
Combination Therapy of Novel Oncolytic Adenovirus with Anti-PD1 Resulted in Enhanced Anti-Cancer Effect in Syngeneic Immunocompetent Melanoma Mouse Model

 , , , , and
, , , , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines, Anti-PD1 Antibodies
2.2. Generation and Production of Oncolytic Adenoviruses
2.3. Restriction Enzyme Assay (REA)
2.4. Whole Genome Sequencing
2.5. Western Blot
2.6. CAR and DSG2 Expression in Melanoma Cell Lines
2.7. Cell Viability: MTS Cytotoxicity Assay
2.8. Immunogenicity of Tumor Cell Death In Vitro and Ex Vivo
2.9. Evaluation of the Concentration of the ICOSL and CD40L Produced by the Virus
2.10. In Vivo Efficacy Studies
2.11. Statistical Analysis
3. Results
3.1. Cloning, Characterization, Confirmation of Genetic Stability and Identity of the Double Transgene Vector Expressing Co-Stimulatory Transgenes (ICOSL and CD40L)
3.1.1. Whole Genome Sequencing of the Vector AdV-D24-ICOSL-CD40L
3.1.2. Expression of CD40L and ICOSL by the Vector AdV-D24-ICOSL-CD40L
3.2. Expression of Coxsackie-Adenovirus Receptor (CAR) and Desmoglein-2 (DSG-2) Receptors in Human Melanoma Cell Lines MUG Mel-1 and MUG Mel-2
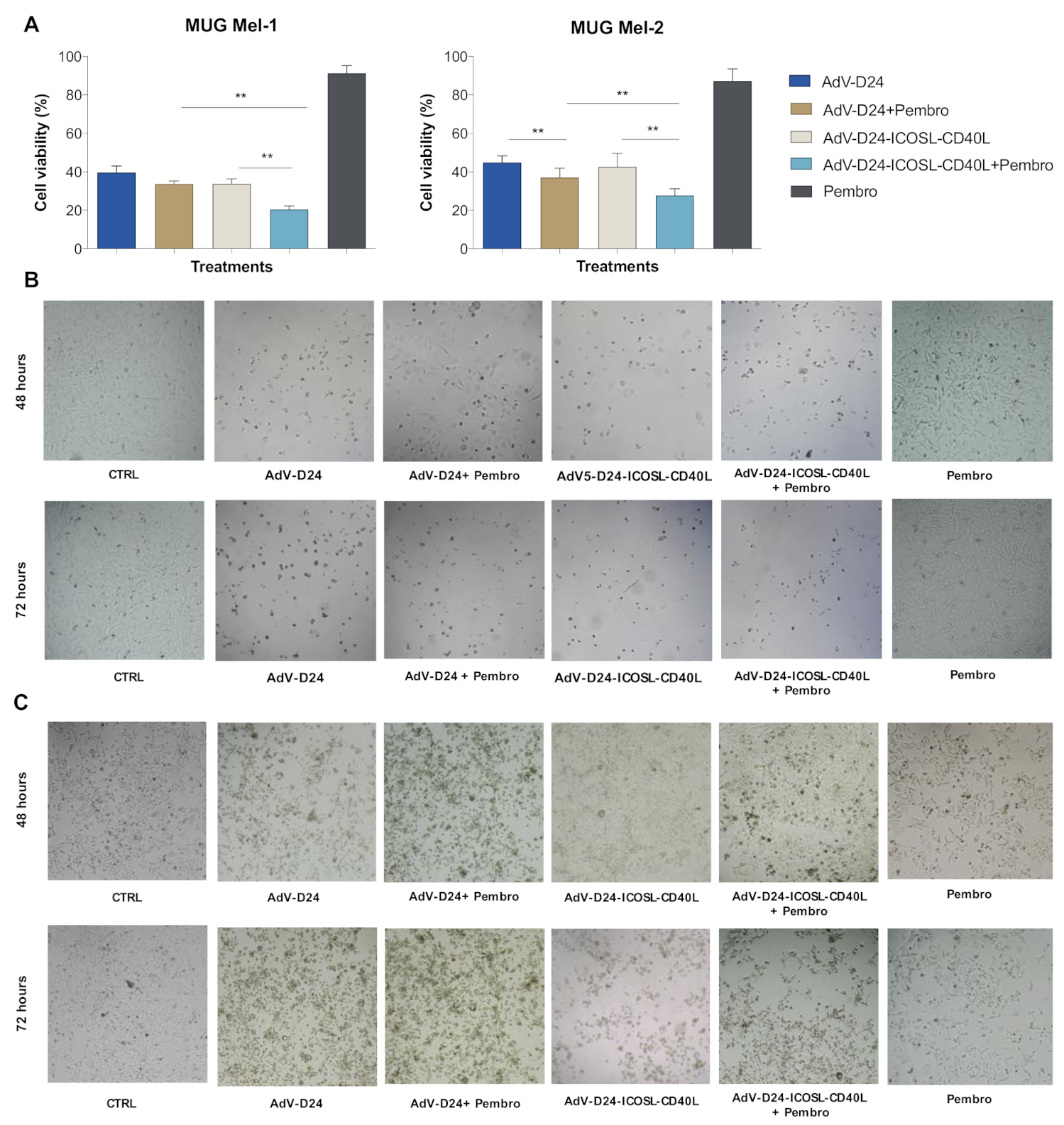
3.3. Evaluation of Cell Viability by MTS Assay (Cell Cytotoxicity Assay)
3.4. Immunogenic Cell Death Assessment
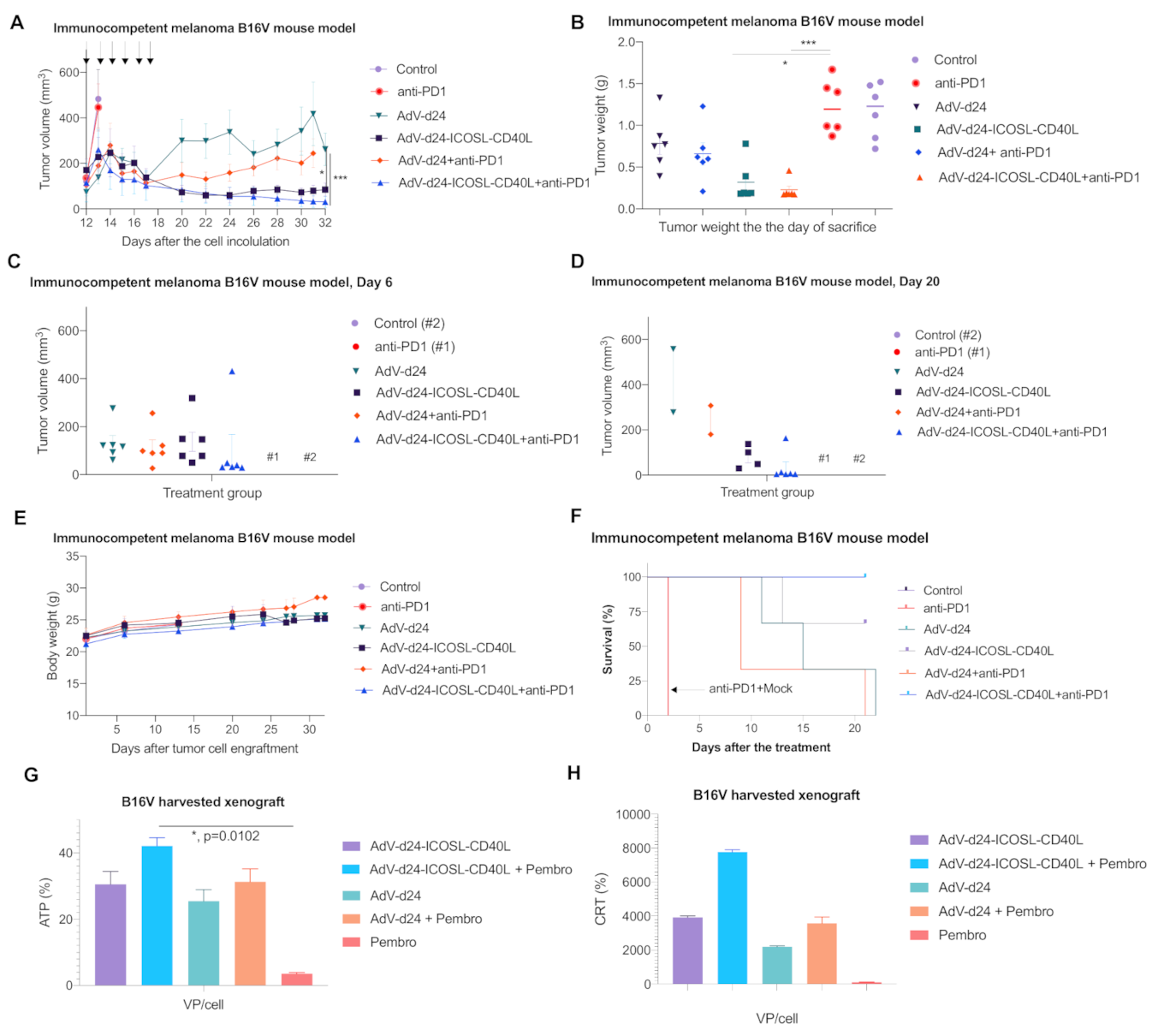
3.5. Antitumor Efficacy of AdV-D24-ICOSL-CD40L and the Combination Therapy with Anti PD-1 Antibody in Murine Melanoma B16V Allograft Immunocompetent C57BL/6 Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yde, S.S.; Sjoegren, P.; Heje, M.; Stolle, L.B. Mucosal Melanoma: A Literature Review. Curr. Oncol. Rep. 2018, 20, 28. [Google Scholar] [CrossRef] [PubMed]
- Rastrelli, M.; Tropea, S.; Rossi, C.R.; Alaibac, M. Melanoma: Epidemiology, risk factors, pathogenesis, diagnosis and classification. In Vivo 2014, 28, 1005–1011. [Google Scholar] [PubMed]
- Elder, D.E.; Bastian, B.C.; Cree, I.A.; Massi, D.; Scolyer, R.A. The 2018 World Health Organization Classification of Cutaneous, Mucosal, and Uveal Melanoma: Detailed Analysis of 9 Distinct Subtypes Defined by Their Evolutionary Pathway. Arch. Pathol. Lab. Med. 2020, 144, 500–522. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- World Health Organization. World Cancer Report (PDF); WHO: Geneva, Switzerland, 2014. [Google Scholar]
- Matthews, N.H.; Li, W.Q.; Qureshi, A.A.; Weinstock, M.A.; Cho, E. Epidemiology of Melanoma. In Cutaneous Melanoma: Etiology and Therapy; Ward, W.H., Farma, J.M., Eds.; Codon Publications: Brisbane, Australia, 2017. [Google Scholar] [CrossRef]
- LaRocca, C.J.; Warner, S.G. Oncolytic viruses and checkpoint inhibitors: Combination therapy in clinical trials. Clin. Transl. Med. 2018, 7, 35. [Google Scholar] [CrossRef]
- Munhoz, R.R.; Postow, M.A. Clinical Development of PD-1 in Advanced Melanoma. Cancer J. 2018, 24, 7–14. [Google Scholar] [CrossRef]
- Imbert, C.; Montfort, A.; Fraisse, M.; Marcheteau, E.; Gilhodes, J.; Martin, E.; Bertrand, F.; Marcellin, M.; Burlet-Schiltz, O.; Peredo, A.G.; et al. Resistance of melanoma to immune checkpoint inhibitors is overcome by targeting the sphingosine kinase-1. Nat. Commun. 2020, 11, 437. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef]
- Chiu, M.; Armstrong, E.J.L.; Jennings, V.; Foo, S.; Crespo-Rodriguez, E.; Bozhanova, G.; Patin, E.C.; McLaughlin, M.; Mansfield, D.; Baker, G.; et al. Combination therapy with oncolytic viruses and immune checkpoint inhibitors. Exp. Opin. Biol. Ther. 2020, 20, 635–652. [Google Scholar] [CrossRef]
- O’Bryan, S.M.; Mathis, J.M. Oncolytic Virotherapy for Breast Cancer Treatment. Curr. Gene Ther. 2018, 18, 192–205. [Google Scholar] [CrossRef]
- Russell, S.J.; Peng, K.W.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef]
- Kuryk, L.; Moller, A.W.; Jaderberg, M. Abscopal effect when combining oncolytic adenovirus and checkpoint inhibitor in a humanized NOG mouse model of melanoma. J. Med. Virol. 2019, 91, 1702–1706. [Google Scholar] [CrossRef]
- Lin, C.Z.; Xiang, G.L.; Zhu, X.H.; Xiu, L.L.; Sun, J.X.; Zhang, X.Y. Advances in the mechanisms of action of cancer-targeting oncolytic viruses. Oncol. Lett. 2018, 15, 4053–4060. [Google Scholar] [CrossRef]
- Liu, T.C.; Kirn, D. Gene therapy progress and prospects cancer: Oncolytic viruses. Gene Ther. 2008, 15, 877–884. [Google Scholar] [CrossRef]
- Maroun, J.; Munoz-Alia, M.; Ammayappan, A.; Schulze, A.; Peng, K.W.; Russell, S. Designing and building oncolytic viruses. Future Virol. 2017, 12, 193–213. [Google Scholar] [CrossRef]
- Lichty, B.D.; Breitbach, C.J.; Stojdl, D.F.; Bell, J.C. Going viral with cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 559–567. [Google Scholar] [CrossRef]
- Liu, Z.; Ravindranathan, R.; Kalinski, P.; Guo, Z.S.; Bartlett, D.L. Rational combination of oncolytic vaccinia virus and PD-L1 blockade works synergistically to enhance therapeutic efficacy. Nat. Commun. 2017, 8, 14754. [Google Scholar] [CrossRef]
- Wang, X.; Teng, F.; Kong, L.; Yu, J. PD-L1 expression in human cancers and its association with clinical outcomes. Oncotargets Ther. 2016, 9, 5023–5039. [Google Scholar] [CrossRef]
- Bommareddy, P.K.; Patel, A.; Hossain, S.; Kaufman, H.L. Talimogene Laherparepvec (T-VEC) and Other Oncolytic Viruses for the Treatment of Melanoma. Am. J. Clin. Derm. 2017, 18, 1–15. [Google Scholar] [CrossRef]
- Alsaab, H.O.; Sau, S.; Alzhrani, R.; Tatiparti, K.; Bhise, K.; Kashaw, S.K.; Iyer, A.K. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front. Pharm. 2017, 8, 561. [Google Scholar] [CrossRef] [PubMed]
- Rothermel, L.D.; Zager, J.S. Engineered oncolytic viruses to treat melanoma: Where are we now and what comes next? Expert Opin. Biol. Ther. 2018, 18, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.I.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2017, 170, 1109–1119.e10. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Huang, J.; Tong, A.; Yang, H. Oncolytic Viruses for Cancer Therapy: Barriers and Recent Advances. Mol. Ther. Oncolytics 2019, 15, 234–247. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.K.; Hong, J.; Yun, C.O. Oncolytic Viruses and Immune Checkpoint Inhibitors: Preclinical Developments to Clinical Trials. Int. J. Mol. Sci. 2020, 21, 8627. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Funchain, P.; Song, J.M.; Rayman, P.; Tannenbaum, C.; Ko, J.; McNamara, M.; Marcela Diaz-Montero, C.; Gastman, B. Talimogene Laherparepvec combined with anti-PD-1 based immunotherapy for unresectable stage III-IV melanoma: A case series. J. Immunother. Cancer 2018, 6, 36. [Google Scholar] [CrossRef]
- Wang, B.; Jiang, H.; Zhou, T.; Ma, N.; Liu, W.; Wang, Y.; Zuo, L. Expression of ICOSL is associated with decreased survival in invasive breast cancer. PeerJ 2019, 7, e6903. [Google Scholar] [CrossRef]
- Aspord, C.; Leccia, M.T.; Charles, J.; Plumas, J. Plasmacytoid dendritic cells support melanoma progression by promoting Th2 and regulatory immunity through OX40L and ICOSL. Cancer Immunol. Res. 2013, 1, 402–415. [Google Scholar] [CrossRef]
- Flies, D.B.; Chen, L. The new B7s: Playing a pivotal role in tumor immunity. J. Immunother. 2007, 30, 251–260. [Google Scholar] [CrossRef]
- Kuryk, L.; Moller, A.-S.W.; Jaderberg, M. Quantification and functional evaluation of CD40L production from the adenovirus vector ONCOS-401. Cancer Gene Ther. 2018, 26, 26–31. [Google Scholar] [CrossRef]
- Liu, X.; Bai, X.F.; Wen, J.; Gao, J.X.; Liu, J.; Lu, P.; Wang, Y.; Zheng, P.; Liu, Y. B7H costimulates clonal expansion of, and cognate destruction of tumor cells by, CD8(+) T lymphocytes in vivo. J. Exp. Med. 2001, 194, 1339–1348. [Google Scholar] [CrossRef]
- Zamarin, D.; Holmgaard, R.B.; Ricca, J.; Plitt, T.; Palese, P.; Sharma, P.; Merghoub, T.; Wolchok, J.D.; Allison, J.P. Intratumoral modulation of the inducible co-stimulator ICOS by recombinant oncolytic virus promotes systemic anti-tumour immunity. Nat. Commun. 2017, 8, 14340. [Google Scholar] [CrossRef]
- Ara, A.; Ahmed, K.A.; Xiang, J. Multiple effects of CD40-CD40L axis in immunity against infection and cancer. Immunotargets Ther. 2018, 7, 55–61. [Google Scholar] [CrossRef]
- Diaconu, I.; Cerullo, V.; Hirvinen, M.L.; Escutenaire, S.; Ugolini, M.; Pesonen, S.K.; Bramante, S.; Parviainen, S.; Kanerva, A.; Loskog, A.S.; et al. Immune response is an important aspect of the antitumor effect produced by a CD40L-encoding oncolytic adenovirus. Cancer Res. 2012, 72, 2327–2338. [Google Scholar] [CrossRef]
- Koski, A.; Kangasniemi, L.; Escutenaire, S.; Pesonen, S.; Cerullo, V.; Diaconu, I.; Nokisalmi, P.; Raki, M.; Rajecki, M.; Guse, K.; et al. Treatment of Cancer Patients With a Serotype 5/3 Chimeric Oncolytic Adenovirus Expressing GMCSF. Mol. Ther. 2010, 18, 1874–1884. [Google Scholar] [CrossRef]
- Sweeney, J.A.; Hennessey, J.P., Jr. Evaluation of accuracy and precision of adenovirus absorptivity at 260 nm under conditions of complete DNA disruption. Virology 2002, 295, 284–288. [Google Scholar] [CrossRef]
- Guo, H.; Jiang, D.; Zhou, T.; Cuconati, A.; Block, T.M.; Guo, J.T. Characterization of the intracellular deproteinized relaxed circular DNA of hepatitis B virus: An intermediate of covalently closed circular DNA formation. J. Virol. 2007, 81, 12472–12484. [Google Scholar] [CrossRef]
- Arad, U. Modified Hirt procedure for rapid purification of extrachromosomal DNA from mammalian cells. Biotechniques 1998, 24, 760–762. [Google Scholar] [CrossRef]
- Marelli, G.; Howells, A.; Lemoine, N.R.; Wang, Y. Oncolytic Viral Therapy and the Immune System: A Double-Edged Sword against Cancer. Front. Immunol. 2018, 9, 866. [Google Scholar] [CrossRef]
- Russell, L.; Peng, K.W.; Russell, S.J.; Diaz, R.M. Oncolytic Viruses: Priming Time for Cancer Immunotherapy. BioDrugs 2019, 33, 485–501. [Google Scholar] [CrossRef]
- Pesonen, S.; Diaconu, I.; Kangasniemi, L.; Ranki, T.; Kanerva, A.; Pesonen, S.K.; Gerdemann, U.; Leen, A.M.; Kairemo, K.; Oksanen, M.; et al. Oncolytic immunotherapy of advanced solid tumors with a CD40L-expressing replicating adenovirus: Assessment of safety and immunologic responses in patients. Cancer Res. 2012, 72, 1621–1631. [Google Scholar] [CrossRef] [PubMed]
- Fu, T.; He, Q.; Sharma, P. The ICOS/ICOSL Pathway Is Required for Optimal Antitumor Responses Mediated by Anti-CTLA-4 Therapy. Cancer Res. 2011, 71, 5445–5454. [Google Scholar] [CrossRef] [PubMed]
- Watts, T.H.; Bertram, E.M.; Bukczynski, J.; Wen, T. T Cell Costimulatory Molecules in Anti-Viral Immunity: Potential Role in Immunotherapeutic Vaccines. Can. J. Infect. Dis. 2003, 14, 221–229. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Heise, C.; Hermiston, T.; Johnson, L.; Brooks, G.; Sampson-Johannes, A.; Williams, A.; Hawkins, L.; Kirn, D. An adenovirus E1A mutant that demonstrates potent and selective systemic anti-tumoral efficacy. Nat. Med. 2000, 6, 1134–1139. [Google Scholar] [CrossRef]
- Murakami, M.; Ugai, H.; Belousova, N.; Pereboev, A.; Dent, P.; Fisher, P.B.; Everts, M.; Curiel, D.T. Chimeric adenoviral vectors incorporating a fiber of human adenovirus 3 efficiently mediate gene transfer into prostate cancer cells. Prostate 2010, 70, 362–376. [Google Scholar] [CrossRef]
- Kim, K.H.; Ryan, M.J.; Estep, J.E.; Miniard, B.M.; Rudge, T.L.; Peggins, J.O.; Broadt, T.L.; Wang, M.; Preuss, M.A.; Siegal, G.P.; et al. A new generation of serotype chimeric infectivity-enhanced conditionally replicative adenovirals: The safety profile of ad5/3-Delta24 in advance of a phase I clinical trial in ovarian cancer patients. Hum. Gene Ther. 2011, 22, 821–828. [Google Scholar] [CrossRef]
- Kuryk, L.; Moller, A.W. Chimeric oncolytic Ad5/3 virus replicates and lyses ovarian cancer cells through desmoglein-2 cell entry receptor. J. Med. Virol. 2020, 92, 1309–1315. [Google Scholar] [CrossRef]
- Kuryk, L.; Haavisto, E.; Garofalo, M.; Capasso, C.; Hirvinen, M.; Pesonen, S.; Ranki, T.; Vassilev, L.; Cerullo, V. Synergistic anti-tumor efficacy of immunogenic adenovirus ONCOS-102 (Ad5/3-D24-GM-CSF) and standard of care chemotherapy in preclinical mesothelioma model. Int. J. Cancer 2016, 139, 1883–1893. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef]
- Green, D.R.; Ferguson, T.; Zitvogel, L.; Kroemer, G. Immunogenic and tolerogenic cell death. Nat. Rev. Immunol. 2009, 9, 353–363. [Google Scholar] [CrossRef]
- McKenna, M.K.; Rosewell-Shaw, A.; Suzuki, M. Modeling the Efficacy of Oncolytic Adenoviruses In Vitro and In Vivo: Current and Future Perspectives. Cancers 2020, 12, 619. [Google Scholar] [CrossRef]
- Olson, B.; Li, Y.; Lin, Y.; Liu, E.T.; Patnaik, A. Mouse Models for Cancer Immunotherapy Research. Cancer Discov. 2018, 8, 1358–1365. [Google Scholar] [CrossRef]
- Kuryk, L.; Moller, A.W.; Garofalo, M.; Cerullo, V.; Pesonen, S.; Alemany, R.; Jaderberg, M. Antitumor-specific T-cell responses induced by oncolytic adenovirus ONCOS-102 (AdV5/3-D24-GM-CSF) in peritoneal mesothelioma mouse model. J. Med. Virol. 2018, 90, 1669–1673. [Google Scholar] [CrossRef]
- Kuryk, L.; Haavisto, E.; Garofalo, M.; Capasso, C.; Hirvinen, M.; Pesonen, S.; Ranki, T.; Vassilev, L.; Cerullo, V. Synergistic Anti-Tumor Efficacy of Immunogenic Adenovirus ONCOS-102 and Standard of Care Chemotherapy in Preclinical Mesothelioma Model. Mol. Ther. 2016, 24, S262. [Google Scholar] [CrossRef]
- Ranki, T.; Pesonen, S.; Hemminki, A.; Partanen, K.; Kairemo, K.; Alanko, T.; Lundin, J.; Linder, N.; Turkki, R.; Ristimaki, A.; et al. Phase I study with ONCOS-102 for the treatment of solid tumors—An evaluation of clinical response and exploratory analyses of immune markers. J. Immunother. Cancer 2016, 4, 17. [Google Scholar] [CrossRef]
- American Cancer Society. Cancer Facts and Figures; American Cancer Society: Atlanta, GA, USA, 2018. [Google Scholar]
- Ferlay, J.; Soerjomataram, I.; Ervik, M.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Globocan 2012: Estimated Cancer Incidence, Mortality and Prevalence Worldwide in 2012. Int. Agency Res. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Filley, A.C.; Dey, M. Immune System, Friend or Foe of Oncolytic Virotherapy? Front. Oncol. 2017, 7, 106. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef]
- Lugowska, I.; Teterycz, P.; Rutkowski, P. Immunotherapy of melanoma. Contemp. Oncol. 2018, 22, 61–67. [Google Scholar] [CrossRef]
- Eggermont, A.M.M.; Crittenden, M.; Wargo, J. Combination Immunotherapy Development in Melanoma. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 197–207. [Google Scholar] [CrossRef]
- Michielin, O.; Atkins, M.B.; Koon, H.B.; Dummer, R.; Ascierto, P.A. Evolving impact of long-term survival results on metastatic melanoma treatment. J. Immunother. Cancer 2020, 8, e000948. [Google Scholar] [CrossRef] [PubMed]
- Smalley, K.S.; Eroglu, Z.; Sondak, V.K. Combination Therapies for Melanoma: A New Standard of Care? Am. J. Clin. Derm. 2016, 17, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Liu, X.; Yang, J.; Zhang, M.; Jin, H.; Ma, X.; Shi, H. Combination of Immunotherapy With Targeted Therapy: Theory and Practice in Metastatic Melanoma. Front. Immunol. 2019, 10, 990. [Google Scholar] [CrossRef] [PubMed]
- Dharmadhikari, N.; Mehnert, J.M.; Kaufman, H.L. Oncolytic virus immunotherapy for melanoma. Curr. Treat. Options Oncol. 2015, 16, 326. [Google Scholar] [CrossRef] [PubMed]
- Kuryk, L.; Bertinato, L.; Staniszewska, M.; Pancer, K.; Wieczorek, M.; Salmaso, S.; Caliceti, P.; Garofalo, M. From Conventional Therapies to Immunotherapy: Melanoma Treatment in Review. Cancers 2020, 12, 3057. [Google Scholar] [CrossRef] [PubMed]
- Peter, M.; Kuhnel, F. Oncolytic Adenovirus in Cancer Immunotherapy. Cancers 2020, 12, 3354. [Google Scholar] [CrossRef] [PubMed]
- Oh, C.M.; Chon, H.J.; Kim, C. Combination Immunotherapy Using Oncolytic Virus for the Treatment of Advanced Solid Tumors. Int. J. Mol. Sci. 2020, 21, 7743. [Google Scholar] [CrossRef]
- Merlino, G.; Herlyn, M.; Fisher, D.E.; Bastian, B.C.; Flaherty, K.T.; Davies, M.A.; Wargo, J.A.; Curiel-Lewandrowski, C.; Weber, M.J.; Leachman, S.A.; et al. The state of melanoma: Challenges and opportunities. Pigment. Cell Melanoma Res. 2016, 29, 404–416. [Google Scholar] [CrossRef]
- Rughani, M.G.; Gupta, A.; Middleton, M.R. New treatment approaches in melanoma: Current research and clinical prospects. Adv. Med. Oncol. 2013, 5, 73–80. [Google Scholar] [CrossRef]
- Chu, R.L.; Post, D.E.; Khuri, F.R.; Van Meir, E.G. Use of replicating oncolytic adenoviruses in combination therapy for cancer. Clin. Cancer Res. 2004, 10, 5299–5312. [Google Scholar] [CrossRef]
- Goradel, N.H.; Mohajel, N.; Malekshahi, Z.V.; Jahangiri, S.; Najafi, M.; Farhood, B.; Mortezaee, K.; Negahdari, B.; Arashkia, A. Oncolytic adenovirus: A tool for cancer therapy in combination with other therapeutic approaches. J. Cell Physiol. 2019, 234, 8636–8646. [Google Scholar] [CrossRef]
- Kuryk, L.; Moller, A.-S.W.; Jaderberg, M. The Combinatory Treatment of the Oncolytic Adenovirus ONCOS-102 with Anti PD-1 (Keytruda (R)) Show Synergistic Anti-Tumor Effect in Humanized A2058 Melanoma huNOG Mouse Model. Mol. Ther. 2018, 26, 200–201. [Google Scholar]
- Chaurasiya, S.; Fong, Y.; Warner, S.G. Optimizing Oncolytic Viral Design to Enhance Antitumor Efficacy: Progress and Challenges. Cancers 2020, 12, 1699. [Google Scholar] [CrossRef]
- Beatty, M.S.; Curiel, D.T. Chapter two–Adenovirus strategies for tissue-specific targeting. Adv. Cancer Res. 2012, 115, 39–67. [Google Scholar] [CrossRef]
- Hensen, L.C.M.; Hoeben, R.C.; Bots, S.T.F. Adenovirus Receptor Expression in Cancer and Its Multifaceted Role in Oncolytic Adenovirus Therapy. Int. J. Mol. Sci. 2020, 21, 6828. [Google Scholar] [CrossRef]
- Kuryk, L.; Moller, A.W.; Jaderberg, M. Combination of immunogenic oncolytic adenovirus ONCOS-102 with anti-PD-1 pembrolizumab exhibits synergistic antitumor effect in humanized A2058 melanoma huNOG mouse model. Oncoimmunology 2019, 8, e1532763. [Google Scholar] [CrossRef]
- Marchini, A.; Daeffler, L.; Pozdeev, V.I.; Angelova, A.; Rommelaere, J. Immune Conversion of Tumor Microenvironment by Oncolytic Viruses: The Protoparvovirus H-1PV Case Study. Front. Immunol. 2019, 10, 1848. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Z. The history and advances in cancer immunotherapy: Understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell Mol. Immunol. 2020, 17, 807–821. [Google Scholar] [CrossRef]
- Saito, R.; Kobayashi, T.; Kashima, S.; Matsumoto, K.; Ogawa, O. Faithful preclinical mouse models for better translation to bedside in the field of immuno-oncology. Int. J. Clin. Oncol. 2020, 25, 831–841. [Google Scholar] [CrossRef]
- Models for Immuno-oncology Research. Cancer Cell 2020, 38, 145–147. [CrossRef] [PubMed]
- Robinson, M.; Li, B.; Ge, Y.; Ko, D.; Yendluri, S.; Harding, T.; VanRoey, M.; Spindler, K.R.; Jooss, K. Novel immunocompetent murine tumor model for evaluation of conditionally replication-competent (oncolytic) murine adenoviral vectors. J. Virol. 2009, 83, 3450–3462. [Google Scholar] [CrossRef] [PubMed]
- Halldén, G.; Hill, R.; Wang, Y.; Anand, A.; Liu, T.-C.; Lemoine, N.R.; Francis, J.; Hawkins, L.; Kirn, D. Novel immunocompetent murine tumor models for the assessment of replication-competent oncolytic adenovirus efficacy. Mol. Ther. 2003, 8, 412–424. [Google Scholar] [CrossRef]
- Jogler, C.; Hoffmann, D.; Theegarten, D.; Grunwald, T.; Uberla, K.; Wildner, O. Replication properties of human adenovirus in vivo and in cultures of primary cells from different animal species. J. Virol. 2006, 80, 3549–3558. [Google Scholar] [CrossRef]
- Bramante, S.; Kaufmann, J.K.; Veckman, V.; Liikanen, I.; Nettelbeck, D.M.; Hemminki, O.; Vassilev, L.; Cerullo, V.; Oksanen, M.; Heiskanen, R.; et al. Treatment of melanoma with a serotype 5/3 chimeric oncolytic adenovirus coding for GM-CSF: Results in vitro, in rodents and in humans. Int. J. Cancer 2015, 137, 1775–1783. [Google Scholar] [CrossRef]
- Targovax Announces Impressive Objective Responses as Well as Effects on Non-Injected Lesions in ONCOS-102 Trial in Anti-PD1 Refractory Melanoma Patients. Available online: https://www.targovax.com/en/targovax-announces-impressive-objective-responses-as-well-as-effects-on-non-injected-lesions-in-oncos-102-trial-in-anti-pd1-refractory-melanoma-patients/ (accessed on 26 January 2021).
- Frohlich, A.; Hoffmann, F.; Niebel, D.; Egger, E.; Kukuk, G.M.; Toma, M.; Sirokay, J.; Bieber, T.; Landsberg, J. Talimogene Laherparepvec in Advanced Mucosal Melanoma of the Urethra Upon Primary Resistance on Immune Checkpoint Inhibition: A Case Report. Front. Oncol. 2020, 10, 611. [Google Scholar] [CrossRef]
- Hamid, O.; Ismail, R.; Puzanov, I. Intratumoral Immunotherapy-Update 2019. Oncologist 2020, 25, e423–e438. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Tumor Cell Engraftment | Day 1 | Day 2 | Day 3 | Day 4 | Day 5 | Day 6 |
|---|---|---|---|---|---|---|---|
| 1. Mock | Tumor inoculation: 2 tumors per mouse, one tumor/flank. Each flank was engrafted with 1 × 106 B16V cells in 50–100 µL (6 tumors per group) | PBS i.t. PBS i.v. | PBS i.t. PBS i.v. | PBS i.t. PBS i.v. | PBS i.t. PBS i.v. | PBS i.t. PBS i.v. | PBS i.t. PBS i.v. |
| 2. AdV-D24-ICOS-CD40L | Virus i.t. | Virus i.t. | Virus i.t. | Virus i.t. | Virus i.t. | Virus i.t. | |
| 3. AdV-D24-WT | Virus i.t. | Virus i.t. | Virus i.t. | Virus i.t. | Virus i.t. | Virus i.t. | |
| 4. Anti PD-1 | 200 µg i.v. | 200 µg i.v. | 200 µg i.v. | 200 µg i.v. | 200 µg i.v. | 200 µg i.v. | |
| 5. AdV-D24-ICOS-CD40L + anti PD-1 | Virus i.t. + 200 µg i.v. | Virus i.t. + 200 µg i.v. | Virus i.t. + 200 µg i.v. | Virus i.t. + 200 µg i.v. | Virus i.t. + 200 µg i.v. | Virus i.t. + 200 µg i.v. | |
| 6. AdV-D24-WT + anti PD-1 | Virus i.t. + 200 µg i.v. | Virus i.t. + 200 µg i.v. | Virus i.t. + 200 µg i.v. | Virus i.t. + 200 µg i.v. | Virus i.t. + 200 µg i.v. | Virus i.t. + 200 µg i.v. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garofalo, M.; Bertinato, L.; Staniszewska, M.; Wieczorek, M.; Salmaso, S.; Schrom, S.; Rinner, B.; Pancer, K.W.; Kuryk, L. Combination Therapy of Novel Oncolytic Adenovirus with Anti-PD1 Resulted in Enhanced Anti-Cancer Effect in Syngeneic Immunocompetent Melanoma Mouse Model. Pharmaceutics 2021, 13, 547. https://doi.org/10.3390/pharmaceutics13040547
Garofalo M, Bertinato L, Staniszewska M, Wieczorek M, Salmaso S, Schrom S, Rinner B, Pancer KW, Kuryk L. Combination Therapy of Novel Oncolytic Adenovirus with Anti-PD1 Resulted in Enhanced Anti-Cancer Effect in Syngeneic Immunocompetent Melanoma Mouse Model. Pharmaceutics. 2021; 13(4):547. https://doi.org/10.3390/pharmaceutics13040547
Chicago/Turabian StyleGarofalo, Mariangela, Laura Bertinato, Monika Staniszewska, Magdalena Wieczorek, Stefano Salmaso, Silke Schrom, Beate Rinner, Katarzyna Wanda Pancer, and Lukasz Kuryk. 2021. "Combination Therapy of Novel Oncolytic Adenovirus with Anti-PD1 Resulted in Enhanced Anti-Cancer Effect in Syngeneic Immunocompetent Melanoma Mouse Model" Pharmaceutics 13, no. 4: 547. https://doi.org/10.3390/pharmaceutics13040547
APA StyleGarofalo, M., Bertinato, L., Staniszewska, M., Wieczorek, M., Salmaso, S., Schrom, S., Rinner, B., Pancer, K. W., & Kuryk, L. (2021). Combination Therapy of Novel Oncolytic Adenovirus with Anti-PD1 Resulted in Enhanced Anti-Cancer Effect in Syngeneic Immunocompetent Melanoma Mouse Model. Pharmaceutics, 13(4), 547. https://doi.org/10.3390/pharmaceutics13040547