1. Introduction
Colorectal cancer, including both cancer of the colon and cancer of the rectum, is the third most frequently diagnosed cancer type in developed countries, with a steady increase in incidence and mortality in the past few years. It mostly affects the male population and is heavily linked with lifestyle choices, such as high consumption of processed meat, smoking tobacco, alcohol consumption, and obesity. If discovered in early stages, the prognostic is positive, with almost full recovery after surgery; however, more than 25% of cases are discovered in late stages [
1,
2], when local affection of the lymph nodes is present as well as possible distant metastasis. In this case, the prognostic is reserved and the primary treatment option is chemotherapy or targeted therapies with a limited increase in patients’ survival [
3].
MicroRNAs (miRNAs) are short non-coding RNAs (ncRNAs), with 19–25 nucleotides in length that can impair protein translation by binding through a seed region of 3–5 nucleotides of the messenger RNA (mRNA) [
4,
5,
6]. miRNAs are associated with a broad spectrum of pathological states, including cancer [
2,
7,
8]. Their ability to target multiple genes at the same time and also their aberrant expression in malignant pathologies, make these small non-coding sequences attractive therapeutic targets in oncology [
9,
10], including colon cancer. Despite intensive preclinical research, a limited number of studies involving miRNA-based therapeutics in cancer have shift to the clinical testing stage [
11]. Among the reasons, could be the fact that numerous studies with miRNAs in cancer are analyzing the direct correlation of expression between a specific miRNA and a targeted mRNA, frequently disregarding the mutational status of the targeted gene and its role in the context of modified genetics. Considering that a tumor suppressor gene can acquire mutations with oncogenic roles outside of the miRNA binding sites (e.g.,
TP53), the tumor suppressor value of a miRNA can switch to oncogenic and vice versa in respect to the genetic integrity of the target genes. Moreover, the large plethora of targeted genes of a specific miRNA and the signaling connection between them in different cellular context is most of the time disregarded in favor of a restricted pool of targeted genes that do not mirror the whole spectrum of pathological changes from the cellular level. This is the case of miR-125b-5p that has a controversial status in cancer, including colon cancer [
12]. The discrepancies between studies showing its oncogenic or tumor suppressor role could be related to the specific function of the targeted genes in respect to their mutational status. Previous studies have shown that high expression of miR-125b in colon cancer is associated with negative prognosis in the context of a direct inhibition of
TP53 network [
13]. Other studies showed that miR-125b suppresses colorectal cancer cells invasion and proliferation through inhibition of PDZ-binding motif (
TAZ) [
14].
To tackle these contradictory results, we decided to investigate the role of miR-125b in colon cancer in reliance of the mutation status of
TP53 target gene. Genetic alteration of
TP53 is present in approximately half of all colorectal cancers. While mutations in
TP53 are not necessarily essential for patients undergoing curable resection, they are decisive for those treated with chemotherapy with a negative impact upon survival [
15]. For example, in vitro studies showed that the response to 5-fluorouracil (5-FU)-based chemotherapy is complete only in colon cancer models with wild-type
TP53; however, retrospective studies or incorporation of
TP53 mutation status in clinical studies could demonstrate the translational value of this dependency [
15].
Considering the opposing roles of TP53 in colon cancer based on the mutational status, tumor suppressor in the case of wild-type forms and oncogenic for some mutant forms, and also the significant percent of patients with TP53 mutation, we employed two different cellular models of colon cancer to investigate the therapeutic role of miR-125b: wild-type and mutant TP53 colon cancer cells. Although in our study miR-125b presented a uniform downregulated level in both patient tumor samples and TP53 wild-type and mutant colon cancer cell lines, the therapeutic role of miRNA replacement was observed only in the case of mutant TP53 study models, mutation identified as pathogenic. Therefore, replacement of miR-125b could be an advantageous strategy for those patients that present oncogenic driven mutation in the TP53 genes, patients that are also the most susceptible to lymphatic invasion, uncomplete therapeutic response and poor survival.
This study is meant to demonstrate, besides the specific activity of miR-125b in colon cancer, that experimental therapeutics with miRNAs should involve a close look at the mutational status of the target genes, and how these genetic abnormalities are changing the spectrum of the other targeted transcripts, even in the case of a uniform dysregulation of the miRNA in large patient cohorts.
2. Methods and Materials
2.1. Tissue Samples Analysis of miR-125b Expression
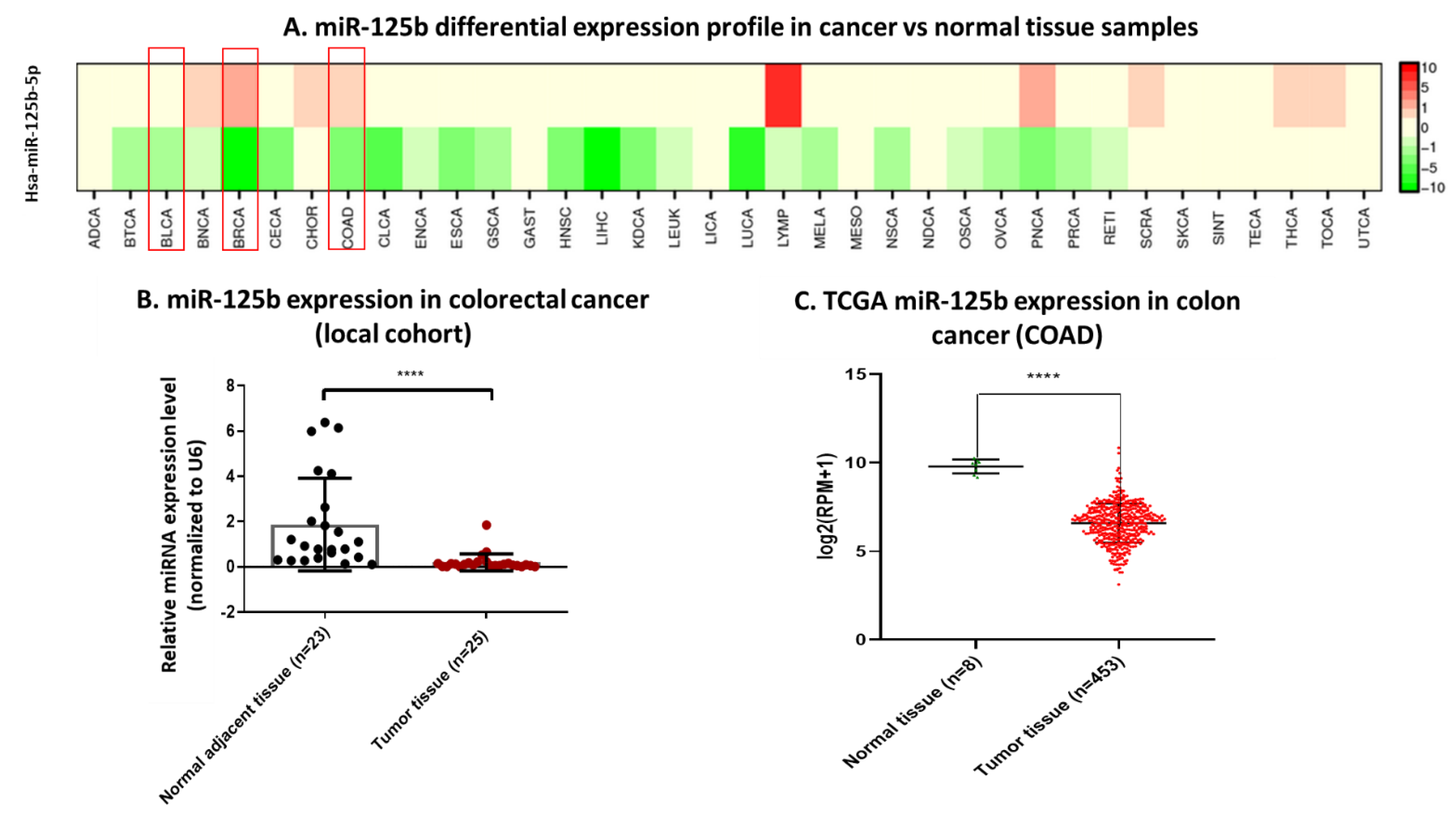
miR-125b expression was analyzed in three cancer types from tumor and normal adjacent tissue samples from the “Prof. Dr. Ion Chiricuta” Institute of Oncology, Cluj-Napoca, Romania. The included patients were diagnosed with double positive breast cancer (21 normal adjacent tissue samples and 44 tumor tissue samples), bladder cancer (39 normal adjacent tissue samples and 37 tumor tissue samples), and colon cancer (23 normal adjacent tissue samples and 25 tumor tissue samples). The tissue samples were taken from the surgically removed tissue during routine surgery and did not pose additional risk for the patients. All patients signed an informed consent for inclusion in the study that was approved by the Ethical Committee of the Institute. The protocol for use of human samples in the current experiment was approved by the Ethical Committee of the Institute and by the Ethical Committee of the Iuliu Hatieganu University of Medicine and Pharmacy, Cluj-Napoca, Romania.
2.2. TCGA Analysis of miRNA/mRNA Expression
The validation of the miRNA expression was made using the RNASeq counts data available at The Tumor Genome Atlas (TCGA) database retrieved from University of California Santa Cruz Cancer Browser. RNASeq counts data for miR-125b expression was organized for each of the pathologies of interest (breast cancer, bladder cancer and colon cancer) and the samples separated in normal and tumoral tissue, depending on the TCGA sample code. The counts (normal tissue versus tumor tissue) were exported in GraphPad Prism (© 2021 GraphPad Software, San Diego, CA, USA) where the Mann–Whitney U test was applied to compare the two groups. For a more complete comparison of miR-125b expression across different malignant pathologies (comparison between normal tissue and tumor tissue), the dbDEMC (database of Differentially Expressed MiRNAs in human Cancers) was used. This database contains integrated data from Gene Expression Omnibus (GEO) and The Cancer Genome Atlas (TCGA), as well as manually curated experimental reports on 2224 miRNAs with differential expression in cancer [
16].
For evaluation of gene expression between normal,
TP53 wild-type colon cancer and
TP53 mutant colon cancer samples, we used the UALCAN database (
http://ualcan.path.uab.edu/cgi-bin/ualcan-res.pl accessed on 15 October 2020), which is based on gene expression extracted from TCGA RNA Seq data correlated with the clinical data. In this database, the name of each gene was introduced first, then its expression based on the
TP53 mutation status was chosen. The results were presented as a box plot graph.
2.3. Cell Culture and Spheroid Formation
Two colon cancer cell lines were used for the study: HCT-116 (ATCC
® CCL-247™, Manassas, VA, USA) and DLD-1 (ATCC
® CCL-221™, Manassas, VA, USA) and one normal fibroblast cell line: CCD-18Co (ATCC
® CRL-1459™, Manassas, VA, USA). The cancer cell lines are originating from colorectal patients, although the transformed cells are from the colon biopsy. HCT-116 cell line was cultured in Mc Coy’s media (Merck KGaA, Darmstadt, Germany) supplemented with 10% fetal bovine serum (FBS), DLD-1 cell line was cultured in RPMI media (Merck KGaA, Darmstadt, Germany) supplemented with 10% FBS, while CCD-18Co cell line was cultured in MEM media (Merck KGaA, Darmstadt, Germany) supplemented with 10% FBS according to the provider protocol. All cell lines were passaged 2–3 times a week and were incubated at 37 °C with 5% CO
2. The 3D spheroids of DLD-1 were obtained through the hanging drops technique as previously exemplified [
17].
2.4. Cell Transfection
HCT-116 and DLD-1 cell lines were transfected with miRNA mimic and inhibitor using Lipofectamine 2000 (Invitrogen), diluted in OptiMEM media (Gibco, Thermo Fisher Scientific, Carlsbad, CA, USA) following the producer instructions. The control cell groups were transfected with empty Lipofectamine 2000 in the same concentration. For hsa-miR-125b mimic (Invitrogen, Thermo Fisher Scientific, Carlsbad, CA, USA) and hsa-miR-125b inhibitor (Invitrogen, Thermo Fisher Scientific, Carlsbad, CA, USA) transfection it was used 100 nM of miRNA mimic and inhibitor (based on literature data), as follows: the oligonucleotides were diluted in OptiMEM (Merck KGaA, Darmstadt, Germany) and then incubated for 5 min at RT with Lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA, USA) diluted also in OptiMEM. After 6 h, the OptiMEM media was removed and replaced with the corresponding culture media, and then 48 h after transfection, the cells were ready for functional and molecular assays.
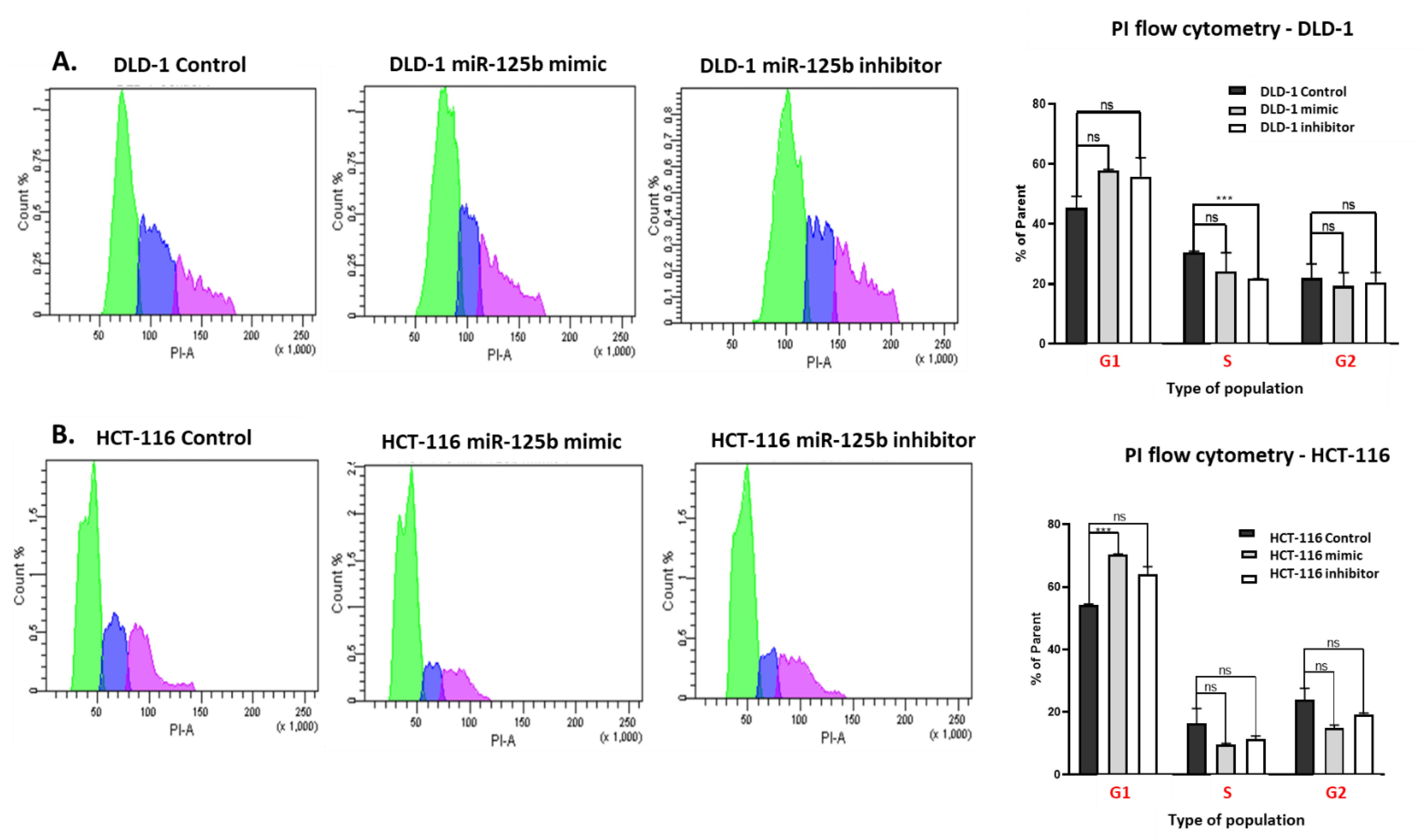
2.5. Flow Cytometry Analysis on Cell Cycle and Apoptosis/Necrosis
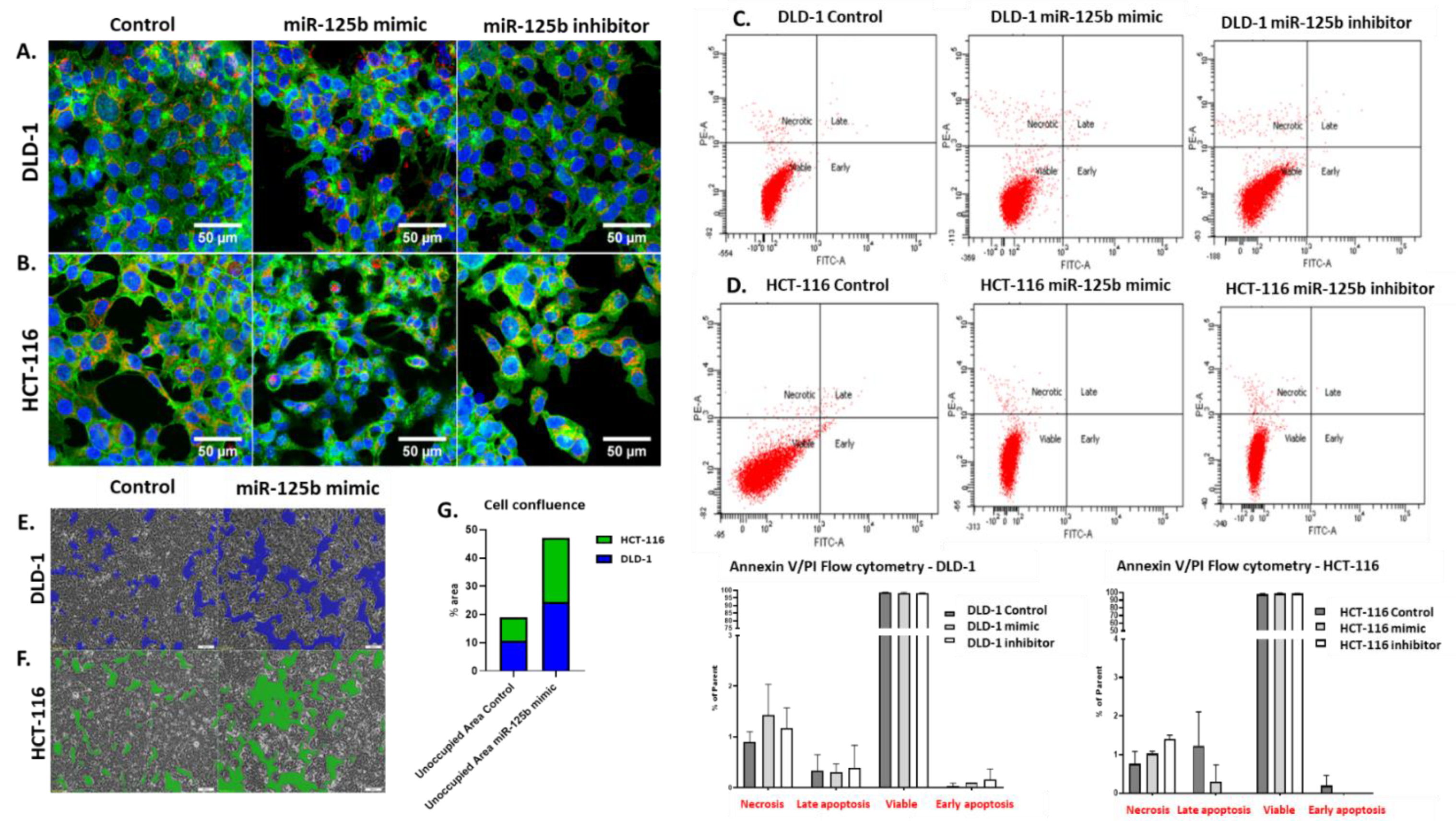
The miRNA transfected and the control cell suspension was washed three times with PBS 1X (Merck KGaA, Darmstadt, Germany) and fixed with pre-chilled 70% ethanol for 50 min for the analysis of cell cycle progression. Afterward, the cells were rewashed with PBS 1X and treated with RNase buffer (40 µg/mL RNase and 0.2% BSA) (Thermo Fisher Scientific, Waltham, MA, USA) for 15 min, at RT. At the end of this time, the propidium iodine (Thermo Fisher Scientific, Waltham, MA, USA) was added in dark conditions at a final concentration of 10 µg/mL in 500μL buffer and incubated at RT for another 30 min.
For apoptosis/necrosis ratio analysis at flow cytometry, the miRNA transfected, and control cells were trypsinized, resuspended in complete media and washed 3 times with PBS 1X. After the washing step the cells were centrifuged at 1200 rpm for 5 min and mixed with Annexin V Binding buffer (Invitrogen, Thermo Fisher Scientific, Carlsbad, CA, USA). The cells were then stained with 5 μL Annexin V (Thermo Fisher Scientific, Waltham, MA, USA) and 5 μL propidium iodide (Merck KGaA, Darmstadt, Germany) for 15 min.
The samples were read at the flow cytometer BD FACS Canto II instrument (BD, San Jose, CA, USA). The initial gaiting was fixed based on the control for each cell line. The data was analyzed with FACS Diva version 6.0 software (BD, San Jose, CA, USA).
2.6. Wound Healing Assay
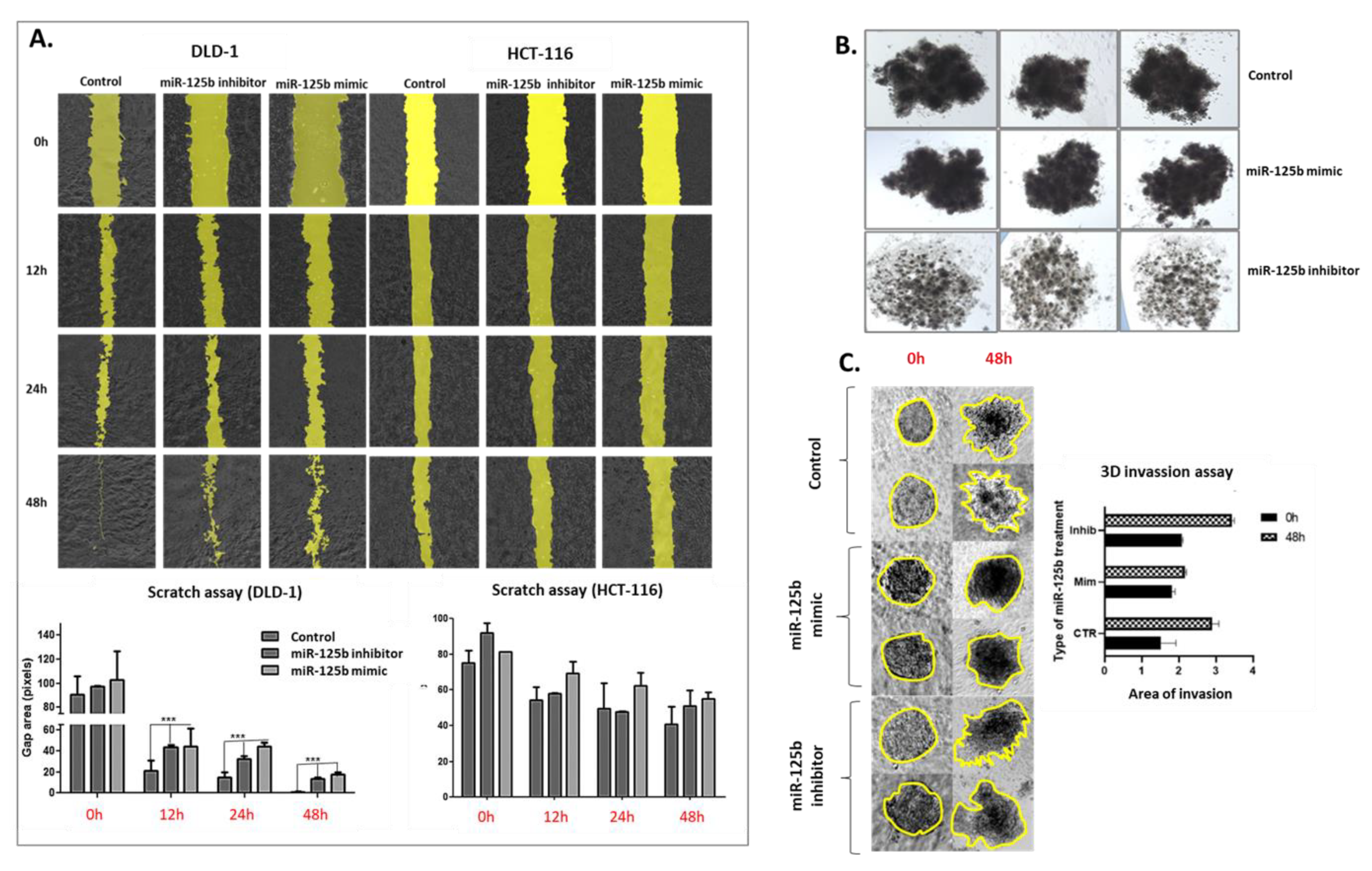
The miRNA transfected and control cells were plated into a 24-well plate designed with a silicon insert (Ibidi, GmbH, Blossom Biotechnologies Inc., Gräfelfing, Germany). The cells were plated at 40,000 cells/well and left to adhere for 24 h. After 24 h, the inserts were removed, and pictures were taken at 0 h, 12 h, 24 h, and 48 h. The covering of the scratch area was analyzed with the help of MiToBo plug-in in Image J software.
2.7. Colony Assay
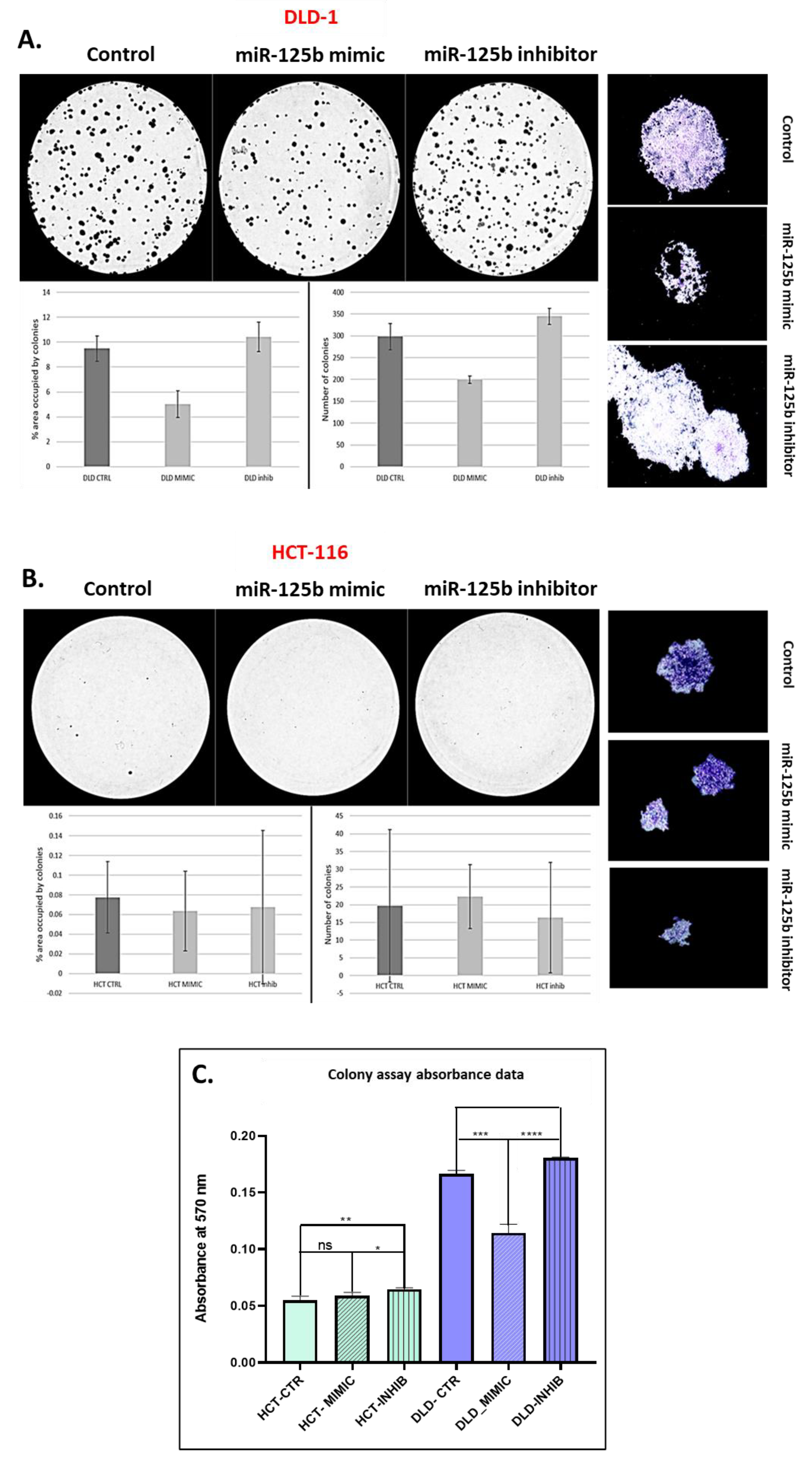
After 48 h from transfection the transfected and control cells were trypsinized and diluted until reaching 1000 cells/mL, then distributed in triplicates in a 6 well plate, 500 cells/well. The colonies were left to form for three weeks, then they were fixated with 80% methanol, stained with 0.5% crystal violet, and then washed until only the colonies remained stained. After the plate was dry, pictures of each well were taken with the help of a Canon camera. The number of colonies and their respective size, as well as the image processing was done with Image J software.
2.8. Extracellular Matrix 3D Invasion Assay
The protocol for extracellular matrix 3D invasion assay was adapted from the 3D invasion assay by Berens E.B. et al. [
18]. After the spheroids from transfected and control cells were formed through the hanging drop technique as previously reminded, they were recovered and immersed in a 1/1 Geltrex stock solution (Thermo Fisher Scientific, Waltham, MA, USA) and 0.25% Collagen type I solution (Thermo Fisher Scientific, Waltham, MA, USA). Photos were taken at 0 h and 48 h. The exterior limits of the invasion were marked and measured with the help of Image J software.
2.9. Confocal Fluorescence Microscopy
HCT-116 and DLD-1 cell suspensions were pipetted in chamber slide and left to adhere. After 24 h, the cells were transfected with empty or miR-125b mimic/inhibitor-containing Lipofectamine 2000 as previously stated; 48 h post-transfection the cells were triple-strained for nucleus, mitochondria, and cytoskeleton based on a coloration protocol previously developed in our laboratory [
19]. First, the cells were incubated for 4 h with MitoTracker (Thermo Fisher Scientific, Waltham, MA, USA), then fixed with 4% paraformaldehyde and treated with Triton X detergent for 15 min. Following detergent treatment and washing three times with PBS, the actin filaments were stained with Phalloidin-FITC (Cytoskeleton, lnc, Denver, CO, USA) for 15 min, washed again with PBS and finally stained with DAPI (Thermo Fisher Scientific, Waltham, MA, USA) for the nucleus for 1 min. The pictures were taken with the help of Olympus FLUOVIEW FV1200 laser scanning fluorescence confocal microscope (Olympus, Tokyo, Japan).
2.10. RNA Isolation and RT-qPCR for miRNA and mRNA Expression
Transfected and control cells were resuspended in 800 µL of TRI Reagent (Sigma Aldrich, Saint Louis, MO, USA) and immediately froze in liquid nitrogen. The RNA was extracted from TRI Reagent based on the chloroform-ethanol extraction method and quantified with NanoDrop-1100. For reverse transcription of miRNA sequences, we used the TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, US), whereas, for reverse transcription of mRNA, we used High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). The RT-PCR reaction was done with SsoAdvanced Universal Probes Supermix (Bio-Rad, Hercules, CA, USA) for miRNA evaluation and SYB Select Master Mix (Bio-Rad, Hercules, CA, USA) for the expression of protein-coding genes. The miRNA expression and mRNA expression were done with the ViiA™ 7 System (Thermo Fischer Scientific, Waltham, MA, USA).
2.11. DNA Isolation and NGS Analysis
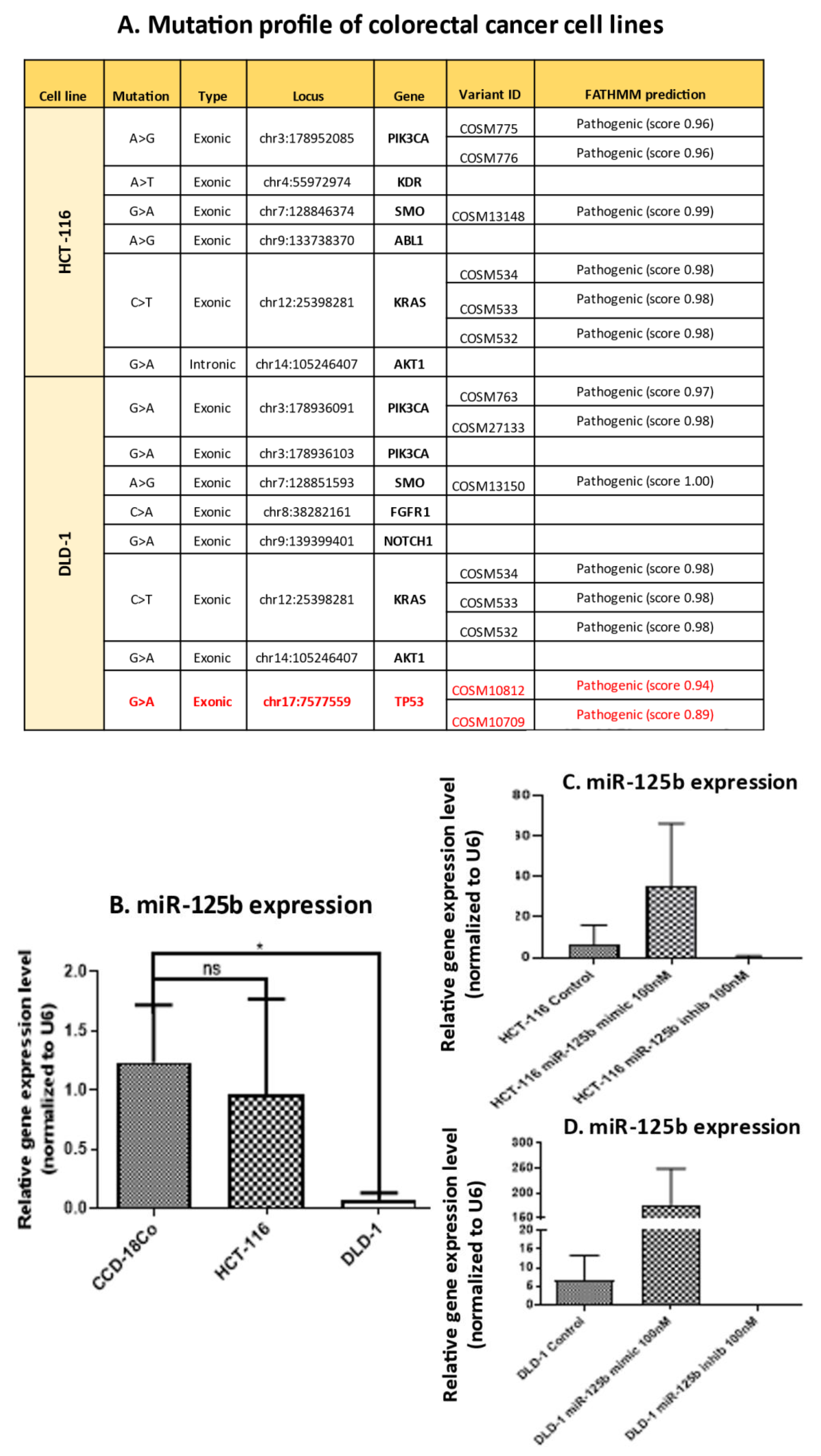
DNA from HCT-116 and DLD-1 colon cancer cell lines was extracted using the Purelink Genomic DNA minikit (Thermofisher Scientific, Waltham, MA, USA) following the manufacturer instruction. 20 ng of DNA were used for sequencing using the Ion AmpliSeq Cancer Hotspot Panel v2 (ThermoFischer Scientific, Waltham, MA, USA) and the Ion AmpliSeq Library 2.0 kit (Thermofisher Scientific, Waltham, MA, USA). The Ion AmpliSeq Cancer Hotspot Panel v2 consists of primers for hotspot evaluation in the following genes: ABL1, AKT1,ALK, APC, ATM, BRAF, CDH1, CDK2A, CSF1R, CTNNB1, EGFR, ERBB2, ERBB4, EZH2, FBXW7, FGFR1, FGFR2, FGFR3, FLT3, GNA11, GNAQ, GNAS, HNF1A, IDH1, IDH2, JAK2, JAK3, KDR, KIT, KRAS, MET, MLH1, MPL, NOTCH1, NPM1, NRAS, PDGFRA, PIK3CA, PTEN, PTPN11, RB1, RET, SMAD4, SMARB1, SMO, STK11, TP53, and VHL. After library preparation the samples were purified using the AMpure XP Beads (Beckman Coulter, Inc., Brea, CA, USA). The purified libraries were quantified using the fluorometer Qubit 2.0 (Sigma Aldrich, Saint Louis, MO, USA) and the Qubit HS DNA kit (Sigma Aldrich, Saint Louis, MO, USA). For template synthesis, libraries were diluted to 100 pM and multiplex 2 libraries on an Ion 316 Chip (ThermoFischer Scientific, Waltham, MA, USA). The sequencing process was performed on the Ion Torrent PGM Machine (ThermoFischer Scientific, Waltham, MA, USA) using the Ion PGM HI-Q Sequencing 200 kit (ThermoFischer Scientific, Waltham, MA, USA). The data obtained after sequencing were analyzed using the Torrent Suit 5.6 and Ion Reporter 5.6 software for data trimming, alignment and variant calling. The obtained variants were filtered using the following conditions: p value ≤ 0.05, coverage ≥ 500.
2.12. Statistical Analysis
All results were evaluated statistically in GraphPad Prism software, with the help of two-tailed t-test and nonparametric tests. Statistical significance was considered a p-value greater than 0.05. Mann–Whitney U test was applied in the case of TCGA and local cohorts screening for miR-125b and ANOVA test to assess the difference between the three groups in the case of the scratch assay. The rest of the experiments were analyzed through paired experimental design, using paired t test.
4. Discussion
miR-125b has a controversial role in malignant disease, with significant differences among different cancer types, depending on the targeted genes and their role in a particular cancer development. However, specific reasons behind these contrasting roles are yet to be identified [
12]. In bladder cancer, miR-125b was reported to be generally down-regulated, where overexpression of this miRNA inhibits the capacity of cancer cells to form colonies or xenografts in immunocompromised mice [
23]. Moreover, through regulation of Matrix metalloproteinase 13 (
MMP13), miR-125b is also mediating the invasion and metastasis process [
24]. The downregulation of miR-125b was also confirmed in the local cohort of bladder cancer tissue samples versus normal adjacent ones. For the case of breast cancer, the promoter region of miR-125b gene is methylated; thus, this miRNA is also generally reported as under expressed in the reminded pathology [
25]. In this case the function of miR-125b is especially related to cellular proliferation, by indirect downregulation of Ras-mitogen activated protein kinase (
MAPK) and inhibition of apoptosis through mammalian target of rapamycin (
mTOR) pathway [
12,
26,
27]. In our local cohort of double positive breast cancer tissue, as well as in our analysis of TCGA RNASeq data, the literature data was confirmed.
In colorectal cancer, reports of miR-125b upregulation are associated with poor prognosis, but without an identified mechanism [
12]. An analysis of 89 colorectal cancer cases concluded that miR-125b upregulation is associated with advanced tumor stage and invasion of local tissue. The role of miR-125b was linked to inhibition of the tumor suppressor gene,
TP53 [
13], but without considering the possible mutational alterations that can intervene within these gene in colorectal malignancies. In our analysis, miR-125b was down-regulated in colon cancer tissue samples in both the local cohort and TCGA RNASeq data. A recently published study reported miR-125 down-regulation in colon cancer cell lines and proved that the activity of this miRNA is related to impaired apoptosis and invasion, through
TAZ gene targeting [
14]. miR-125b is also down-regulated in colon cancer chemoresistant cell lines, as a result of Toll-like receptor (
TLR) 2/6 and
TRL5 activation. miR-125b mimic re-sensitized the cells and reversed the overactivation of the invasion process [
28]. Moreover, miR-125b hypermethylation has also been reported in colon cancer, process generally associated with decreased expression [
29].
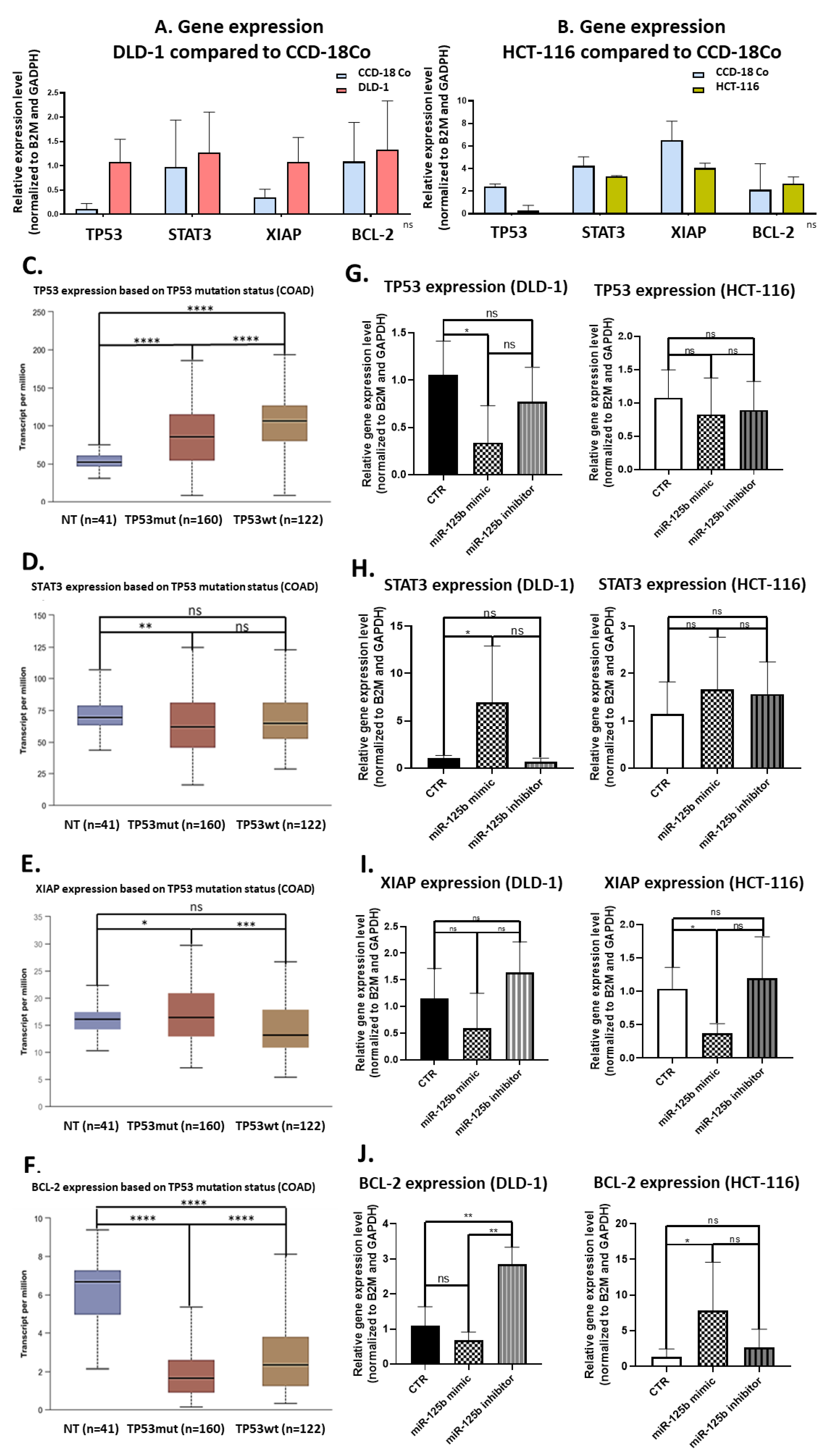
For the present study, in connection to the previous ones, and also to the fact that the function of TP53 (target of miR-125b) in cancer is highly dependent on its mutational status, the decision was made to test whether miR-125b role in colon cancer is dependent on the mutation status of TP53. miR-125b is downregulated in both colon cancer cell lines (DLD-1 and HCT-116) and tissue samples, data confirmed by TCGA RNASeq values for COAD cohort. Moreover, this miRNA is targeting TP53 that is upregulated in COAD samples from TCGA database. TP53 is highly expressed in DLD-1 cell line that carries high mutation burden including an exonic mutation in TP53 gene and is also associated with a more aggressive phenotype in comparison with HCT-116 colon cell line that is TP53 wild-type; the expression of TP53 in HCT-116 was found at low levels compared to the normal cell line. miR-125b mimic or inhibitor transfection in both colon cancer cell line did not significantly affect the percentage of apoptotic/necrotic cells, or cell cycle progression. However, the mimic transfected cells have a lower confluence and increased morphological stress in the cytoskeleton architecture. The therapeutic role of miR-125b replacement in DLD-1 cell line was observed in the case of impaired colony forming capacity and invasion and ability to form spheroid structures; in the case of the inhibitor the malignant potential of the TP53 mutated cells was enhanced. Addition of miR-125b in HCT-116 cell line did not impose a significant effect upon these hallmarks. These results prove that miR-125b has a tumor suppressor role in TP53 mutated colon cancer cells, while it does not significantly affect the behavior of TP53 wild-type cells.
At molecular level, increased levels of miR-125b determined a significant suppression of
TP53 in DLD-1 cells, while minimal changes were observed in HCT-116 compared to control samples. The downregulation of
TP53 was further associated with an contradictory increase in
STAT3 expression, gene that is also a target of miR-125b, demonstrating a possible novel inter- dependency between
TP53 and
STAT3 and also a non-direct modulation of
STAT3 by miR-125b (although
STAT3 is confirmed as a target of miR-125b). This result is favoring the malignant development, considering that
STAT3 plays an oncogenic role in colon cancer [
30].
XIAP gene, a direct target of miR-125b was found as inhibited after miR-125b replacement and upregulated within miR-125b inhibitor treated cells. A similar profile was found in both colon cancer cells, confirming the role of miR-125b as a direct modulator of
XIAP and implicitly an inhibitor of cancer growth considering the oncogenic role of
XIAP gene in colon malignancies [
21]. miR-125b causes
BCL-2 down-regulation in miR-125b mimic treated DLD-1 cells and up-regulation in miR-125b inhibitor treated DLD-1 cells. What is further interesting is that
BCL-2 was overexpressed in HCT-116 transfected with miR-125b mimic.
BCL-2 overexpression is considered a favorable prognostic factor in colon cancer [
31], especially if associated with wild-type
TP53 status [
32].
In conclusion, our results show the importance of differentiating between TP53 mutation status when assessing the role of miR-125b in colon cancer, and not only. The in vitro results showed that in colon cancer cell lines, mutated TP53 is inhibited by exogenous miR-125b causing inhibition of colony formation ability, invasion, and metastasis, while wild-type TP53 is not affected, concomitant with minimal phenotypical differences. Further research, on clinical samples and in vivo experiments are needed to investigate the dual role of miR-125b-TP53 in colon and colorectal cancer.



 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






