
α-Galactosidase A Augmentation by Non-Viral Gene Therapy: Evaluation in Fabry Disease Mice

 , , , , , and
, , , , , and 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of SLNs and Vector
2.3. Characterization of the Vector: Size, Polydispersity Index and ζ Potential Measurements
2.4. Animal Model
2.5. Animal Experimentation
- -
- Wild-type (+/0 for α-Gal A);
- -
- Non-treated Fabry mice (−/0 for α-Gal A);
- -
- Fabry mice treated with a single administration of the naked pDNA and sacrificed at day 3;
- -
- Fabry mice treated with a single administration of the naked pDNA and sacrificed at day 7;
- -
- Fabry mice treated with the multiple-dosage regimen of the naked pDNA and sacrificed 7 days after the last administration;
- -
- Fabry mice treated with a single administration of the SLN-based vector and sacrificed at day 3;
- -
- Fabry mice treated with a single administration of the SLN-based vector and sacrificed at day 7;
- -
- Fabry mice treated with the multiple-dosage regimen of the SLN-based vector and sacrificed 7 days after the last administration.
2.6. α-Galactosidase A Activity Assay
2.7. Liver Transaminase Activity Assay
2.8. Data Analysis and Statistics
3. Results
3.1. Characterization of the Vector: Size, Polydispersity Index and ζ Potential Measurements
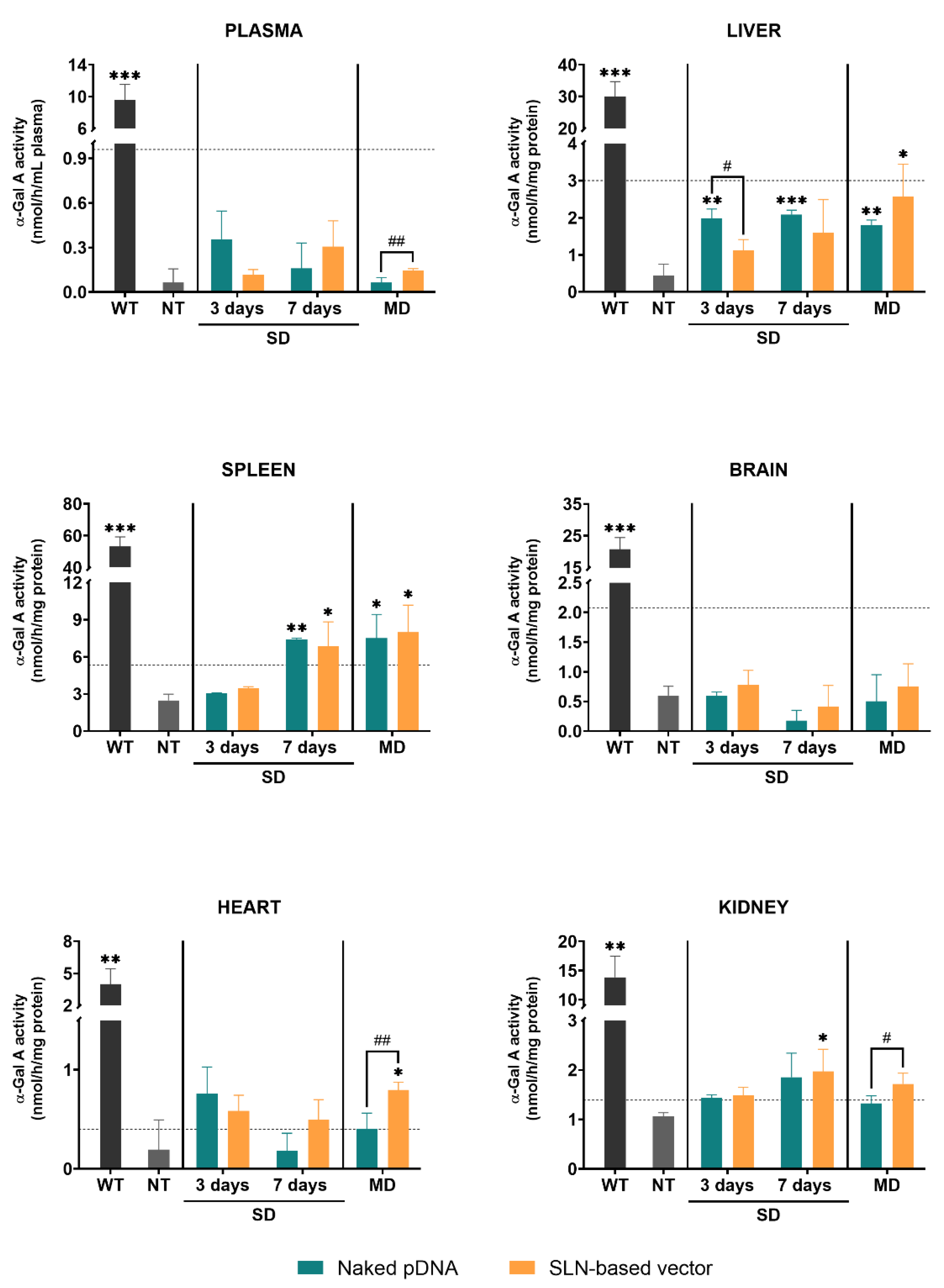
3.2. α-Galactosidase A Activity Assay
3.3. Liver Transaminase Activity Assay
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, W.W.; Li, L.; Li, D.; Liu, J.; Li, X.; Li, W.; Xu, X.; Zhang, M.J.; Chandler, L.A.; Lin, H.; et al. The First Approved Gene Therapy Product for Cancer Ad-p53 (Gendicine): 12 Years in the Clinic. Hum. Gene Ther. 2018, 29, 160–179. [Google Scholar] [CrossRef]
- Wang, F.; Qin, Z.; Lu, H.; He, S.; Luo, J.; Jin, C.; Song, X. Clinical translation of gene medicine. J. Gene Med. 2019, 21, 1–8. [Google Scholar] [CrossRef]
- Gene Therapy Clinical Trials Worldwide, Provided by the Journal of Gene Medicine, John Wileys and Sons LTD. 2021. Available online: https://a873679.fmphost.com/fmi/webd/GTCT (accessed on 24 March 2021).
- Buck, J.; Grossen, P.; Cullis, P.R.; Huwyler, J.; Witzigmann, D. Lipid-based DNA therapeutics: Hallmarks of non-viral gene delivery. ACS Nano 2019, 13, 3754–3782. [Google Scholar] [CrossRef] [PubMed]
- Bernardes, T.P.; Foresto, R.D.; Kirsztajn, G.M. Fabry disease: Genetics, pathology, and treatment. Rev. Assoc. Med. Bras. 2020, 66, 10–16. [Google Scholar] [CrossRef]
- Miller, J.J.; Kanack, A.J.; Dahms, N.M. Progress in the understanding and treatment of Fabry disease. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129437. [Google Scholar] [CrossRef] [PubMed]
- McCafferty, E.H.; Scott, L.J. Migalastat: A Review in Fabry Disease. Drugs 2019, 79, 543–554. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Science Medicines Health. Galafold. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/galafold (accessed on 6 May 2021).
- van der Veen, S.J.; Hollak, C.E.M.; van Kuilenburg, A.B.P.; Langeveld, M. Developments in the treatment of Fabry disease. J. Inherit. Metab. Dis. 2020, 1–14. [Google Scholar] [CrossRef]
- Felis, A.; Whitlow, M.; Kraus, A.; Warnock, D.G.; Wallace, E. Current and Investigational Therapeutics for Fabry Disease. Kidney Int. Reports 2020, 5, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Sands, M.S.; Davidson, B.L. Gene therapy for lysosomal storage diseases. Mol. Ther. 2006, 13, 839–849. [Google Scholar] [CrossRef]
- Lenders, M.; Brand, E. Fabry Disease: The Current Treatment Landscape. Drugs 2021, 81, 635–645. [Google Scholar] [CrossRef]
- del Pozo-Rodríguez, A.; Rodríguez-Gascón, A.; Rodríguez-Castejón, J.; Vicente-Pascual, M.; Gómez-Aguado, I.; Battaglia, L.S.; Solinís Aspiazu, M.Á. Gene Therapy. In Advances in Biochemical Engineering/Biotechnology; Silva, A.C., Moreira, J.N., Lobo, J.M.S., Almeida, H., Eds.; Springer International Publishing: Cham, Germany, 2020; pp. 321–368. [Google Scholar] [CrossRef]
- Carvalho, M.; Sepodes, B.; Martins, A.P. Regulatory and Scientific Advancements in Gene Therapy: State-of-the-Art of Clinical Applications and of the Supporting European Regulatory Framework. Front. Med. 2017, 4, 182. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Gascón, A.; del Pozo-Rodríguez, A.; Isla, A.; Solinís Aspiazu, M.Á. Vaginal gene therapy. Adv. Drug Deliv. Rev. 2015, 92, 71–83. [Google Scholar] [CrossRef] [PubMed]
- del Pozo-Rodríguez, A.; Solinís Aspiazu, M.Á.; Rodríguez-Gascón, A. Applications of lipid nanoparticles in gene therapy. Eur. J. Pharm. Biopharm. 2016, 109, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Kristen, A.V.; Ajroud-Driss, S.; Conceição, I.; Gorevic, P.; Kyriakides, T.; Obici, L. Patisiran, an RNAi therapeutic for the treatment of hereditary transthyretin-mediated amyloidosis. Neurodegener. Dis. Manag. 2019, 9, 5–23. [Google Scholar] [CrossRef]
- Wu, Z.; Li, T. Nanoparticle-Mediated Cytoplasmic Delivery of Messenger RNA Vaccines: Challenges and Future Perspectives. Pharm. Res. 2021, 38, 473–478. [Google Scholar] [CrossRef]
- Zhu, X.; Yin, L.; Theisen, M.; Zhuo, J.; Siddiqui, S.; Levy, B.; Presnyak, V.; Frassetto, A.; Milton, J.; Salerno, T.; et al. Systemic mRNA Therapy for the Treatment of Fabry Disease: Preclinical Studies in Wild-Type Mice, Fabry Mouse Model, and Wild-Type Non-human Primates. Am. J. Hum. Genet. 2019, 104, 625–637. [Google Scholar] [CrossRef]
- DeRosa, F.; Smith, L.; Shen, Y.; Huang, Y.; Pan, J.; Xie, H.; Yahalom, B.; Heartlein, M.W. Improved Efficacy in a Fabry Disease Model Using a Systemic mRNA Liver Depot System as Compared to Enzyme Replacement Therapy. Mol. Ther. 2019, 27, 878–889. [Google Scholar] [CrossRef]
- Delgado, D.; Rodríguez-Gascón, A.; del Pozo-Rodríguez, A.; Echevarría, E.; Ruiz de Garibay, A.P.; Rodríguez, J.M.; Solinís Aspiazu, M.Á. Dextran-protamine-solid lipid nanoparticles as a non-viral vector for gene therapy: In vitro characterization and in vivo transfection after intravenous administration to mice. Int. J. Pharm. 2012, 425, 35–43. [Google Scholar] [CrossRef]
- Delgado, D.; del Pozo-Rodríguez, A.; Solinís Aspiazu, M.Á.; Bartkowiak, A.; Rodríguez-Gascón, A. New gene delivery system based on oligochitosan and solid lipid nanoparticles: “In vitro” and “in vivo” evaluation. Eur. J. Pharm. Sci. 2013, 50, 484–491. [Google Scholar] [CrossRef]
- Ohshima, T.; Murray, G.J.; Swaim, W.D.; Longenecker, G.; Quirk, J.M.; Cardarelli, C.O.; Sugimoto, Y.; Pastan, I.; Gottesman, M.M.; Brady, R.O.; et al. α-Galactosidase A deficient mice: A model of fabry disease. Proc. Natl. Acad. Sci. USA 1997, 94, 2540–2544. [Google Scholar] [CrossRef]
- del Pozo-Rodríguez, A.; Delgado, D.; Solinís Aspiazu, M.Á.; Rodríguez-Gascón, A.; Pedraz, J.L. Solid lipid nanoparticles: Formulation factors affecting cell transfection capacity. Int. J. Pharm. 2007, 339, 261–268. [Google Scholar] [CrossRef]
- Gómez-Aguado, I.; Rodríguez-Castejón, J.; Vicente-Pascual, M.; Rodríguez-Gascón, A.; del Pozo-Rodríguez, A.; Solinís Aspiazu, M.Á. Nucleic Acid Delivery by Solid Lipid Nanoparticles Containing Switchable Lipids: Plasmid DNA vs. Messenger RNA. Molecules 2020, 25, 5995. [Google Scholar] [CrossRef] [PubMed]
- Ruiz de Garibay, A.P.; Delgado, D.; del Pozo-Rodríguez, A.; Solinís Aspiazu, M.Á.; Rodríguez-Gascón, A. Multicomponent nanoparticles as nonviral vectors for the treatment of fabry disease by gene therapy. Drug Des. Devel. Ther. 2012, 6, 303–310. [Google Scholar] [CrossRef]
- Ruiz de Garibay, A.P.; Solinís Aspiazu, M.Á.; del Pozo-Rodríguez, A.; Apaolaza, P.S.; Shen, J.S.; Rodríguez-Gascón, A. Solid lipid nanoparticles as non-viral vectors for gene transfection in a cell model of fabry disease. J. Biomed. Nanotechnol. 2015, 11, 500–511. [Google Scholar] [CrossRef]
- Apaolaza, P.S.; del Pozo-Rodríguez, A.; Torrecilla, J.; Rodríguez-Gascón, A.; Rodríguez, J.M.; Friedrich, U.; Weber, B.H.F.; Solinís Aspiazu, M.Á. Solid lipid nanoparticle-based vectors intended for the treatment of X-linked juvenile retinoschisis by gene therapy: In vivo approaches in Rs1h-deficient mouse model. J. Control. Release 2015, 217, 273–283. [Google Scholar] [CrossRef] [PubMed]
- del Pozo-Rodríguez, A.; Delgado, D.; Solinís Aspiazu, M.Á.; Pedraz, J.L.; Echevarría, E.; Rodríguez, J.M.; Rodríguez-Gascón, A. Solid lipid nanoparticles as potential tools for gene therapy: In vivo protein expression after intravenous administration. Int. J. Pharm. 2010, 385, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, G.; Maruyama, H.; Ishii, S.; Shimotori, M.; Kameda, S.; Kono, T.; Miyazaki, J.I.; Kulkarni, A.B.; Gejyo, F. Naked plasmid DNA-based α-galactosidase a gene transfer partially reduces systemic accumulation of globotriaosylceramide in Fabry mice. Mol. Biotechnol. 2008, 38, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Ishii, S.; Yoshioka, H.; Mannen, K.; Kulkarni, A.B.; Fan, J.Q. Transgenic mouse expressing human mutant α-galactosidase A in an endogenous enzyme deficient background: A biochemical animal model for studying active-site specific chaperone therapy for Fabry disease. Biochim. Biophys. Acta Mol. Basis Dis. 2004, 1690, 250–257. [Google Scholar] [CrossRef] [PubMed]
- Nowak, A.; Huynh-Do, U.; Krayenbuehl, P.A.; Beuschlein, F.; Schiffmann, R.; Barbey, F. Fabry disease genotype, phenotype, and migalastat amenability: Insights from a national cohort. J. Inherit. Metab. Dis. 2020, 43, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Rastall, D.P.W.; Amalfitano, A. Recent advances in gene therapy for lysosomal storage disorders. Appl. Clin. Genet. 2015, 8, 157–169. [Google Scholar] [CrossRef]
- Fan, J.-Q.; Ishii, S. Active-site-specific chaperone therapy for Fabry disease. FEBS J. 2007, 274, 4962–4971. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Hirai, Y.; Migita, M.; Seino, Y.; Fukuda, Y.; Sakuraba, H.; Kase, R.; Kobayashi, T.; Hashimoto, Y.; Shimada, T. Long-term systemic therapy of Fabry disease in a knockout mouse by adeno-associated virus-mediated muscle-directed gene transfer. Proc. Natl. Acad. Sci. USA 2002, 99, 13777–13782. [Google Scholar] [CrossRef]
- Moghimi, S.M.; Hunter, A.C.; Andresen, T.L. Factors Controlling Nanoparticle Pharmacokinetics: An Integrated Analysis and Perspective. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 481–503. [Google Scholar] [CrossRef] [PubMed]
- Beloqui, A.; Solinís Aspiazu, M.Á.; Delgado, A.; Évora, C.; del Pozo-Rodríguez, A.; Rodríguez-Gascón, A. Biodistribution of Nanostructured Lipid Carriers (NLCs) after intravenous administration to rats: Influence of technological factors. Eur. J. Pharm. Biopharm. 2013, 84, 309–314. [Google Scholar] [CrossRef]
- Alroy, J.; Sabnis, S.; Kopp, J.B. Renal Pathology in Fabry Disease. J. Am. Soc. Nephrol. 2002, 13, S134–S138. [Google Scholar] [CrossRef] [PubMed]
- Branton, M.H.; Schiffmann, R.; Sabnis, S.G.; Murray, G.J.; Quirk, J.M.; Altarescu, G.; Goldfarb, L.; Brady, R.O.; Balow, J.E.; Austin, H.A.; et al. Natural history of fabry renal disease: Influence of α-galactosidase a activity and genetic mutations on clinical course. Medicine (Baltimore) 2002, 81, 122–138. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, O.; Gago, M.F.; Miltenberger-Miltenyi, G.; Sousa, N.; Cunha, D. Review fabry disease therapy: State-of-the-art and current challenges. Int. J. Mol. Sci. 2021, 22, 1–16. [Google Scholar]
- Cesta, M.F. Normal Structure, Function, and Histology of the Spleen. Toxicol. Pathol. 2006, 34, 455–465. [Google Scholar] [CrossRef]
- Chrastina, A.; Massey, K.A.; Schnitzer, J.E. Overcoming in vivo barriers to targeted nanodelivery. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnology 2011, 3, 421–437. [Google Scholar] [CrossRef]
- Ruponen, M.; Honkakoski, P.; Rönkkö, S.; Pelkonen, J.; Tammi, M.; Urtti, A. Extracellular and intracellular barriers in non-viral gene delivery. J. Control. Release 2003, 93, 213–217. [Google Scholar] [CrossRef]
- Kawabata, K.; Takakura, Y.; Hashida, M. The Fate of Plasmid DNA After Intravenous Injection in Mice: Involvement of Scavenger Receptors in Its Hepatic Uptake. Pharm. Res. An Off. J. Am. Assoc. Pharm. Sci. 1995, 12, 825–830. [Google Scholar] [CrossRef]
- Hisazumi, J.; Kobayashi, N.; Nishikawa, M.; Takakura, Y. Significant role of liver sinusoidal endothelial cells in hepatic uptake and degradation of naked plasmid DNA after intravenous injection. Pharm. Res. 2004, 21, 1223–1228. [Google Scholar] [CrossRef] [PubMed]
- Van Hoecke, L.; Roose, K. How mRNA therapeutics are entering the monoclonal antibody field. J. Transl. Med. 2019, 17, 54. [Google Scholar] [CrossRef] [PubMed]
- Oh, R.C.; Hustead, T.R.; Ali, S.M.; Pantsari, M.W. Mildly Elevated Liver Transaminase Levels: Causes and Evaluation. Am. Fam. Physician 2017, 96, 709–715. [Google Scholar] [PubMed]
- Reagan, W.J.; Yang, R.-Z.; Park, S.; Goldstein, R.; Brees, D.; Gong, D.-W. Metabolic Adaptive ALT Isoenzyme Response in Livers of C57/BL6 Mice Treated with Dexamethasone. Toxicol. Pathol. 2012, 40, 1117–1127. [Google Scholar] [CrossRef] [PubMed]
- Harms, M.; Müller-Goymann, C.C. Solid lipid nanoparticles for drug delivery. J. Drug Deliv. Sci. Technol. 2011, 21, 89–99. [Google Scholar] [CrossRef]
- Jacobs, F.; Gordts, S.C.; Muthuramu, I.; De Geest, B. The liver as a target organ for gene therapy: State of the art, challenges, and future perspectives. Pharmaceuticals 2012, 5, 1372–1392. [Google Scholar] [CrossRef]
- Chen, F.; Huang, G.; Huang, H. Sugar ligand-mediated drug delivery. Future Med. Chem. 2019, 12, 161–171. [Google Scholar] [CrossRef]
- Cocozza, S.; Russo, C.; Pontillo, G.; Pisani, A.; Brunetti, A. Neuroimaging in Fabry disease: Current knowledge and future directions. Insights Imaging 2018, 9, 1077–1088. [Google Scholar] [CrossRef] [PubMed]
- Muntoni, E.; Martina, K.; Marini, E.; Giorgis, M.; Lazzarato, L.; Salaroglio, I.; Riganti, C.; Lanotte, M.; Battaglia, L. Methotrexate-Loaded Solid Lipid Nanoparticles: Protein Functionalization to Improve Brain Biodistribution. Pharmaceutics 2019, 11, 65. [Google Scholar] [CrossRef] [PubMed]
- Przybylska, M.; Wu, I.-H.; Zhao, H.; Ziegler, R.J.; Tousignant, J.D.; Desnick, R.J.; Scheule, R.K.; Cheng, S.H.; Yew, N.S. Partial correction of the α-galactosidase A deficiency and reduction of glycolipid storage in Fabry mice using synthetic vectors. J. Gene Med. 2004, 6, 85–92. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Castejón, J.; Alarcia-Lacalle, A.; Gómez-Aguado, I.; Vicente-Pascual, M.; Solinís Aspiazu, M.Á.; del Pozo-Rodríguez, A.; Rodríguez-Gascón, A. α-Galactosidase A Augmentation by Non-Viral Gene Therapy: Evaluation in Fabry Disease Mice. Pharmaceutics 2021, 13, 771. https://doi.org/10.3390/pharmaceutics13060771
Rodríguez-Castejón J, Alarcia-Lacalle A, Gómez-Aguado I, Vicente-Pascual M, Solinís Aspiazu MÁ, del Pozo-Rodríguez A, Rodríguez-Gascón A. α-Galactosidase A Augmentation by Non-Viral Gene Therapy: Evaluation in Fabry Disease Mice. Pharmaceutics. 2021; 13(6):771. https://doi.org/10.3390/pharmaceutics13060771
Chicago/Turabian StyleRodríguez-Castejón, Julen, Ana Alarcia-Lacalle, Itziar Gómez-Aguado, Mónica Vicente-Pascual, María Ángeles Solinís Aspiazu, Ana del Pozo-Rodríguez, and Alicia Rodríguez-Gascón. 2021. "α-Galactosidase A Augmentation by Non-Viral Gene Therapy: Evaluation in Fabry Disease Mice" Pharmaceutics 13, no. 6: 771. https://doi.org/10.3390/pharmaceutics13060771
APA StyleRodríguez-Castejón, J., Alarcia-Lacalle, A., Gómez-Aguado, I., Vicente-Pascual, M., Solinís Aspiazu, M. Á., del Pozo-Rodríguez, A., & Rodríguez-Gascón, A. (2021). α-Galactosidase A Augmentation by Non-Viral Gene Therapy: Evaluation in Fabry Disease Mice. Pharmaceutics, 13(6), 771. https://doi.org/10.3390/pharmaceutics13060771