
Divergent Regulation of OCT and MATE Drug Transporters by Cadmium Exposure

 , and
, and
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Antibodies and Reagents
2.2. Cell Culture and Transfection
2.3. Mouse Primary Renal Tubular Cell Isolation and Culture
2.4. Cell Surface Biotinylation
2.5. Immunoblotting Analysis
2.6. Analysis of Protein Degradation Half-Life
2.7. Cellular Uptake Assay and Cd Exposure
2.8. Animal Studies
2.9. Statistical Analysis
3. Results
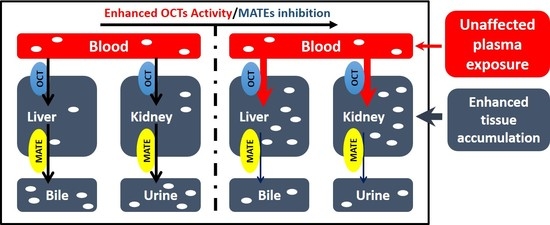
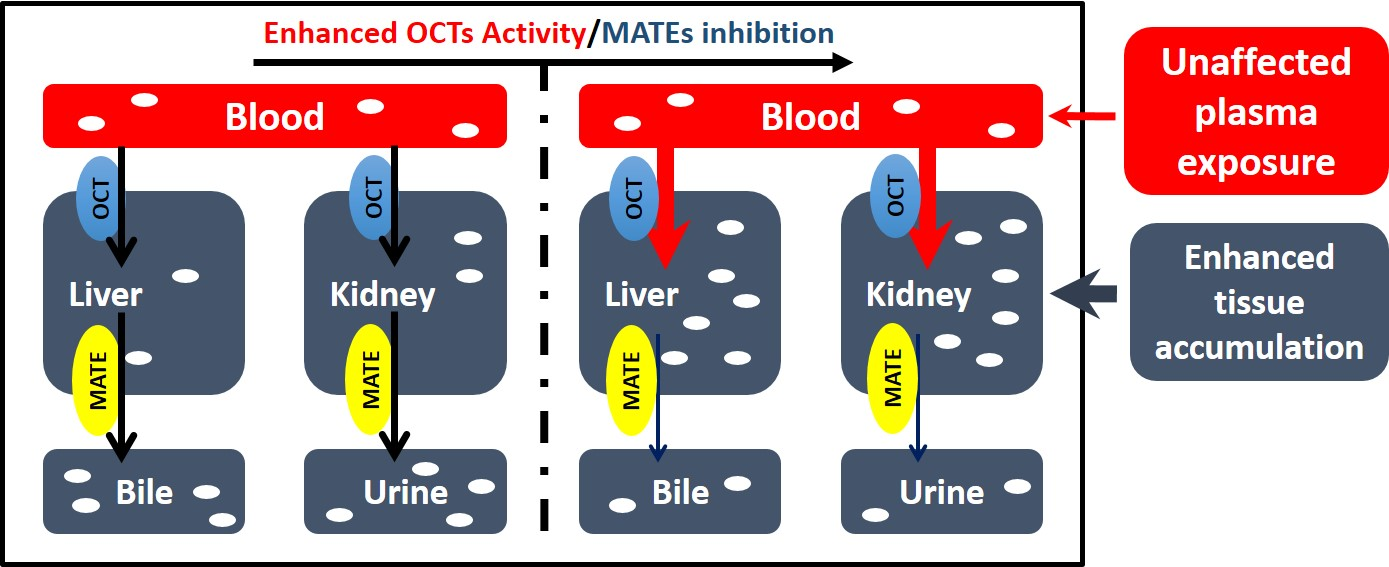
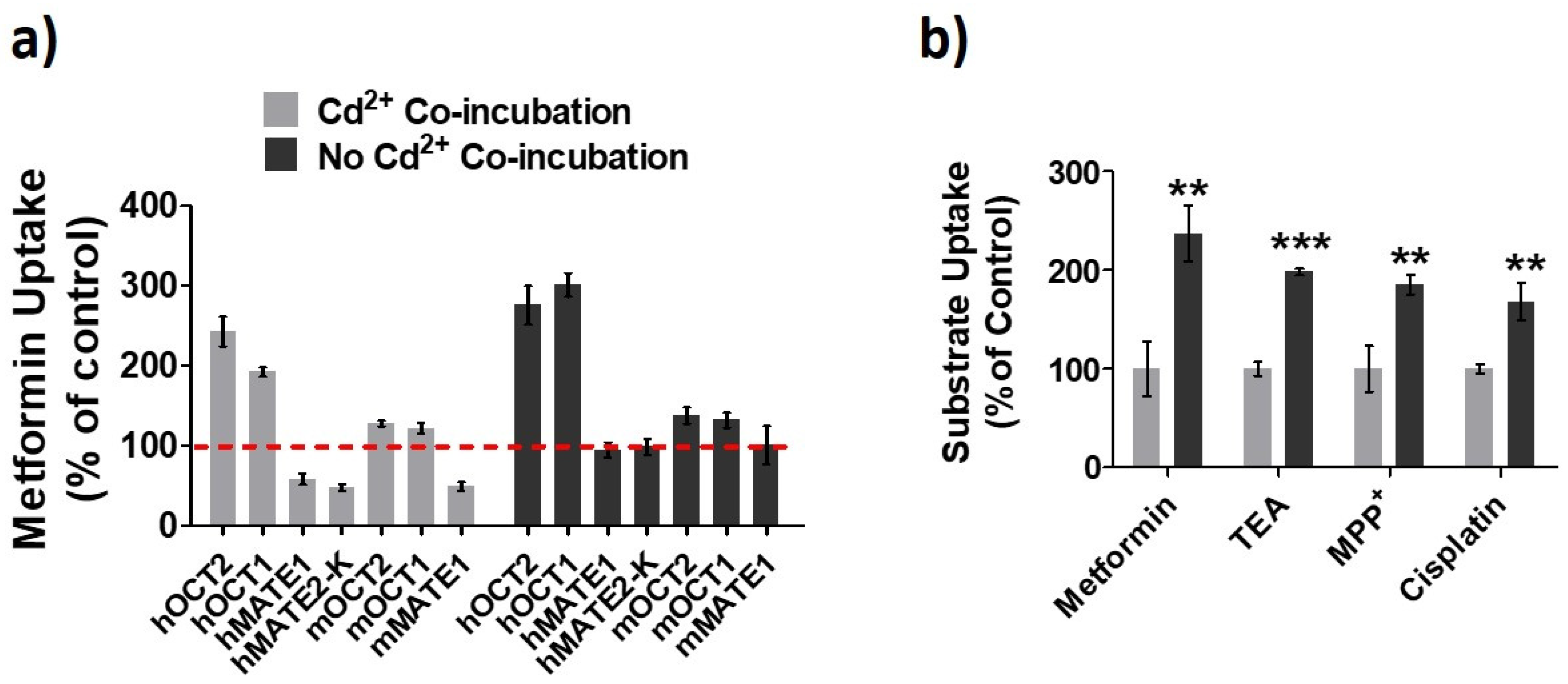
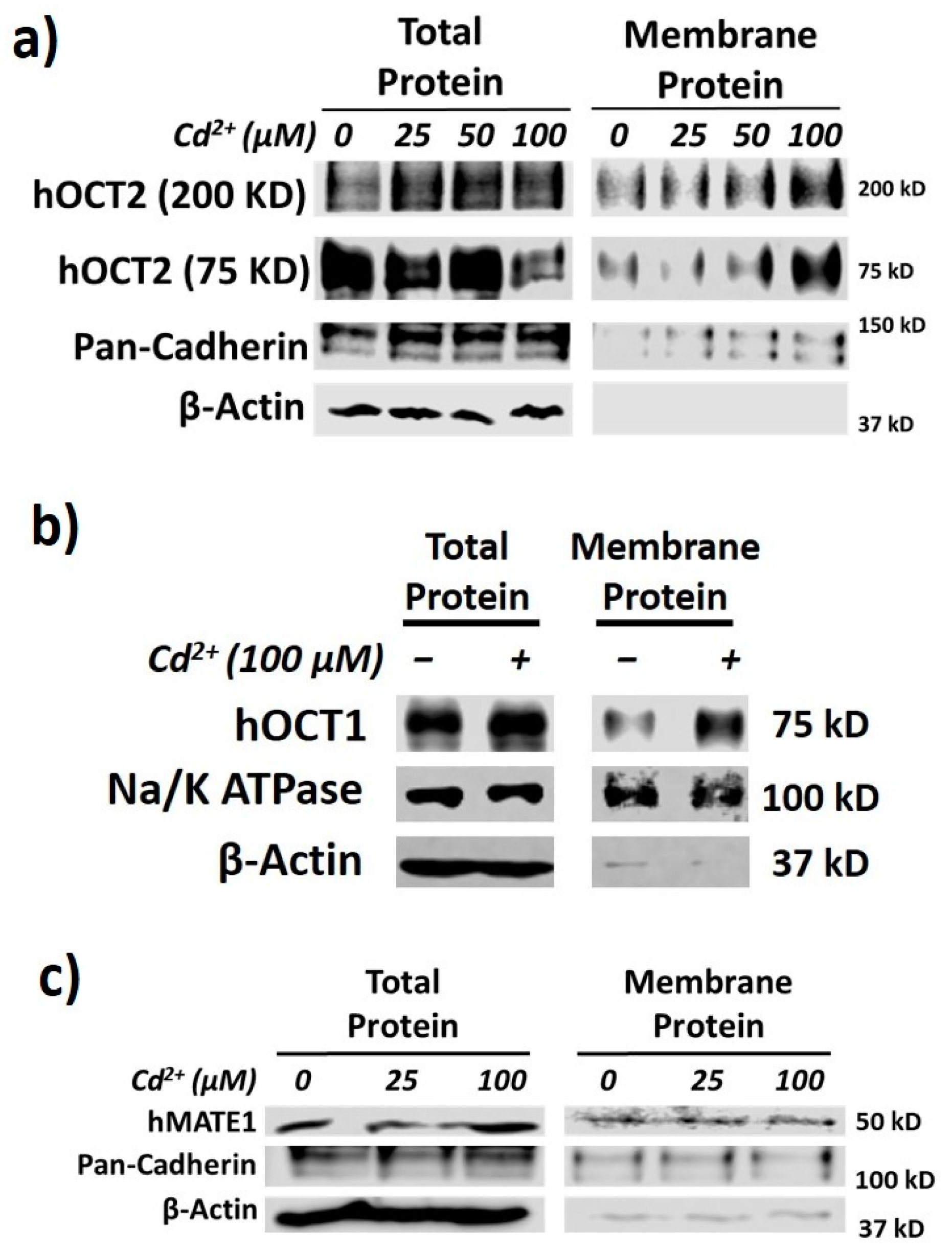
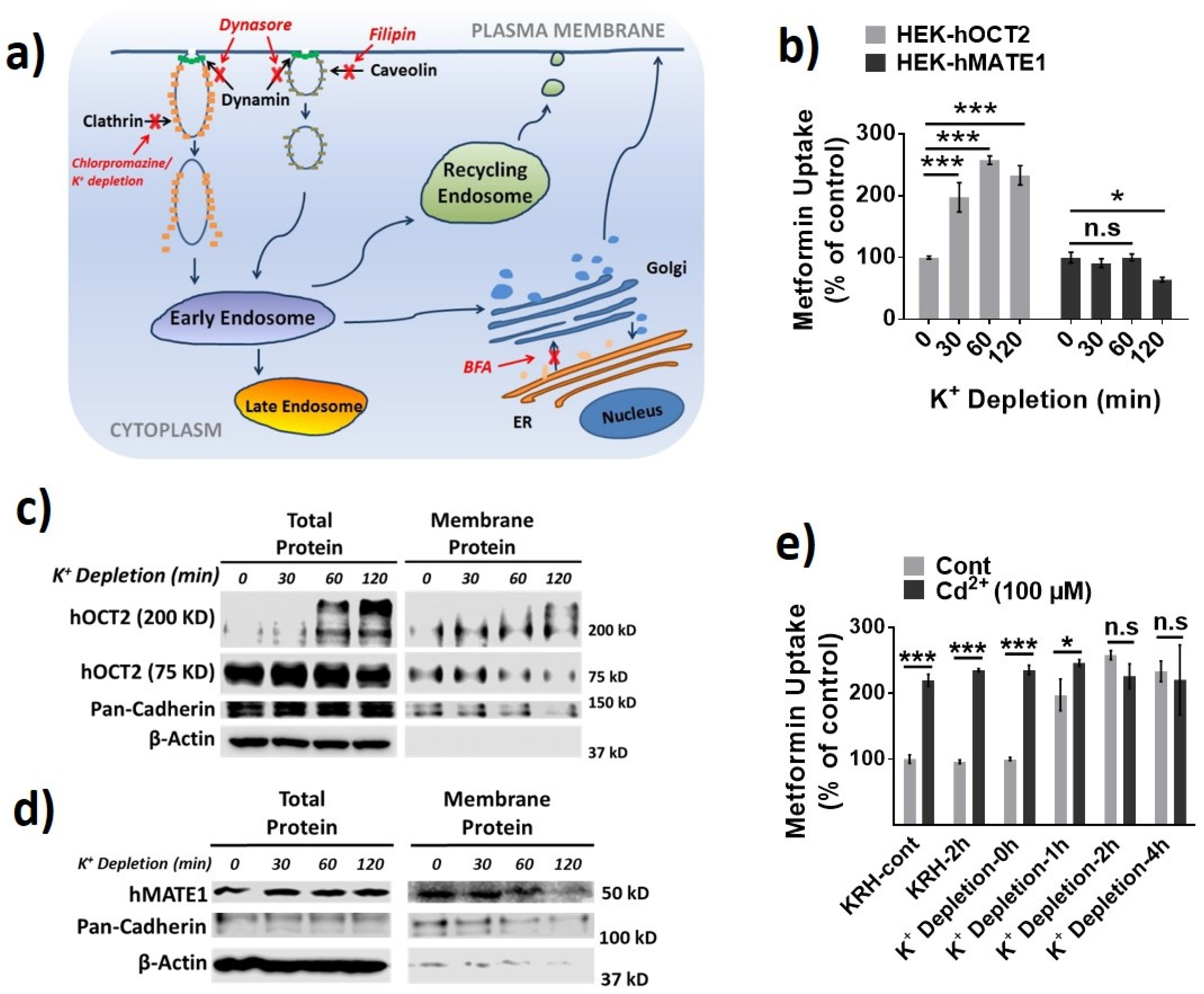
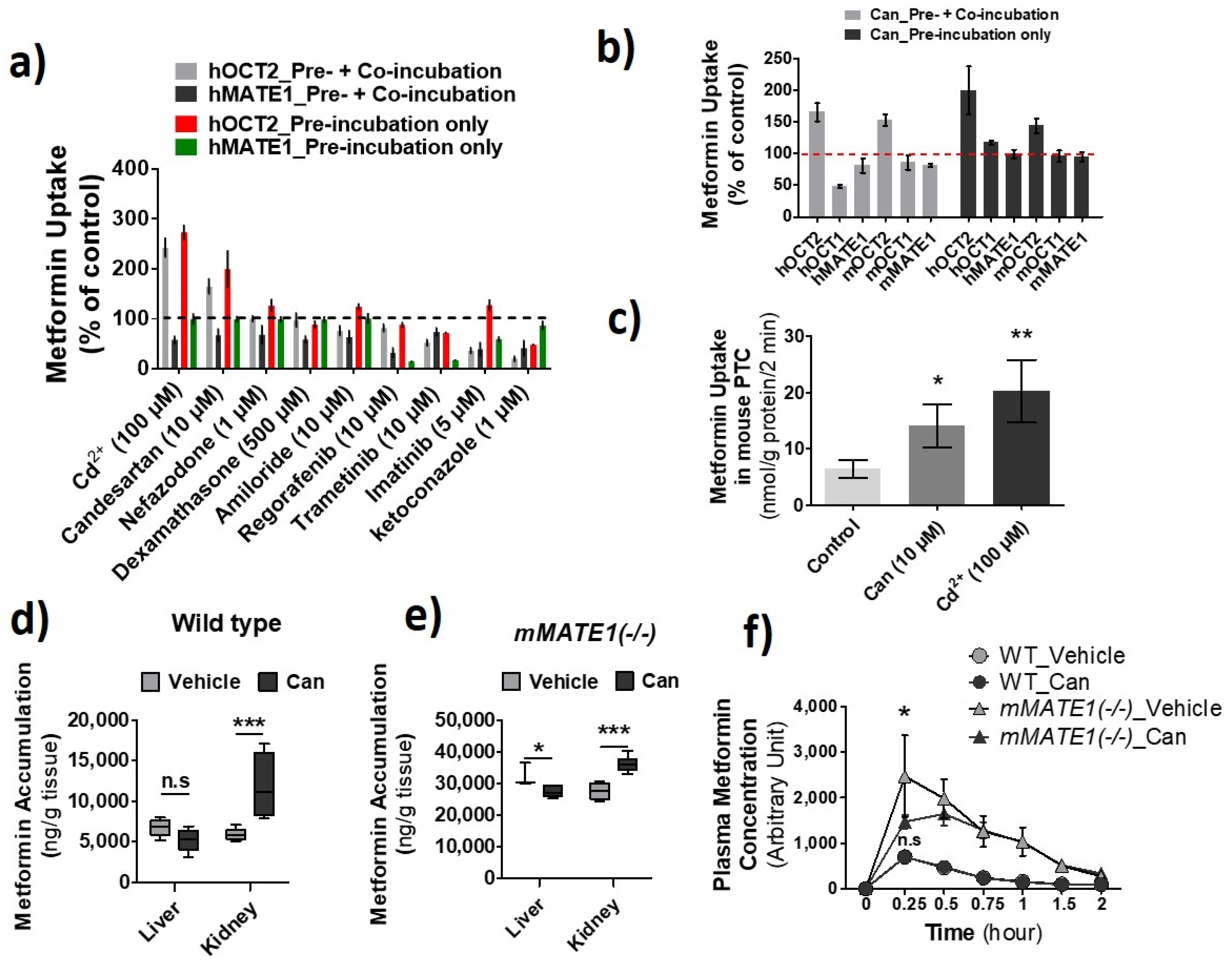
3.1. Effects of Cd2+ Exposure on the Activities of OCTs and MATEs
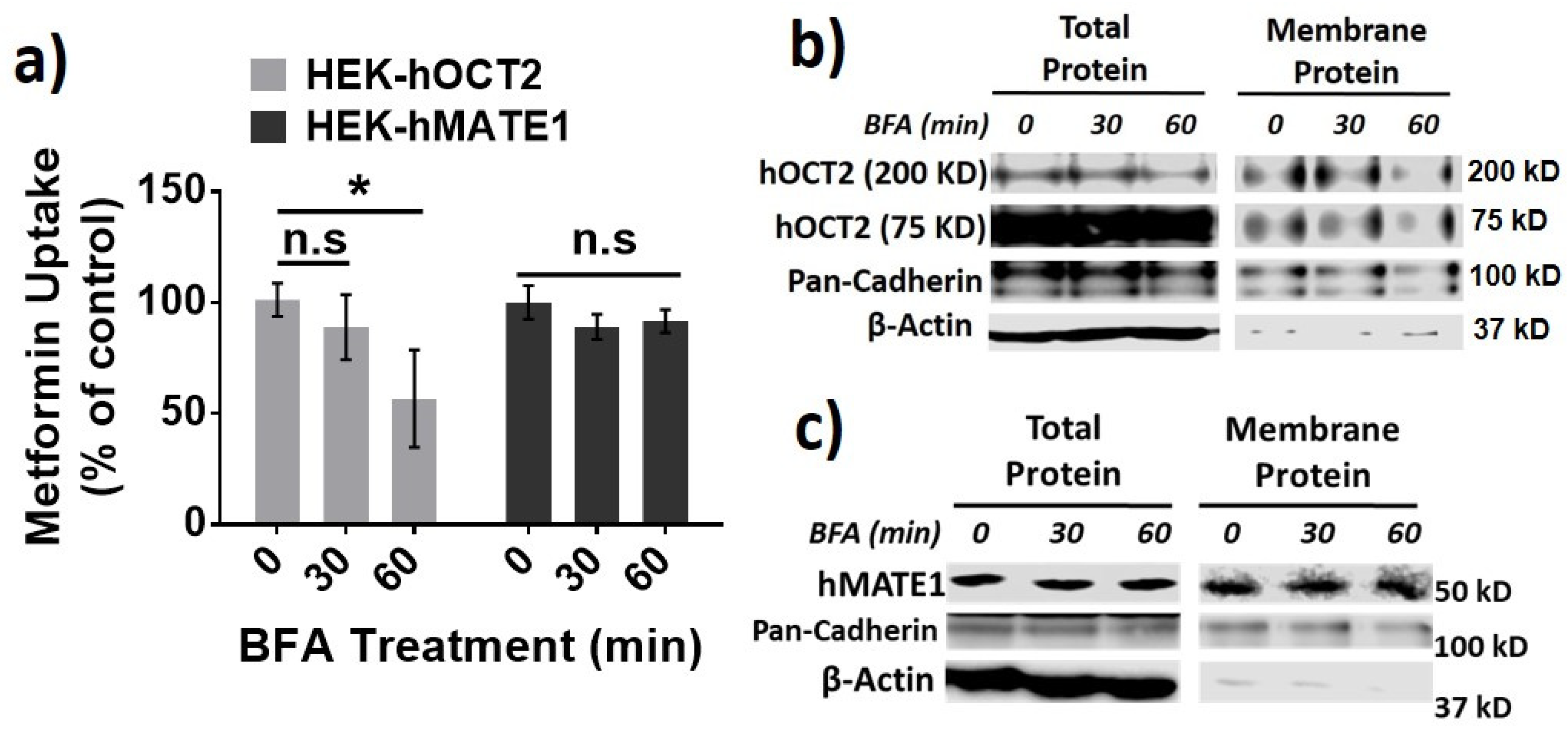
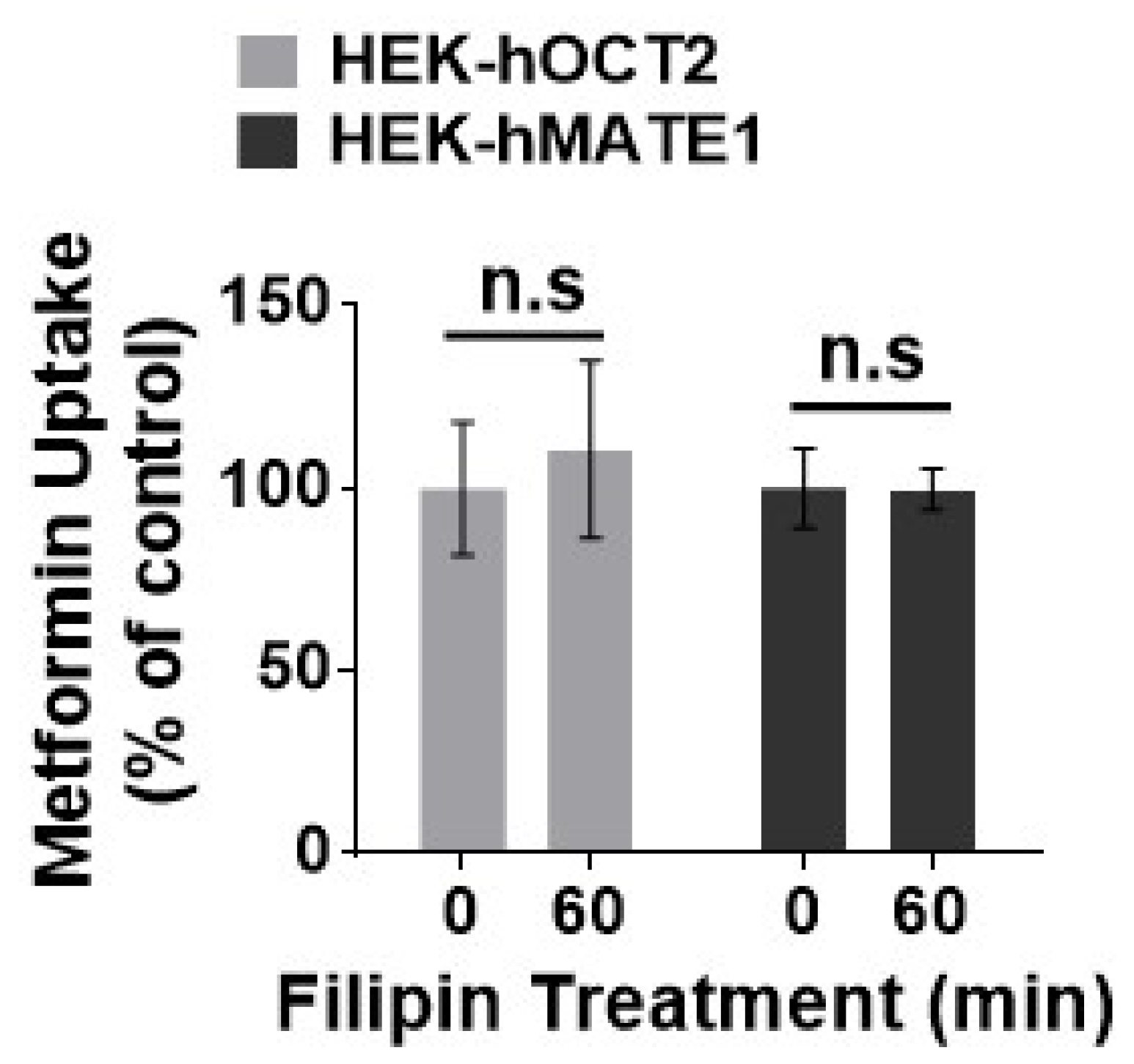
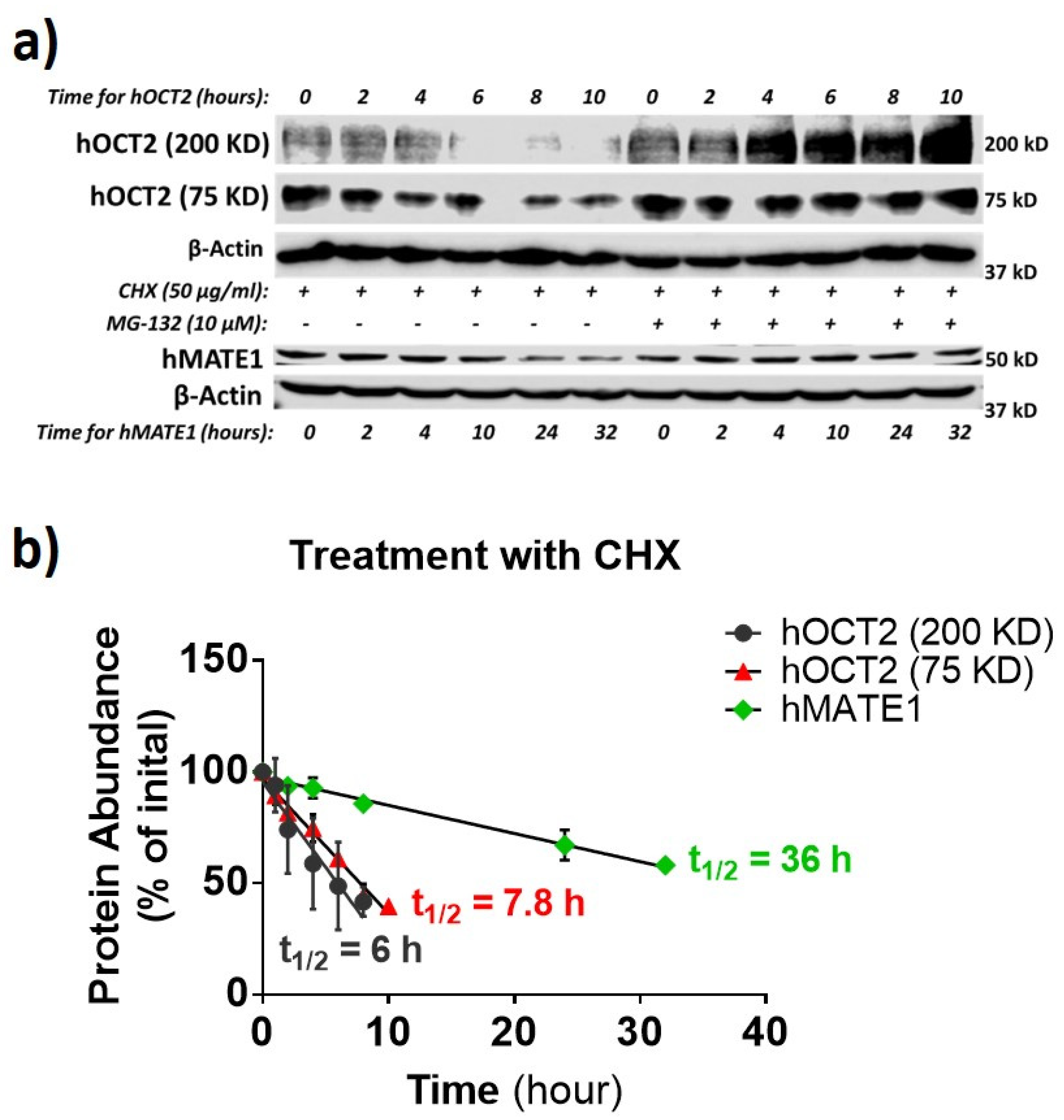
3.2. hOCT2 Was More Active in Cellular Trafficking Than hMATE1
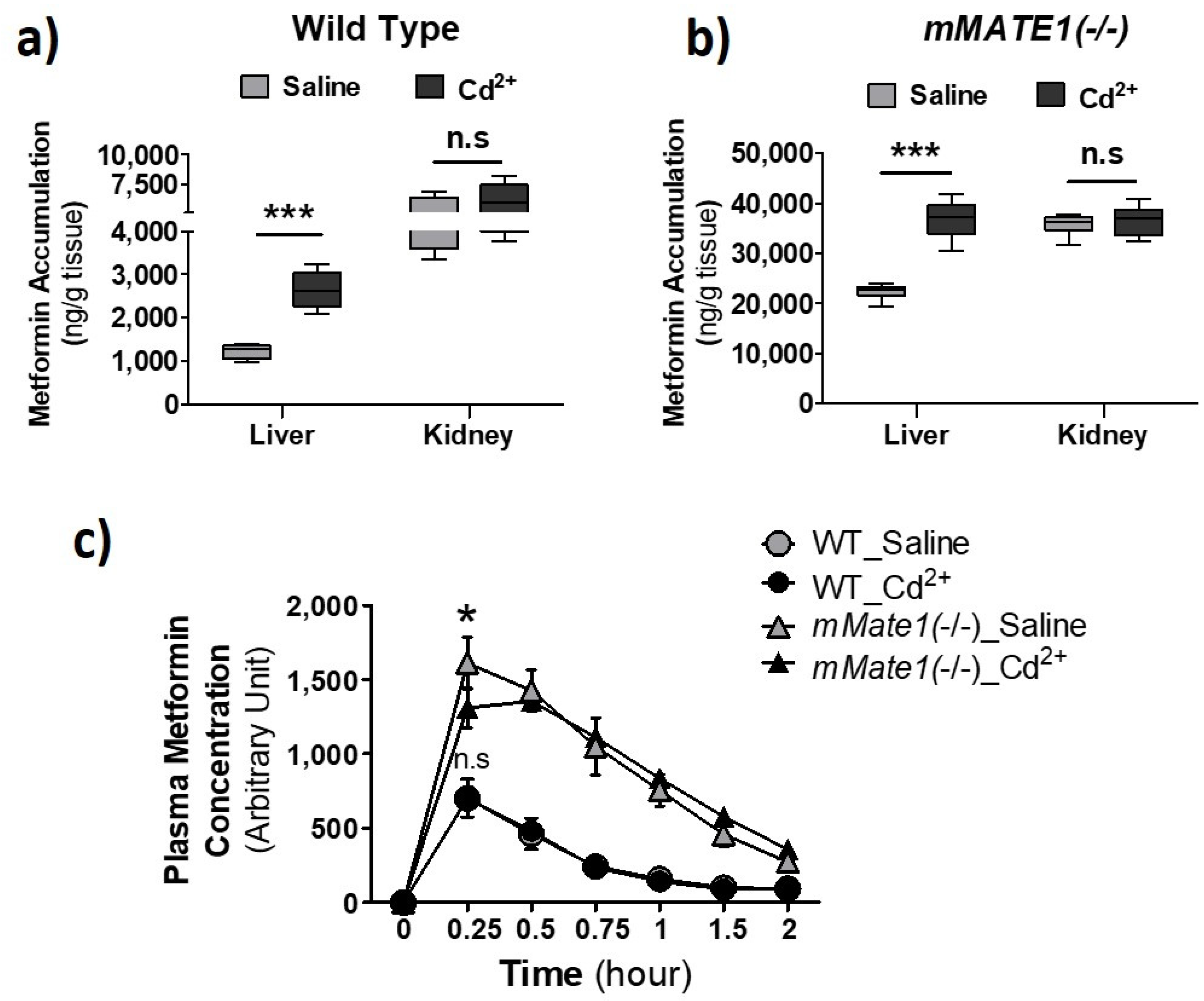
3.3. Cd2+ Exposure Altered the Pharmacokinetics (PK) of Metformin in Mice
3.4. Increase of OCT2 Activity by Candesartan
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Giacomini, K.M.; Huang, S.M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; Hillgren, K.M.; et al. Membrane transporters in drug development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Wang, J. Renal drug transporters and their significance in drug—Drug interactions. Acta Pharm. Sin. B 2016, 6, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Taskar, K.S.; Zamek-Gliszczynski, M.J. Importance of Hepatic Transporters in Clinical Disposition of Drugs and Their Metabolites. J. Clin. Pharmacol. 2016, 56, S23–S39. [Google Scholar] [CrossRef] [PubMed]
- Morrissey, K.M.; Stocker, S.L.; Wittwer, M.B.; Xu, L.; Giacomini, K.M. Renal Transporters in Drug Development. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 503–529. [Google Scholar] [CrossRef]
- Burckhardt, G. Drug transport by Organic Anion Transporters (OATs). Pharmacol. Ther. 2012, 136, 106–130. [Google Scholar] [CrossRef]
- Nies, A.T.; Koepsell, H.; Damme, K.; Schwab, M. Organic Cation Transporters (OCTs, MATEs), In Vitro and In Vivo Evidence for the Importance in Drug Therapy. Handb. Exp. Pharmacol. 2010, 105–167. [Google Scholar] [CrossRef]
- Chen, Y.; Li, S.; Brown, C.; Cheatham, S.; Castro, R.A.; Leabman, M.K.; Urban, T.J.; Chen, L.; Yee, S.W.; Choi, J.H.; et al. Effect of genetic variation in the organic cation transporter 2 on the renal elimination of metformin. Pharmacogenet. Genom. 2009, 19, 497–504. [Google Scholar] [CrossRef]
- Song, I.S.; Shin, H.J.; Shim, E.J.; Jung, I.S.; Kim, W.Y.; Shon, J.H.; Shin, J.G. Genetic Variants of the Organic Cation Transporter 2 Influence the Disposition of Metformin. Clin. Pharmacol. Ther. 2008, 84, 559–562. [Google Scholar] [CrossRef]
- Filipski, K.K.; Mathijssen, R.H.; Mikkelsen, T.S.; Schinkel, A.H.; Sparreboom, A. Contribution of Organic Cation Transporter 2 (OCT2) to Cisplatin-Induced Nephrotoxicity. Clin. Pharmacol. Ther. 2009, 86, 396–402. [Google Scholar] [CrossRef]
- Becker, M.L.; Visser, L.E.; van Schaik, R.H.; Hofman, A.; Uitterlinden, A.G.; Stricker, B.H. Genetic variation in the multidrug and toxin extrusion 1 transporter protein influences the glucose-lowering effect of metformin in patients with diabetes: A preliminary study. Diabetes 2009, 58, 745–749. [Google Scholar] [CrossRef]
- Li, Q.; Guo, D.; Dong, Z.; Zhang, W.; Zhang, L.; Huang, S.M.; Polli, J.E.; Shu, Y. Ondansetron can enhance cisplatin-induced nephrotoxicity via inhibition of multiple toxin and extrusion proteins (MATEs). Toxicol. Appl. Pharmacol. 2013, 273, 100–109. [Google Scholar] [CrossRef]
- Hillgren, K.M.; Keppler, D.; Zur, A.; Giacomini, K.M.; Stieger, B.; Cass, E.C.; Zhang, L. Emerging Transporters of Clinical Importance: An Update from the International Transporter Consortium. Clin. Pharmacol. Ther. 2013, 94, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Biermann, J.; Lang, D.; Gorboulev, V.; Koepsell, H.; Sindić, A.; Schröter, R.; Zvirbliene, A.; Pavenstädt, H.; Schlatter, E.; Ciarimboli, G. Characterization of regulatory mechanisms and states of human organic cation transporter 2. Am. J. Physiol. Physiol. 2006, 290, C1521–C1531. [Google Scholar] [CrossRef] [PubMed]
- Çetinkaya, I.; Ciarimboli, G.; Yalçinkaya, G.; Mehrens, T.; Velic, A.; Hirsch, J.R.; Gorboulev, V.; Koepsell, H.; Schlatter, E. Regulation of human organic cation transporter hOCT2 by PKA, PI3K, and calmodulin-dependent kinases. Am. J. Physiol. 2003, 284, F293–F302. [Google Scholar] [CrossRef] [PubMed]
- Wilde, S.; Schlatter, E.; Koepsell, H.; Edemir, B.; Reuter, S.; Pavenstädt, H.; Neugebauer, U.; Schröter, R.; Brast, S.; Ciarimboli, G. Calmodulin-associated post-translational regulation of rat organic cation transporter 2 in the kidney is gender dependent. Cell. Mol. Life Sci. 2009, 66, 1729–1740. [Google Scholar] [CrossRef]
- Guckel, D.; Ciarimboli, G.; Pavenstädt, H.; Schlatter, E. Regulation of Organic Cation Transport in Isolated Mouse Proximal Tubules Involves Complex Changes in Protein Trafficking and Substrate Affinity. Cell. Physiol. Biochem. 2012, 30, 269–281. [Google Scholar] [CrossRef]
- Sprowl, J.A.; Ong, S.S.; Gibson, A.A.; Hu, S.; Du, G.; Lin, W.; Li, L.; Bharill, S.; Ness, R.A.; Stecula, A.; et al. A phosphotyrosine switch regulates organic cation transporters. Nat. Commun. 2016, 7, 10880. [Google Scholar] [CrossRef]
- Yang, H.; Tang, J.; Guo, D.; Zhao, Q.; Wen, J.; Zhang, Y.; Obianom, O.N.; Zhou, S.; Zhang, W.; Shu, Y. Cadmium exposure enhances organic cation transporter 2 trafficking to the kidney membrane and exacerbates cisplatin nephrotoxicity. Kidney Int. 2020, 97, 765–777. [Google Scholar] [CrossRef]
- Xu, D.; Wang, H.; You, G. An Essential Role of Nedd4-2 in the Ubiquitination, Expression, and Function of Organic Anion Transporter-3. Mol. Pharm. 2015, 13, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Hong, M.; Duan, P.; Pan, Z.; Ma, J.; You, G. Organic Anion Transporter OAT1 Undergoes Constitutive and Protein Kinase C-regulated Trafficking through a Dynamin- and Clathrin-dependent Pathway. J. Biol. Chem. 2008, 283, 32570–32579. [Google Scholar] [CrossRef] [PubMed]
- Soodvilai, S.; Nantavishit, J.; Muanprasat, C.; Chatsudthipong, V. Renal organic cation transporters mediated cadmium-induced nephrotoxicity. Toxicol. Lett. 2011, 204, 38–42. [Google Scholar] [CrossRef]
- Thévenod, F.; Ciarimboli, G.; Leistner, M.; Wolff, N.A.; Lee, W.-K.; Schatz, I.; Keller, T.; Al-Monajjed, R.; Gorboulev, V.; Koepsell, H. Substrate- and Cell Contact-Dependent Inhibitor Affinity of Human Organic Cation Transporter 2: Studies with Two Classical Organic Cation Substrates and the Novel Substrate Cd2+. Mol. Pharm. 2013, 10, 3045–3056. [Google Scholar] [CrossRef]
- Yang, H.; Guo, D.; Obianom, O.N.; Su, T.; Polli, J.E.; Shu, Y. Multidrug and toxin extrusion proteins mediate cellular transport of cadmium. Toxicol. Appl. Pharmacol. 2017, 314, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Shu, Y. Cadmium Transporters in the Kidney and Cadmium-Induced Nephrotoxicity. Int. J. Mol. Sci. 2015, 16, 1484–1494. [Google Scholar] [CrossRef]
- Thévenod, F.; Lee, W.-K. Cadmium and cellular signaling cascades: Interactions between cell death and survival pathways. Arch. Toxicol. 2013, 87, 1743–1786. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Yang, H.; Li, Q.; Bae, H.J.; Obianom, O.; Zeng, S.; Su, T.; Polli, J.E.; Shu, Y. Selective Inhibition on Organic Cation Transporters by Carvedilol Protects Mice from Cisplatin-Induced Nephrotoxicity. Pharm. Res. 2018, 35, 204. [Google Scholar] [CrossRef]
- Li, Q.; Yang, H.; Peng, X.; Guo, D.; Dong, Z.; Polli, J.E.; Shu, Y. Ischemia/Reperfusion-Inducible Protein Modulates the Function of Organic Cation Transporter 1 and Multidrug and Toxin Extrusion 1. Mol. Pharm. 2013, 10, 2578–2587. [Google Scholar] [CrossRef][Green Version]
- Kingston, R.E.; Chen, C.A.; Rose, J.K. Calcium Phosphate Transfection. Curr. Protoc. Mol. Biol. 2003, 63. [Google Scholar] [CrossRef]
- Ding, W.; Yousefi, K.; Shehadeh, L.A. Isolation, Characterization, And High Throughput Extracellular Flux Analysis of Mouse Primary Renal Tubular Epithelial Cells. J. Vis. Exp. 2018, e57718. [Google Scholar] [CrossRef] [PubMed]
- Vesey, D.A.; Qi, W.; Chen, X.; Pollock, C.A.; Johnson, D.W. Isolation and Primary Culture of Human Proximal Tubule Cells. Adv. Struct. Safety Stud. 2008, 466, 19–24. [Google Scholar] [CrossRef]
- Brast, S.; Grabner, A.; Sucic, S.; Sitte, H.H.; Hermann, E.; Pavenstadt, H.; Schlatter, E.; Ciarimboli, G. The cysteines of the extracellular loop are crucial for trafficking of human organic cation transporter 2 to the plasma membrane and are involved in oligomerization. FASEB J. 2012, 26, 976–986. [Google Scholar] [CrossRef]
- Stewart, D.P.; Koss, B.; Bathina, M.; Perciavalle, R.M.; Bisanz, K.; Opferman, J.T. Ubiquitin-Independent Degradation of Antiapoptotic MCL-1. Mol. Cell. Biol. 2010, 30, 3099–3110. [Google Scholar] [CrossRef]
- Filipski, K.; Loos, W.; Verweij, J.; Sparreboom, A. Interaction of Cisplatin with the Human Organic Cation Transporter 2. Clin. Cancer Res. 2008, 14, 3875–3880. [Google Scholar] [CrossRef]
- Hacker, K.; Maas, R.; Kornhuber, J.; Fromm, M.F.; Zolk, O. Substrate-Dependent Inhibition of the Human Organic Cation Transporter OCT2: A Comparison of Metformin with Experimental Substrates. PLoS ONE 2015, 10, e0136451. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Kusuhara, H.; Yokochi, M.; Toyoshima, J.; Inoue, K.; Yuasa, H.; Sugiyama, Y. Time profiles of cationic compounds (TEA, Metformin, MPP, ASP, MIBG) uptake by hOCT1, hOCT2, hMATE1, and hMATE2-K. J. Pharmacol. Exp. Ther. 2012, 340, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Nies, A.T.; Hofmann, U.; Resch, C.; Schaeffeler, E.; Rius, M.; Schwab, M. Proton Pump Inhibitors Inhibit Metformin Uptake by Organic Cation Transporters (OCTs). PLoS ONE 2011, 6, e22163. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Peng, X.; Yang, H.; Wang, H.; Shu, Y. Deficiency of Multidrug and Toxin Extrusion 1 Enhances Renal Accumulation of Paraquat and Deteriorates Kidney Injury in Mice. Mol. Pharm. 2011, 8, 2476–2483. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.M.; Brown, M.S.; Goldstein, J.L.; Anderson, R.G. Depletion of intracellular potassium arrests coated pit formation and receptor-mediated endocytosis in fibroblasts. Cell 1983, 33, 273–285. [Google Scholar] [CrossRef]
- Bershadsky, A.D.; Futerman, A.H. Disruption of the Golgi apparatus by brefeldin A blocks cell polarization and inhibits directed cell migration. Proc. Natl. Acad. Sci. USA 1994, 91, 5686–5689. [Google Scholar] [CrossRef] [PubMed]
- Schnitzer, J.E.; Oh, P.; Pinney, E.; Allard, J. Filipin-sensitive caveolae-mediated transport in endothelium: Reduced transcytosis, scavenger endocytosis, and capillary permeability of select macromolecules. J. Cell Biol. 1994, 127, 1217–1232. [Google Scholar] [CrossRef]
- Park, W.H.; Han, Y.H.; Moon, H.J.; You, B.R. The effect of MG132, a proteasome inhibitor on HeLa cells in relation to cell growth, reactive oxygen species and GSH. Oncol. Rep. 2009, 22, 215–221. [Google Scholar] [CrossRef]
- Schneider-Poetsch, T.; Ju, J.; Eyler, E.D.; Dang, Y.; Bhat, S.; Merrick, W.C.; Green, R.; Shen, B.; Liu, O.J. Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat. Chem. Biol. 2010, 6, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Schulze, U.; Brast, S.; Grabner, A.; Albiker, C.; Snieder, B.; Holle, S.; Schlatter, E.; Schröter, R.; Pavenstädt, H.; Herrmann, E.; et al. Tetraspanin CD63 controls basolateral sorting of organic cation transporter 2 in renal proximal tubules. FASEB J. 2017, 31, 1421–1433. [Google Scholar] [CrossRef]
- Macia, E.; Ehrlich, M.; Massol, R.; Boucrot, E.; Brunner, C.; Kirchhausen, T. Dynasore, a Cell-Permeable Inhibitor of Dynamin. Dev. Cell 2006, 10, 839–850. [Google Scholar] [CrossRef]
- Vercauteren, D.; Vandenbroucke, E.R.; Jones, A.T.; Rejman, J.; Demeester, J.; De Smedt, S.C.; Sanders, N.N.; Braeckmans, K. The Use of Inhibitors to Study Endocytic Pathways of Gene Carriers: Optimization and Pitfalls. Mol. Ther. 2010, 18, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Świergosz-Kowalewska, R. Cadmium distribution and toxicity in tissues of small rodents. Microsc. Res. Tech. 2001, 55, 208–222. [Google Scholar] [CrossRef]
- Thévenod, F.; Lee, W.-K. Toxicology of Cadmium and Its Damage to Mammalian Organs. Metal Ions Life Sci. 2012, 11, 415–490. [Google Scholar] [CrossRef]
- Thomas, R.G.; Wilson, J.S.; London, J.E. Multispecies retention parameters for cadmium. Environ. Res. 1980, 23, 191–207. [Google Scholar] [CrossRef]
- Zalups, R.K.; Ahmad, S. Molecular handling of cadmium in transporting epithelia. Toxicol. Appl. Pharmacol. 2003, 186, 163–188. [Google Scholar] [CrossRef]
- Kimura, N.; Masuda, S.; Tanihara, Y.; Ueo, H.; Okuda, M.; Katsura, T.; Inui, K.-I. Metformin is a Superior Substrate for Renal Organic Cation Transporter OCT2 rather than Hepatic OCT1. Drug Metab. Pharmacokinet. 2005, 20, 379–386. [Google Scholar] [CrossRef]
- Jonker, J.W.; Wagenaar, E.; Van Eijl, S.; Schinkel, A.H. Deficiency in the Organic Cation Transporters 1 and 2 (Oct1/Oct2 [Slc22a1/Slc22a2]) in Mice Abolishes Renal Secretion of Organic Cations. Mol. Cell. Biol. 2003, 23, 7902–7908. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, H.; Zhou, S.; Guo, D.; Obianom, O.N.; Li, Q.; Shu, Y. Divergent Regulation of OCT and MATE Drug Transporters by Cadmium Exposure. Pharmaceutics 2021, 13, 537. https://doi.org/10.3390/pharmaceutics13040537
Yang H, Zhou S, Guo D, Obianom ON, Li Q, Shu Y. Divergent Regulation of OCT and MATE Drug Transporters by Cadmium Exposure. Pharmaceutics. 2021; 13(4):537. https://doi.org/10.3390/pharmaceutics13040537
Chicago/Turabian StyleYang, Hong, Shiwei Zhou, Dong Guo, Obinna N. Obianom, Qing Li, and Yan Shu. 2021. "Divergent Regulation of OCT and MATE Drug Transporters by Cadmium Exposure" Pharmaceutics 13, no. 4: 537. https://doi.org/10.3390/pharmaceutics13040537
APA StyleYang, H., Zhou, S., Guo, D., Obianom, O. N., Li, Q., & Shu, Y. (2021). Divergent Regulation of OCT and MATE Drug Transporters by Cadmium Exposure. Pharmaceutics, 13(4), 537. https://doi.org/10.3390/pharmaceutics13040537