=spironolactone&cntry=IN&draw=2 (accessed on 20 February 2021).
- CMP Pharma USA Clinical Study 064-15. An Open Label, Randomized, Two Treatment, Two Period, Two Sequence, Crossover, Single Dose, Oral Pharmacokinetic and Comparative Bioavailability Study of Spironolactone Suspension 25 mg/5 mL of CMP Pharma, USA. Available online: https://clinicaltrials.gov/ct2/show/record/NCT01083290?cond=spironolactone&cntry=IN&draw=2 (accessed on 20 February 2021).
- CAROSPIR (Spironolactone) Oral Suspension Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/209478s000lbl.pdf (accessed on 20 February 2021).
- Takamura, N.; Maruyama, T.; Ahmed, S.; Suenaga, A.; Otagiri, M. Interactions of aldosterone antagonist diuretics with human serum proteins. Pharm. Res. 1997, 14, 522–526. [Google Scholar] [CrossRef]
- Sungaila, I.; Bartle, W.R.; Walker, S.E.; DeAngelis, C.; Uetrecht, J.; Pappas, C.; Vidins, E. Spironolactone pharmacokinetics and pharmacodynamics in patients with cirrhotic ascites. Gastroenterology 1992, 102, 1680–1685. [Google Scholar] [CrossRef]
- Byon, W.; Smith, M.K.; Chan, P.; Tortorici, M.A.; Riley, S.; Dai, H.; Dong, J.; Ruiz-Garcia, A.; Sweeney, K.; Cronenberger, C. Establishing best practices and guidance in population modeling: An experience with an internal population pharmacokinetic analysis guidance. CPT Pharmacomet. Syst. Pharm. 2013, 2, e51. [Google Scholar] [CrossRef]
- Anderson, B.J.; Allegaert, K.; Holford, N.H. Population clinical pharmacology of children: Modelling covariate effects. Eur. J. Pediatr. 2006, 165, 819–829. [Google Scholar] [CrossRef]
- Anderson, B.J.; Holford, N.H. Mechanism-based concepts of size and maturity in pharmacokinetics. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 303–332. [Google Scholar] [CrossRef]
- Anderson, B.J.; Holford, N.H. Mechanistic basis of using body size and maturation to predict clearance in humans. Drug Metab. Pharm. 2009, 24, 25–36. [Google Scholar] [CrossRef]
- Vozmediano, V.; Sologuren, A.; Lukas, J.C.; Leal, N.; Rodriguez, M. Model Informed Pediatric Development Applied to Bilastine: Ontogenic PK Model Development, Dose Selection for First Time in Children and PK Study Design. Pharm. Res. 2017, 34, 2720–2734. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, P.; Schrode, K.; Quinlan, D.; Martin, B.K.; Boreham, D.R.; Rogers, M.S.; Stubbs, K.; Smith, M.; Karim, A. Spironolactone metabolism: Steady-state serum levels of the sulfur-containing metabolites. J. Clin. Pharmacol. 1989, 29, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Hines, R.N.; McCarver, D.G. The ontogeny of human drug-metabolizing enzymes: Phase I oxidative enzymes. J. Pharmacol. Exp. Ther. 2002, 300, 355–360. [Google Scholar] [CrossRef] [PubMed]
- van Groen, B.D.; Nicolaï, J.; Kuik, A.C.; Van Cruchten, S.; van Peer, E.; Smits, A.; Schmidt, S.; de Wildt, S.N.; Allegaert, K.; De Schaepdrijver, L.; et al. Ontogeny of Hepatic Transporters and Drug-Metabolizing Enzymes in Humans and in Nonclinical Species. Pharmacol. Rev. 2021, 73, 597–678. [Google Scholar] [CrossRef]
- Committee, E.S.; Hardy, A.; Benford, D.; Halldorsson, T.; Jeger, M.J.; Knutsen, H.K.; More, S.; Naegeli, H.; Noteborn, H.; Ockleford, C.; et al. Guidance on the risk assessment of substances present in food intended for infants below 16 weeks of age. EFSA J. 2017, 15, e04849. [Google Scholar] [CrossRef]
- Siripuram, V.K.; Wright, D.F.B.; Barclay, M.L.; Duffull, S.B. Deterministic identifiability of population pharmacokinetic and pharmacokinetic-pharmacodynamic models. J. Pharm. Pharm. 2017, 44, 415–423. [Google Scholar] [CrossRef]
- Frattarelli, D.A.; Galinkin, J.L.; Green, T.P.; Johnson, T.D.; Neville, K.A.; Paul, I.M.; Van Den Anker, J.N.; American Academy of Pediatrics Committee. Off-label use of drugs in children. Pediatrics 2014, 133, 563–567. [Google Scholar] [CrossRef]
- Hu, T.M.; Hayton, W.L. Allometric scaling of xenobiotic clearance: Uncertainty versus universality. AAPS PharmSci 2001, 3, E29. [Google Scholar] [CrossRef] [PubMed]
- Rhodin, M.M.; Anderson, B.J.; Peters, A.M.; Coulthard, M.G.; Wilkins, B.; Cole, M.; Chatelut, E.; Grubb, A.; Veal, G.J.; Keir, M.J.; et al. Human renal function maturation: A quantitative description using weight and postmenstrual age. Pediatr. Nephrol. 2009, 24, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Han, P.Y.; Duffull, S.B.; Kirkpatrick, C.M.; Green, B. Dosing in obesity: A simple solution to a big problem. Clin. Pharmacol. Ther. 2007, 82, 505–508. [Google Scholar] [CrossRef] [PubMed]
- West, G.B.; Brown, J.H.; Enquist, B.J. The fourth dimension of life: Fractal geometry and allometric scaling of organisms. Science 1999, 284, 1677–1679. [Google Scholar] [CrossRef]
- Holford, N.; Heo, Y.A.; Anderson, B. A pharmacokinetic standard for babies and adults. J. Pharm. Sci. 2013, 102, 2941–2952. [Google Scholar] [CrossRef]
- Janmahasatian, S.; Duffull, S.B.; Ash, S.; Ward, L.C.; Byrne, N.M.; Green, B. Quantification of lean bodyweight. Clin. Pharm. 2005, 44, 1051–1065. [Google Scholar] [CrossRef]
- Cheng, L.; Wong, H. Food Effects on Oral Drug Absorption: Application of Physiologically-Based Pharmacokinetic Modeling as a Predictive Tool. Pharmaceutics 2020, 12, 672. [Google Scholar] [CrossRef]
- Yanez, J.A.; Remsberg, C.M.; Sayre, C.L.; Forrest, M.L.; Davies, N.M. Flip-flop pharmacokinetics--delivering a reversal of disposition: Challenges and opportunities during drug development. Ther. Deliv. 2011, 2, 643–672. [Google Scholar] [CrossRef]
- Persson, E.M.; Gustafsson, A.S.; Carlsson, A.S.; Nilsson, R.G.; Knutson, L.; Forsell, P.; Hanisch, G.; Lennernas, H.; Abrahamsson, B. The effects of food on the dissolution of poorly soluble drugs in human and in model small intestinal fluids. Pharm. Res. 2005, 22, 2141–2151. [Google Scholar] [CrossRef]
- Hobbins, S.M.; Fowler, R.S.; Rowe, R.D.; Korey, A.G. Spironolactone therapy in infants with congestive heart failure secondary to congenital heart disease. Arch. Dis. Child. 1981, 56, 934–938. [Google Scholar] [CrossRef]
- Wimmer, M.; Bachl, G.; Schlemmer, M.; Stiskal, A. Experiences with aldactone in pediatric cardiology (author’s transl). Padiatr. Padol. 1979, 14, 363–372. [Google Scholar] [PubMed]
- Baylen, B.G.; Johnson, G.; Tsang, R.; Srivastava, L.; Kaplan, S. The occurrence of hyperaldosteronism in infants with congestive heart failure. Am. J. Cardiol. 1980, 45, 305–310. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Description | Reference Listed Drug | N |
|---|---|---|---|
| 063-15 (Pilot) | An open-label, randomized, two-treatment, two-period, two-sequence, crossover, single-dose, oral pharmacokinetic and comparative bioavailability study in healthy, human subjects under fasting conditions | 25 mg Aldactone® Tablets | 14 |
| 064-15 (Pivotal) | An open-label, randomized, two-treatment, two-period, two-sequence, crossover, single-dose, oral pharmacokinetic and comparative bioavailability study in healthy, human adult subjects under fasting conditions | 100 mg Aldactone® Tablets | 56 |
| 084-15 (Food effect) | An open-label, balanced, randomized, single-dose, two-treatment (fed vs. fasting), two-period, two-way cross over, oral food effect study in healthy human adult subjects | 100 mg Aldactone® Tablets | 23 |
| Demographic | 063-15 | 064-15 | 084-15 | All Subjects |
|---|---|---|---|---|
| (N = 14) | (N = 56) | (N = 24) | (N = 94) | |
| Age (years) Mean ± SD (median, range) | 36.1 ± 4.3 (36.5, 31–43) | 29.2 ± 5.5 (28.0, 19–40) | 29.5 ± 7.1 (29.0, 20–44) | 30.3 ± 6.2 (29, 19–44) |
| Height (cm) Mean ± SD (median, range) | 163.7 ± 5.3 (163.5, 156–173) | 167.8 ± 5.7 (167, 157–183) | 168.1 ± 6.5 (168, 156–180) | 167.2 ± 6 (166.5, 156–183) |
| Weight (kg) Mean ± SD (median, range) | 63.1 ± 4.5 (62.2, 57.4–72.5) | 64.4 ± 6.8 (63.6, 53.1–83.1) | 64.1 ± 6.4 (62.8, 55.0–78.1) | 64.1 ± 6.4 (63.1, 53.1–83.1) |
| BMI (kg/m2) Mean ± SD (median, range) | 23.6 ± 1.4 (24, 19.6–24.8) | 22.8 ± 1.7 (23.3, 18.6–24.9) | 22.6 ± 1.6 (22.9, 19.9–24.8) | 22.9 ± 1.7 (23.3, 18.6–24.9) |
| Parameter | Model Results | Bootstrap Results | |||
|---|---|---|---|---|---|
| Value | % RSE | % Shrinkage | Mean | 95% CI | |
| ALAG1 | 0.156 | 0.7 | - | 0.153 | 0.14–0.17 |
| ka(1/h) | 5.22 | 1.1 | - | 4.24 | 4.0–6.3 |
| CL (L/h) | 629 | 3.3 | - | 636.2 | 462.8–794.8 |
| V2 (L) | 517 | 2.0 | - | 540.3 | 469.5–563.5 |
| Q (L/h) | 89.9 | 2.2 | - | 90.7 | 82.3–97.3 |
| V3 (L) | 777 | 1.9 | - | 785.5 | 687.9–866.6 |
| Fm | 0.7 FIX | - | - | 0.7 FIX | |
| CLM1 (L/h) | 217 | 4.1 | - | 228.3 | 146.8–286.7 |
| CLM (L/h) | 17 | 3.6 | - | 17.8 | 11.5–22.2 |
| V4 (L) | 189 | 3.4 | - | 192.8 | 130.1–248.7 |
| Q1 (L/h) | 60 | 3.9 | - | 65.1 | 39.3–80.7 |
| V5 (L) | 448 | 3.0 | - | 470.3 | 296.9–598.2 |
| Between Subject Variability | |||||
| ka (1/h) | 0.9 | 26.2 | 7 | 0.85 | 0.49–1.38 |
| CL (L/h) | 0.166 | 19.8 | 5 | 0.18 | 0.07–0.26 |
| V2 (L) | 0.118 | 23.2 | 10 | 0.112 | 0.07–0.15 |
| CL, V2 (covariance) | 0.112 | 0.02 | - | 0.114 | 0.06–0.16 |
| Q (L/h) | 0.08 | 29.1 | 20 | 0.08 | 0.03–0.12 |
| CLM1 (L/h) | 0.18 | 26.7 | 5 | 0.17 | 0.12–0.24 |
| CLM (L/h) | 0.08 | 25.9 | 12 | 0.07 | 0.04–0.11 |
| V4 (L) | 0.03 | 44.9 | 36 | 0.02 | 0.001–0.059 |
| Q1 (L/h) | 0.07 | 35 | 26 | 0.1 | −0.08–0.22 |
| V5 (L) | 0.09 | 23.1 | 19 | 0.08 | 0.04–0.12 |
| Residual Error | |||||
| EPS1 | 0.08 | 5.5 | 7 | 0.08 | 0.07–0.09 |
| EPS2 | 0.017 | 2.5 | 10 | 0.017 | 0.014–0.020 |
| Age Group | CL | V2 | Q | V3 | ka | V4 | CLM | Q1 | V5 | ALAG1 | Fm | CLM1 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (L/h) | (L) | (L/h) | (L) | (h−1) | (L) | (L/h) | (L/h) | (L) | (h) | (L/h) | ||
| Adults | 629.00 | 517.00 | 89.90 | 777.00 | 5.22 | 189.00 | 17.00 | 60.00 | 448.00 | 0.156 | 0.70 | 217.00 |
| 2 years—female | 174.13 | 93.28 | 24.89 | 140.19 | 5.22 | 34.10 | 4.71 | 16.61 | 80.83 | 0.160 | 0.70 | 60.07 |
| 2 years—male | 165.27 | 87.00 | 23.62 | 130.76 | 5.22 | 31.81 | 4.47 | 15.76 | 75.39 | 0.160 | 0.70 | 57.02 |
| 6 years—female | 260.33 | 159.46 | 37.21 | 239.65 | 5.22 | 58.29 | 7.04 | 24.83 | 138.18 | 0.160 | 0.70 | 89.81 |
| 6 years—male | 259.33 | 158.65 | 37.06 | 238.43 | 5.22 | 58.00 | 7.01 | 24.74 | 137.47 | 0.160 | 0.70 | 89.47 |
| 12 years—female | 433.78 | 315.00 | 62.00 | 473.42 | 5.22 | 115.16 | 11.72 | 41.38 | 272.96 | 0.160 | 0.70 | 149.65 |
| 12 years—male | 439.41 | 320.47 | 62.80 | 481.63 | 5.22 | 117.15 | 11.88 | 41.92 | 277.70 | 0.160 | 0.70 | 151.59 |
| 17 years—female | 589.42 | 474.09 | 84.24 | 712.51 | 5.22 | 173.31 | 15.93 | 56.22 | 410.82 | 0.160 | 0.70 | 203.35 |
| 17 years—male | 522.85 | 404.07 | 74.73 | 607.28 | 5.22 | 147.72 | 14.13 | 49.87 | 350.14 | 0.160 | 0.70 | 180.38 |
| Condition/State | Parameter Affected | % of Change from the Model Estimate | Rationale | Reference(s) |
|---|---|---|---|---|
| Non-cirrhotic under fasting conditions | NA | NA | Model parameter estimates are used for the simulations | |
| Cirrhotic under fasting conditions | CL | ↓ 84.44% | Based on the literature | Sungaila et al. [17] Gardiner et al. [23] |
| CLM1 | ↓ 37.76% | Reduced to achieve Tmax for CAN in literature | Sungaila et al. [17] Gardiner et al. [23] | |
| CLM | ↓ 71.55% | Based on the literature | Sungaila et al. [17] Gardiner et al. [23] | |
| Non-cirrhotic under fed conditions | f | ↑ 100% | Based on fed data | Study 8415 |
| ka | ↓ 93.5% | Based on fed data | Study 8415 | |
| k | changed to new ka | K(original) >> ka, hence a flip-flop PK was considered which matches to the profile in fed data | Study 8415 | |
| Fm | ↓ 60% | Based on fed data | Study 8415 | |
| Cirrhotic under fed conditions | f | ↑ 100% | Based on fed data | Study 8415 |
| ka | ↓ 93.5% | Based on fed data | Study 8415 | |
| CL | ↓ 84.44% | Based on the literature | Sungaila et al. [17] Gardiner et al. [23] | |
| Fm | ↓ 60% | Based on fed data | Study 8415 | |
| CLM1 | ↓ 37.76% | Reduced to achieve Tmax for CAN in literaure | Sungaila et al. [17] Gardiner et al. [23] | |
| CLM | ↓ 71.55% | Based on the literature | Sungaila et al. [17] Gardiner et al. [23] |
| Group 1 (12 to <17 years) (Total N = 6) | ||
|---|---|---|
| Subgroups (N = Number of Subjects) | Sampling Intervals after Dose Administration | Number of Samples per Subject |
| Subgroup 1.1 (N = 3) | 5–30 min (0.08–0.5 h) 1–1.5 h 2–3 h 4–6 h | 4 |
| Subgroup 1.2 (N = 3) | 30 min–1 h 1.5–2 h 3–4 h 6–8 h | 4 |
| 1.1, 1.2 (N = 6) | 46–50 h | 1 |
| 1.1, 1.2 (N = 6) | 166–170 h | 1 |
| Group 2–3 (2 to <12 years) (Total N = 6 per group) | ||
| Subgroups (N = Number of Subjects) | Sampling Intervals after Dose Administration | Number of Samples per Subject |
| Subgroups 2.1 (N=3), 3.1 (N = 3) | 5–45 min (0.08–0.75 h) 1.5–2.5 h 3.5–6 h | 3 |
| Subgroups 2.2 (N=3), 3.2 (N = 3) | 45 min–1.5 h (0.75–1.5 h) 2.5–3.5 h 6–8 h | 3 |
| 2 (N=6), 3 (N = 6) | 46–50 h | 1 |
| 2 (N=6), 3 (N = 6) | 166–170 h | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tatipalli, M.; Siripuram, V.K.; Long, T.; Shuster, D.; Bernstein, G.; Martineau, P.; Cook, K.A.; Cristofoletti, R.; Schmidt, S.; Vozmediano, V. Model-Informed Optimization of a Pediatric Clinical Pharmacokinetic Trial of a New Spironolactone Liquid Formulation. Pharmaceutics 2021, 13, 849. https://doi.org/10.3390/pharmaceutics13060849
Tatipalli M, Siripuram VK, Long T, Shuster D, Bernstein G, Martineau P, Cook KA, Cristofoletti R, Schmidt S, Vozmediano V. Model-Informed Optimization of a Pediatric Clinical Pharmacokinetic Trial of a New Spironolactone Liquid Formulation. Pharmaceutics. 2021; 13(6):849. https://doi.org/10.3390/pharmaceutics13060849
Chicago/Turabian StyleTatipalli, Manasa, Vijay Kumar Siripuram, Tao Long, Diana Shuster, Galina Bernstein, Pierre Martineau, Kim A. Cook, Rodrigo Cristofoletti, Stephan Schmidt, and Valvanera Vozmediano. 2021. "Model-Informed Optimization of a Pediatric Clinical Pharmacokinetic Trial of a New Spironolactone Liquid Formulation" Pharmaceutics 13, no. 6: 849. https://doi.org/10.3390/pharmaceutics13060849
APA StyleTatipalli, M., Siripuram, V. K., Long, T., Shuster, D., Bernstein, G., Martineau, P., Cook, K. A., Cristofoletti, R., Schmidt, S., & Vozmediano, V. (2021). Model-Informed Optimization of a Pediatric Clinical Pharmacokinetic Trial of a New Spironolactone Liquid Formulation. Pharmaceutics, 13(6), 849. https://doi.org/10.3390/pharmaceutics13060849