
Rational Vaccine Design in Times of Emerging Diseases: The Critical Choices of Immunological Correlates of Protection, Vaccine Antigen and Immunomodulation

 , ,
, ,  , ,
, , 
 , , ,
, , , 
 ,
,  , and
, and
Abstract
1. Introduction
2. Vaccine Types for Artificial Adaptive Immunity
2.1. Live Attenuated Vaccines
2.2. Inactivated “Killed” Vaccines
2.3. Subunit Protein and Peptide Protein and Polysaccharide Vaccines
2.4. RNA Vaccines
2.5. DNA Vaccines
2.6. Recombinant Viral Vector Vaccines
2.7. Recombinant Bacterial Vector Vaccines
3. The Immunological Correlate(s) of Protection
3.1. Passive Immunity Transfer or Immunity Depletion
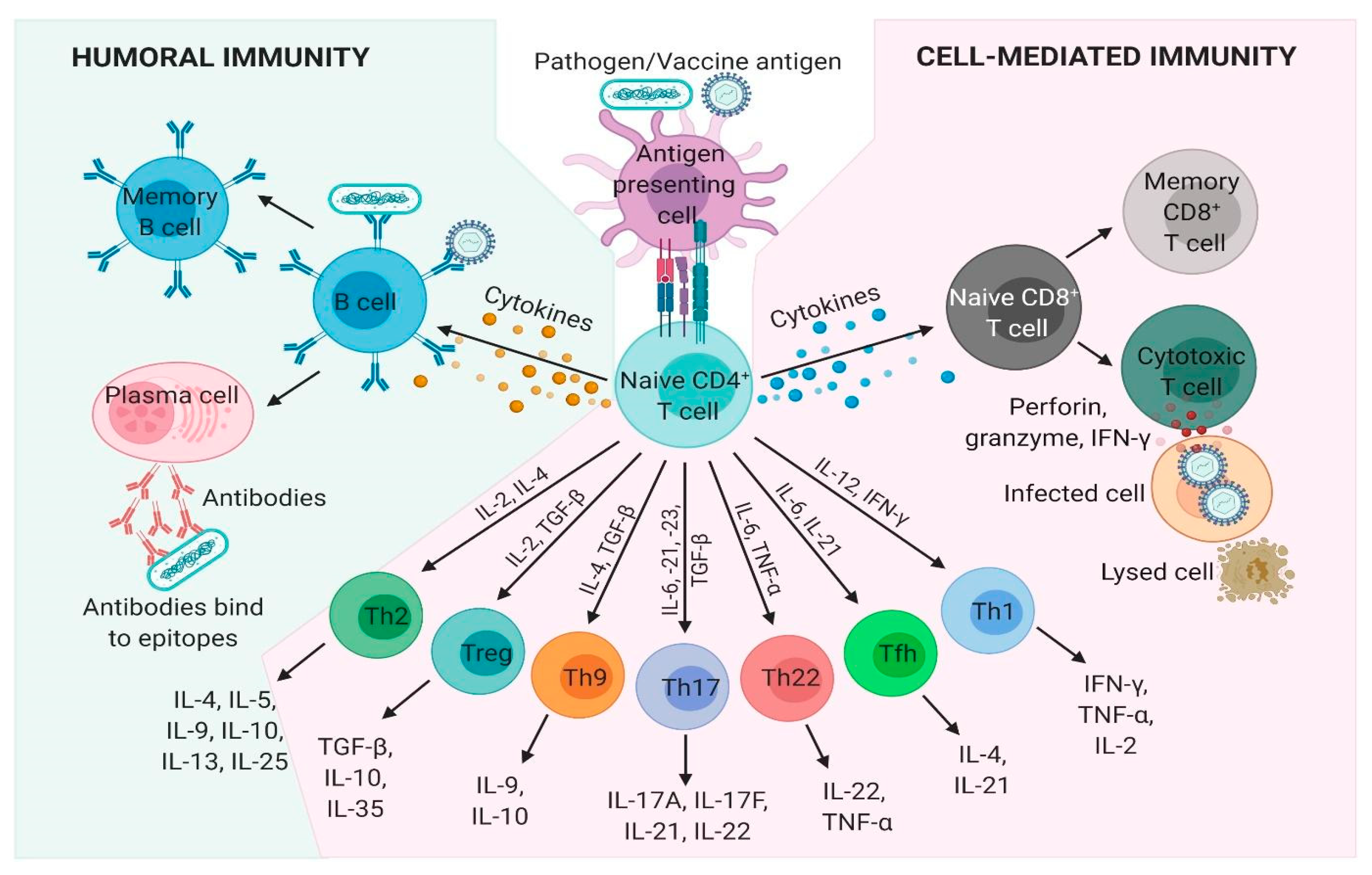
3.2. Types of Protective Immune Responses
3.2.1. Antibody-Based Immunity
3.2.2. Cell-Mediated Immunity
3.2.3. Innate Immunity
4. Choice of Vaccine Antigen
4.1. Rational Antigens for Antibody Immunity
4.2. Rational Antigens for Cell-Mediated Immunity
5. Choice of Immunomodulation
5.1. Rational Vaccine Adjuvant Design
5.2. Rationally Designed Vaccine Adjuvants
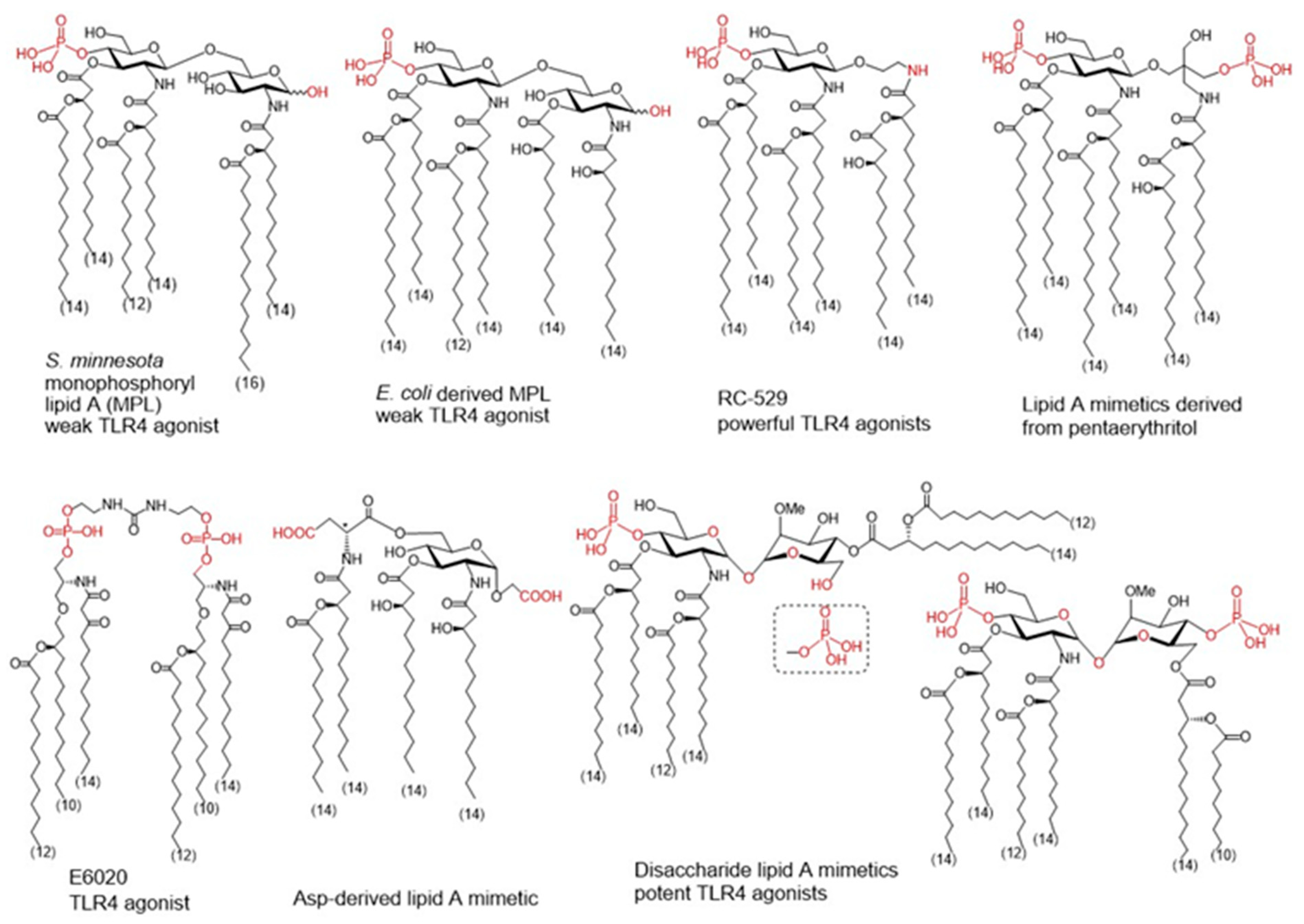
5.2.1. Natural Lipid A as Source of Inspiration for New Adjuvants
5.2.2. Rational Design of Allostatine Adjuvant
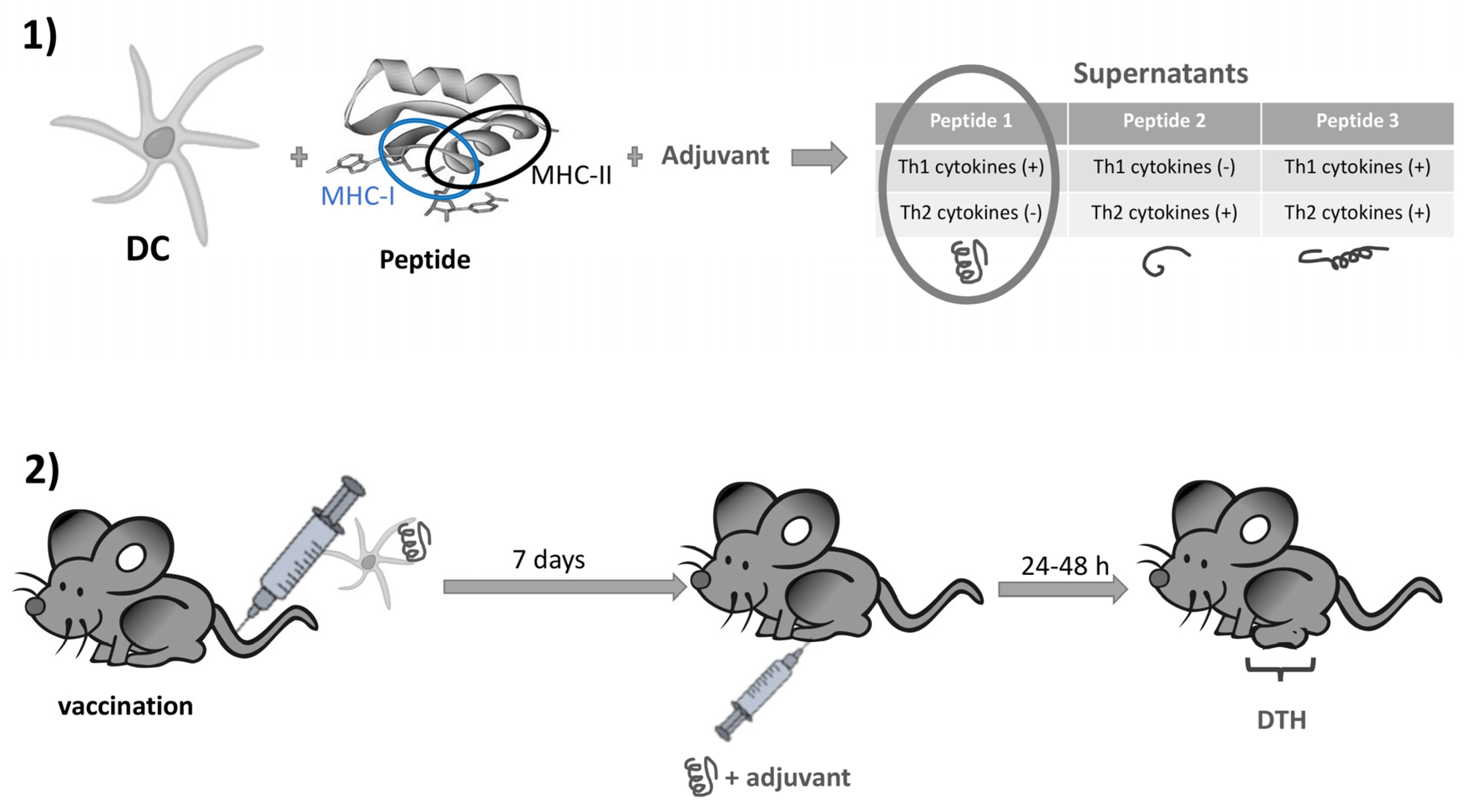
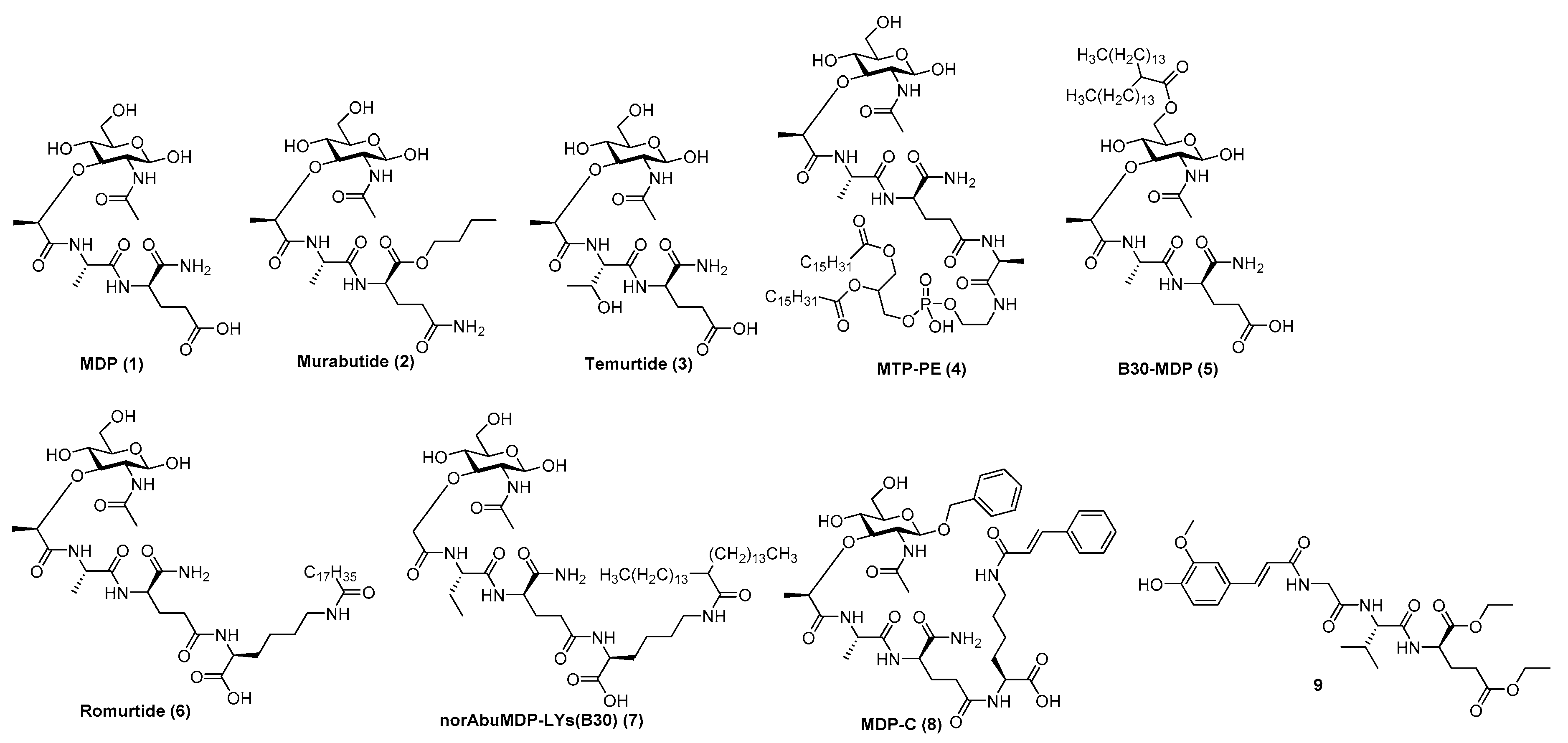
5.2.3. Nod2 Agonists as Rationally Designed Vaccine Adjuvants
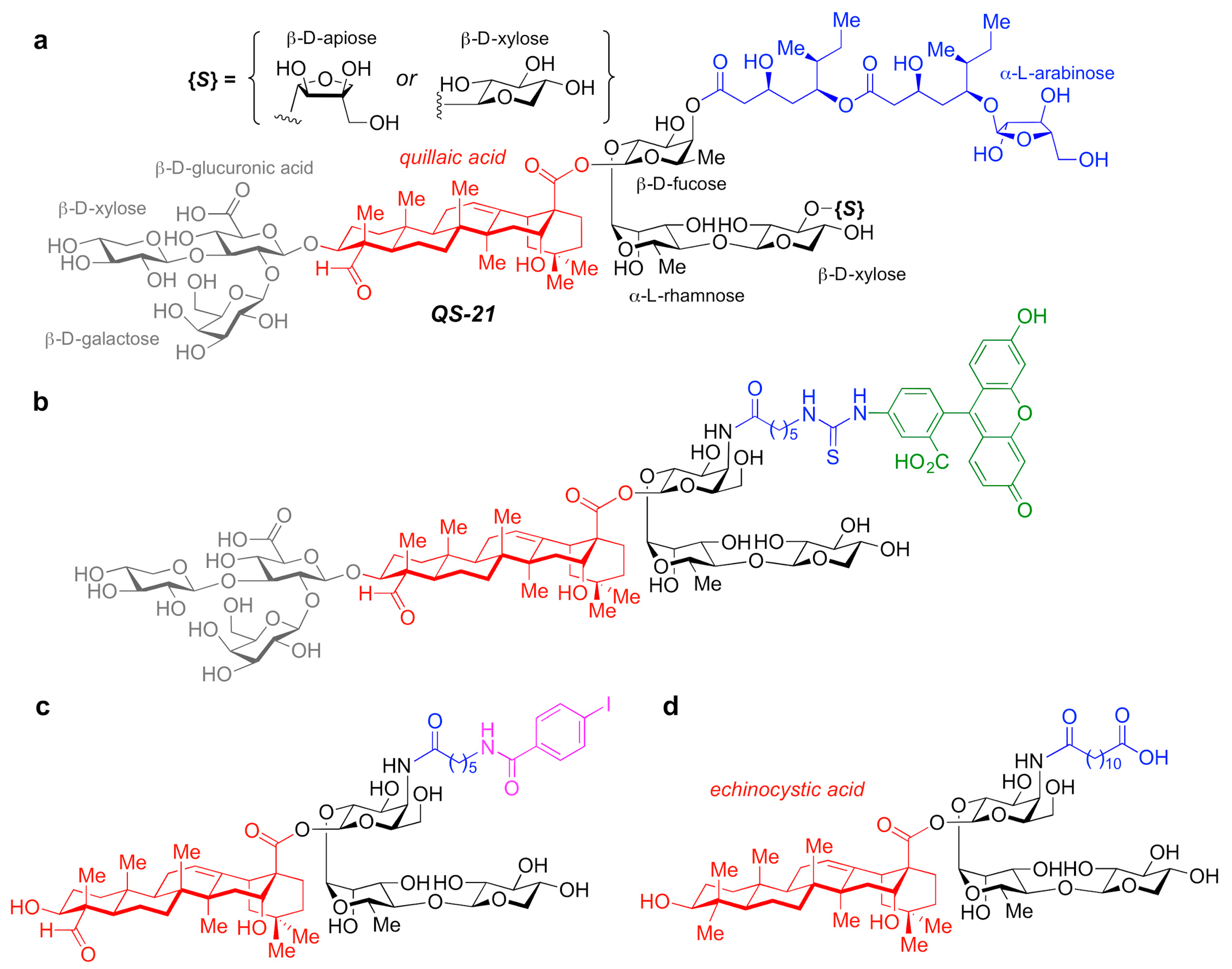
5.2.4. QS-21-Based Synthetic Saponin Adjuvants
5.3. The VLP-Based Vaccine Platform and CpG ODNs as Immunoprotective Vaccine Adjuvants
6. Outlook/Closing Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Robbins, A. Progress towards vaccines we need and do not have. Lancet 1990, 335, 1436–1438. [Google Scholar] [CrossRef]
- Moyer, T.J.; Zmolek, A.C.; Irvine, D.J. Beyond antigens and adjuvants: Formulating future vaccines. J. Clin. Investig. 2016, 126, 799–808. [Google Scholar] [CrossRef]
- Guillot, S.; Otelea, D.; Delpeyroux, F.; Crainic, R. Point mutations involved in the attenuation/neurovirulence alterna-tion in type 1 and 2 oral polio vaccine strains detected by site-specific polymerase chain reaction. Vaccine 1994, 12, 503–507. [Google Scholar] [CrossRef]
- Carter, N.J.; Curran, M.P. Live attenuated influenza vaccine (FluMist(R); Fluenz): A review of its use in the preven-tion of seasonal influenza in children and adults. Drugs 2011, 71, 1591–1622. [Google Scholar] [CrossRef]
- Torresi, J.; Ebert, G.; Pellegrini, M. Vaccines licensed and in clinical trials for the prevention of dengue. Hum. Vaccines Immunother. 2017, 13, 1059–1072. [Google Scholar] [CrossRef]
- McKenna, A.J.; Bygraves, J.A.; Maiden, M.C.; Feavers, I.M. Attenuated typhoid vaccine Salmonella typhi Ty21a: Fingerprinting and quality control. Microbiology 1995, 141, 1993–2002. [Google Scholar] [CrossRef] [PubMed]
- Kopecko, D.J.; Sieber, H.; Ures, J.A.; Fürer, A.; Schlup, J.; Knof, U.; Collioud, A.; Xu, D.; Colburn, K.; Dietrich, G. Genetic stability of vaccine strain Salmonella Typhi Ty21a over 25 years. Int. J. Med. Microbiol. 2009, 299, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.L.; Jeza, V.T.; Pan, Q. Salmonella typhi: From a human pathogen to a vaccine vector. Cell Mol. Immunol. 2008, 5, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Dharmasena, M.N.; Feuille, C.M.; Starke, C.E.; Bhagwat, A.A.; Stibitz, S.; Kopecko, D.J. Development of an Acid-Resistant Salmonella Typhi Ty21a Attenuated Vector For Improved Oral Vaccine Delivery. PLoS ONE 2016, 11, e0163511. [Google Scholar] [CrossRef] [PubMed]
- Van den Ende, C.; Marano, C.; van Ahee, A.; Bunge, E.M.; de Moerlooze, L. The immunogenicity of GSK’s recombinant hepatitis B vaccine in children: A systematic review of 30 years of experience. Expert Rev. Vaccines 2017, 16, 789–809. [Google Scholar] [CrossRef] [PubMed]
- Syed, Y.Y. Recombinant Zoster Vaccine (Shingrix((R))): A Review in Herpes Zoster. Drugs Aging 2018, 35, 1031–1040. [Google Scholar] [CrossRef]
- Dhillon, S.; Pace, D. Meningococcal Quadrivalent Tetanus Toxoid Conjugate Vaccine (MenACWY-TT; Nimenrix ®): A Review. Drugs 2017, 77, 1881–1896. [Google Scholar] [CrossRef]
- Pizza, M.; Scarlato, V.; Masignani, V.; Giuliani, M.M.; Arico, B.; Comanducci, M.; Jennings, G.T.; Baldi, L.; Bartolini, E.; Capec-chi, B.; et al. Identification of vaccine candidates against serogroup B meningococ-cus by whole-genome sequencing. Science 2000, 287, 1816–1820. [Google Scholar] [CrossRef]
- Sette, A.; Rappuoli, R. Reverse vaccinology: Developing vaccines in the era of genomics. Immunity 2010, 33, 530–541. [Google Scholar] [CrossRef]
- Dennehy, R.; McClean, S. Immunoproteomics: The Key to Discovery of New Vaccine Antigens against Bacterial Respiratory Infections. Curr. Protein Pept. Sci. 2012, 13, 807–815. [Google Scholar] [CrossRef]
- López, D.; Barriga, A.; Lorente, E.; Mir, C. Mir Immunoproteomic Lessons for Human Respiratory Syncytial Virus Vaccine Design. J. Clin. Med. 2019, 8, 486. [Google Scholar] [CrossRef] [PubMed]
- McClean, S.; Healy, M.E.; Collins, C.; Carberry, S.; O’Shaughnessy, L.; Dennehy, R.; Adams, A.; Kennelly, H.; Corbett, J.M.; Carty, F.; et al. Linocin and OmpW Are Involved in Attachment of the Cystic Fibrosis-Associated Pathogen Burkholderia cepacia Complex to Lung Epithelial Cells and Protect Mice against Infection. Infect. Immun. 2019, 84, 1424–1437. [Google Scholar] [CrossRef] [PubMed]
- Casey, W.T.; Spink, N.; Cia, F.; Collins, C.; Romano, M.; Berisio, R.; Bancroft, G.J.; McClean, S. Identification of an OmpW homologue in Burkholderia pseudomallei, a protective vaccine antigen against melioidosis. Vaccine 2016, 34, 2616–2621. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [PubMed]
- Karikó, K.; Muramatsu, H.; Ludwig, J.; Weissman, D. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res. 2011, 39, e142. [Google Scholar] [CrossRef]
- Alberer, M.; Gnad-Vogt, U.; Hong, H.S.; Mehr, K.T.; Backert, L.; Finak, G.; Gottardo, R.; Bica, M.A.; Garofano, A.; Koch, S.D.; et al. Safety and immunogenicity of a mRNA rabies vaccine in healthy adults: An open-label, non-randomised, prospective, first-in-human phase 1 clinical trial. Lancet 2017, 390, 1511–1520. [Google Scholar] [CrossRef]
- Gandhi, R.T.; Kwon, D.S.; Macklin, E.A.; Shopis, J.R.; McLean, A.P.; McBrine, N.; Flynn, T.; Peter, L.; Sbrolla, A.; Kauf-mann, D.E.; et al. Immunization of HIV-1-Infected Persons With Autologous Dendritic Cells Transfected With mRNA Encoding HIV-1 Gag and Nef: Results of a Randomized, Placebo-Controlled Clinical Trial. J. Acquir. Immune. Defic. Syndr. 2016, 71, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Jackson, L.A.; Anderson, E.J.; Rouphael, N.G.; Roberts, P.C.; Makhene, M.; Coler, R.N.; McCullough, M.P.; Chappell, J.D.; Denison, M.R.; Stevens, L.J.; et al. An mRNA Vaccine against SARS-CoV-2—Preliminary Report. N. Eng. J. Med. 2020, 383, 1920–1931. [Google Scholar] [CrossRef] [PubMed]
- Oliver, S.E.; Gargano, J.W.; Marin, M.; Wallace, M.; Curran, K.G.; Chamberland, M.; McClung, N.; Campos-Outcalt, D.; Morgan, R.L.; Mbaeyi, S.; et al. The Advisory Committee on Immunization Practices’ Interim Recommendation for Use of Pfizer-BioNTech COVID-19 Vaccine—United States, December 2020. Mmwr. Morb. Mortal. Wkly. Rep. 2020, 69, 1922–1924. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.Y.; Thone, M.N.; Kwon, Y.J. COVID-19 vaccines: The status and perspectives in delivery points of view. Adv. Drug Deliv. Rev. 2021, 170, 1–25. [Google Scholar] [CrossRef]
- Schalk, J.A.C.; Mooi, F.R.; Berbers, G.A.M.; Aerts, L.A.V.; Ovelgönne, H.; Kimman, T.G. Preclinical and Clini-cal Safety Studies on DNA Vaccines. Hum. Vaccines 2006, 2, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Kutzler, M.A.; Weiner, D.B. DNA vaccines: Ready for prime time? Nat. Rev. Genet. 2008, 9, 776–788. [Google Scholar] [CrossRef]
- Dauphin, G.; Zientara, S. West Nile virus: Recent trends in diagnosis and vaccine development. Vaccine 2007, 25, 5563–5576. [Google Scholar] [CrossRef]
- Atherton, M.J.; Morris, J.S.; McDermott, M.R.; Lichty, B.D. Cancer immunology and canine malignant melanoma: A comparative review. Vet. Immunol. Immunopathol. 2016, 169, 15–26. [Google Scholar] [CrossRef]
- MacGregor, R.R.; Boyer, J.D.; Ugen, K.E.; Lacy, K.E.; Gluckman, S.J.; Bagarazzi, M.L.; Chattergoon, M.A.; Baine, Y.; Hig-gins, T.J.; Ciccarelli, R.B.; et al. First human trial of a DNA-based vaccine for treatment of human immunodeficiency virus type 1 infection: Safety and host response. J. Infect. Dis. 1998, 178, 92–100. [Google Scholar] [CrossRef]
- Rouphael, N.G.; Morgan, C.; Li, S.S.; Jensen, R.; Sanchez, B.; Karuna, S.; Swann, E.; Sobieszczyk, M.E.; Frank, I.; Wilson, G.J.; et al. DNA priming and gp120 boosting induces HIV-specific antibodies in a randomized clinical trial. J. Clin. Investig. 2019, 129, 4769–4785. [Google Scholar] [CrossRef] [PubMed]
- Lévy, Y.; Lacabaratz, C. Optimal priming of poxvirus vector (NYVAC)-based HIV vaccine regimens for T cell responses requires three DNA injections. Results of the randomized multicentre EV03/ANRS VAC20 Phase I/II Trial. PLoS Pathog. 2020, 16, e1008522. [Google Scholar] [CrossRef] [PubMed]
- Boppana, S.; Fiore-Gartland, A.; Bansal, A.; Goepfert, P. Cross-Reactive CD8 T cell Responses Elicited by Adenovirus Type 5-Based HIV-1 Vaccines Contributed to Early Viral Evolution in Vaccine Recipients Who Became Infected. J. Virol. 2019, 94. [Google Scholar] [CrossRef]
- Modjarrad, K.; Roberts, C.C.; Mills, K.T.; Castellano, A.R.; Paolino, K.; Muthumani, K.; Reuschel, E.L.; Robb, M.L.; Racine, T.; Oh, M.D.; et al. Safety and immunogenicity of an anti-Middle East respiratory syndrome coronavirus DNA vaccine: A phase 1, open-label, single-arm, dose-escalation trial. Lancet Infect Dis. 2019, 19, 1013–1022. [Google Scholar] [CrossRef]
- Smith, T.R.F.; Patel, A. Immunogenicity of a DNA vaccine candidate for COVID-19. Nat. Commun. 2020, 11, 2601. [Google Scholar] [CrossRef] [PubMed]
- Bouard, D.; Alazard-Dany, N.; Cosset, F.-L. Viral vectors: From virology to transgene expression. Br. J. Pharmacol. 2009, 157, 153–165. [Google Scholar] [CrossRef]
- Van Winkel, C.A.J.; Moreno, A.; Curiel, D.T. Capsid-Incorporation Strategy to Display Antigens for an Alternative Adenoviral Vector Vaccine Approach. Mol. Pharm. 2018, 15, 5446–5453. [Google Scholar] [CrossRef] [PubMed]
- Ura, T.; Okuda, K.; Shimada, M. Developments in Viral Vector-Based Vaccines. Vaccines 2014, 2, 624–641. [Google Scholar] [CrossRef]
- Vellinga, J.; Smith, J.P.; Lipiec, A.; Majhen, D.; Lemckert, A.; van Ooij, M.; Ives, P.; Yallop, C.; Custers, J.; Havenga, M. Challenges in manufacturing adenoviral vectors for global vaccine product deployment. Hum. Gene Ther. 2014, 25, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Ramezanpour, B.; Haan, I.; Osterhaus, A.; Claassen, E. Vector-based genetically modified vaccines: Exploiting Jenner’s legacy. Vaccine 2016, 34, 6436–6448. [Google Scholar] [CrossRef] [PubMed]
- Case, J.B.; Rothlauf, P.W.; Chen, R.E.; Kafai, N.M.; Fox, J.M.; Smith, B.K.; Shrihari, S.; McCune, B.T.; Harvey, I.B.; Keeler, S.P.; et al. Replication-Competent Vesicular Stomatitis Virus Vaccine Vector Protects against SARS-CoV-2-Mediated Pathogenesis in Mice. Cell Host Microbe 2020, 28, 465–474. [Google Scholar] [CrossRef]
- Graham, S.P.; McLean, R.K. Evaluation of the immunogenicity of prime-boost vaccination with the replication-deficient viral vectored COVID-19 vaccine candidate ChAdOx1 nCoV-19. NPJ Vaccines 2020, 5, 69. [Google Scholar] [CrossRef]
- Van Doremalen, N.; Lambe, T.; Spencer, A.; Belij-Rammerstorfer, S.; Purushotham, J.N.; Port, J.R.; Avanzato, V.A.; Bushmaker, T.; Flaxman, A.; Ulaszewska, M.; et al. ChAdOx1 nCoV-19 vaccine prevents SARS-CoV-2 pneumonia in rhesus macaques. Nature 2020, 586, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Folegatti, P.M.; Ewer, K.J.; Aley, P.K.; Angus, B.; Becker, S.; Belij-Rammerstorfer, S.; Bellamy, D.; Bibi, S.; Bittaye, M.; Clut-terbuck, E.A.; et al. Safety and immunogenicity of the ChAdOx1 nCoV-19 vaccine against SARS-CoV-2: A preliminary report of a phase 1/2, single-blind, randomised controlled trial. Lancet 2020, 396, 467–478. [Google Scholar] [CrossRef]
- Zhu, F.C.; Guan, X.H.; Li, Y.H.; Huang, J.Y.; Jiang, T.; Hou, L.H.; Li, J.X.; Yang, B.F.; Wang, L.; Wang, W.J.; et al. Immunogenicity and safety of a recombinant adenovirus type-5-vectored COVID-19 vaccine in healthy adults aged 18 years or older: A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2020, 396, 479–488. [Google Scholar] [CrossRef]
- Zhu, F.C.; Li, Y.H.; Guan, X.H.; Hou, L.H.; Wang, W.J.; Li, J.X.; Wu, S.P.; Wang, B.S.; Wang, Z.; Wang, L.; et al. Safety, tolerability, and immuno-genicity of a recombinant adenovirus type-5 vectored COVID-19 vaccine: A dose-escalation, open-label, non-randomised, first-in-human trial. Lancet 2020, 395, 1845–1854. [Google Scholar] [CrossRef]
- Mercado, N.B.; Zahn, R.; Wegmann, F.; Loos, C.; Chandrashekar, A. Single-shot Ad26 vaccine protects against SARS-CoV-2 in rhesus macaques. Nature 2020, 586, 583–588. [Google Scholar] [CrossRef]
- Redoni, M.; Yacoub, S.; Rivino, L.; Giacobbe, D.R.; Luzzati, R.; Di Bella, S. Dengue: Status of current and under-development vaccines. Rev. Med. Virol. 2020, 30, e2101. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.Q.; Naguib, Y.W.; Dong, Y.; Shi, Y.C.; Bou, S.; Cui, Z. Applications of bacillus Calmette-Guerin and recombinant bacillus Calmette-Guerin in vaccine development and tumor immunotherapy. Expert Rev. Vaccines 2015, 14, 1255–1275. [Google Scholar] [CrossRef]
- Holst, J.; Martin, D.; Arnold, R.; Huergo, C.C.; Oster, P.; O’Hallahan, J.; Rosenqvist, E. Properties and clinical performance of vaccines containing outer membrane vesicles from Neisseria meningitidis. Vaccine 2009, 27S, B3–B12. [Google Scholar] [CrossRef]
- Pizza, M.; Bekkat-Berkani, R.; Rappuoli, R. Vaccines against Meningococcal Diseases. Micororganisms 2020, 8, 1521. [Google Scholar] [CrossRef]
- Van de Waterbeemd, B.; Streefland, M.; Van der Ley, P.; Zomer, B.; Van Dijken, H.; Martens, D.; Wijffels, R.; Van der Pol, L. OMV vaccine against Neisseria meningitidis using genetically engineered strains and a deter-gent-free purification process. Vaccine 2010, 28, 4810–4816. [Google Scholar] [CrossRef]
- Zariri, A.; Pupo, E.; Van Riet, E.; Van Putten, J.P.; Van Der Ley, P. Modulating endotoxin activity by combinatorial bioengineering of meningococcal lipopolysaccharide. Sci. Rep. 2016, 6, 36575. [Google Scholar] [CrossRef] [PubMed]
- Salverda, M.L.; Meinderts, S.M.; Hamstra, H.J.; Wagemakers, A.; Hovius, J.W.; van der Ark, A.; Stork, M.; van der Ley, P. Surface display of a borrelial lipoprotein on meningococcal outer membrane vesicles. Vaccine 2016, 34, 1025–1033. [Google Scholar] [CrossRef]
- Zhu, Y.; An, X.; Zhang, X.; Qiao, Y.; Zheng, T.; Li, X. STING: A master regulator in the cancer-immunity cycle. Mol. Cancer 2019, 18, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Pulendran, B.; Li, S.; Nakaya, H.I. Systems Vaccinology. Immunity 2010, 33, 516–529. [Google Scholar] [CrossRef] [PubMed]
- Rappuoli, R. Bridging the knowledge gaps in vaccine design. Nat. Biotechnol. 2007, 25, 1361–1366. [Google Scholar] [CrossRef]
- Brodin, P.; Jojic, V.; Gao, T.; Bhattacharya, S.; Angel, C.J.L.; Furman, D.; Shen-Orr, S.; Dekker, C.L.; Swan, G.E.; Butte, A.J.; et al. Variation in the Human Immune System Is Largely Driven by Non-Heritable Influences. Cell 2015, 160, 37–47. [Google Scholar] [CrossRef]
- Cheung, P.; Vallania, F.; Warsinske, H.C.; Donato, M.; Schaffert, S.; Chang, S.E.; Dvorak, M.; Dekker, C.L.; Davis, M.M.; Utz, P.J.; et al. Single-Cell Chromatin Modification Profiling Reveals Increased Epigenetic Variations with Aging. Cell 2018, 173, 1385–1397.e14. [Google Scholar] [CrossRef]
- Hagan, T.; Cortese, M.; Rouphael, N.; Boudreau, C.; Linde, C.; Maddur, M.S.; Das, J.; Wang, H.; Guthmiller, J.; Zheng, N.-Y.; et al. Antibiotics-Driven Gut Microbiome Perturbation Alters Immunity to Vaccines in Humans. Cell 2019, 178, 1313–1328.e13. [Google Scholar] [CrossRef]
- Cortese, M.; Sherman, A.C.; Rouphael, N.G.; Pulendran, B. Systems Biological Analysis of Immune Response to Influenza Vaccination. Cold Spring Harb. Perspect. Med. 2020, a038596. [Google Scholar] [CrossRef]
- Chaussabel, D.; Quinn, C.; Shen, J.; Patel, P.; Glaser, C.; Baldwin, N.; Stichweh, D.; Blankenship, D.; Li, L.; Munagala, I.; et al. A modu-lar analysis framework for blood genomics studies: Application to systemic lupus erythematosus. Immunity 2008, 29, 150–164. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Rouphael, N.; Duraisingham, S.S.; Romero-Steiner, S.; Presnell, S.R.; Davis, C.; Schmidt, D.S.; E Johnson, S.; Milton, A.; Rajam, G.; et al. Molecular signatures of antibody responses derived from a systems biology study of five human vaccines. Nat. Immunol. 2014, 15, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Wimmers, F.; Pulendran, B. Emerging technologies for systems vaccinology—Multiomics integration and single-cell (epi)genomic profiling. Curr. Opin. Immunol. 2020, 65, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Sullivan, N.L.; Rouphael, N.; Yu, T.; Banton, S.; Maddur, M.S.; McCausland, M.; Chiu, C.; Canniff, J.; Dubey, S.; et al. Metabolic Phenotypes of Response to Vaccination in Humans. Cell 2017, 169, 862–877.e17. [Google Scholar] [CrossRef]
- Braun, R.O.; Brunner, L.; Wyler, K.; Auray, G.; Garcia-Nicolas, O.; Python, S.; Zumkehr, B.; Gaschen, V.; Stoffel, M.H.; Collin, N.; et al. System immunology-based identification of blood transcrip-tional modules correlating to antibody responses in sheep. NPJ Vaccines 2018, 3, 41. [Google Scholar] [CrossRef]
- Matthijs, A.M.F.; Auray, G.; Jakob, V.; Garcia-Nicolas, O.; Braun, R.O.; Keller, I.; Bruggman, R.; Devriendt, B.; Boyen, F.; Guzman, C.A.; et al. Systems Immu-nology Characterization of Novel Vaccine Formulations for Mycoplasma hyopneumoniae Bacterins. Front. Immunol. 2019, 10, 1087. [Google Scholar] [CrossRef]
- Tsang, J.S.; Dobaño, C.; VanDamme, P.; Moncunill, G.; Marchant, A.; Ben Othman, R.; Sadarangani, M.; Koff, W.C.; Kollmann, T.R. Improving Vaccine-Induced Immunity: Can Baseline Predict Outcome? Trends Immunol. 2020, 41, 457–465. [Google Scholar] [CrossRef]
- Delany, I.; Rappuoli, R.; Seilb, K.L. Vaccines, reverse vaccinology and bacterial pathogenesis. CSH Perspect. 2013, 3, a012476. [Google Scholar] [CrossRef]
- Jefferies, J.M.C.; Macdonald, E.; Faust, S.N. Clarke SC, 13-valent pneumococcal conjugate vaccine (PCV13). Hum. Vaccines 2011, 7, 1012–1018. [Google Scholar] [CrossRef]
- Rappuoli, R.; Bottomley, M.J.; D’Oro, U.; Finco, O.; De Gregorio, E. Reverse vaccinology 2.0: Human immunology instructs vac-cine antigen design. J. Exp. Med. 2016, 213, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Burton, D.R. What Are the Most Powerful Immunogen Design Vaccine Strategies? Reverse Vaccinology 2.0 Shows Great Promise. Cold Spring Harb. Perspect. Biol. 2017, 9, 030262. [Google Scholar] [CrossRef] [PubMed]
- Correia, B.E.; Bates, J.T.; Loomis, R.J.; Baneyx, G.; Carrico, C.; Jardine, J.G.; Rupert, P.B.; Correnti, C.; Kalyuzhniy, O.; Vittal, V.; et al. Proof of principle for epitope-focused vaccine design. Nat. Cell Biol. 2014, 507, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Graham, B.S.; Gilman, M.S.A.; McLellan, J.S. Structure-Based Vaccine Antigen Design. Annu. Rev. Med. 2019, 70, 91–104. [Google Scholar] [CrossRef]
- Anasir, M.A. Chit Laa Poh. Structural Vaccinology for Viral Vaccine Design. Front Microbiol. 2019, 10, 738. [Google Scholar] [CrossRef]
- Malonis, R.J.; Lai, J.R.; Vergnolle, O. Peptide-Based Vaccines: Current Progress and Future Challenges. Chem. Rev. 2020, 120, 3210–3229. [Google Scholar] [CrossRef]
- Alam, S.M.; Dennison, S.M.; Aussedat, B.; Vohra, Y.; Park, P.K.; Fernández-Tejada, A.; Stewart, S.; Jaeger, F.H.; Anasti, K.; Blinn, J.H.; et al. Recognition of synthetic glycopeptides by HIV-1 broadly neutralizing antibodies and their unmutated ancestors. Proc. Natl. Acad. Sci. USA 2013, 110, 18214–18219. [Google Scholar] [CrossRef]
- Kanekiyo, M.; Ellis, D.; King, N.P. New Vaccine Design and Delivery Technologies. J. Infect. Dis. 2019, 219, S88–S96. [Google Scholar] [CrossRef]
- Compañón, I.; Guerreiro, A.; Mangini, V.; Castro-López, J.; Escudero-Casao, M.; Avenoza, A.; Busto, J.H.; Castillón, S.; Jiménez-Barbero, J.; Asensio, J.L.; et al. Structure-based design of potent tumor-associated antigens: Modulation of peptide presentation by single-atom O/S or O/Se substitutions at the glycosidic linkage. J. Am. Chem. Soc. 2019, 141, 4063–4072. [Google Scholar] [CrossRef]
- Lewis, G.K.; DeVico, A.L.; Gallo, R.C. Antibody persistence and T cell balance: Two key factors confronting HIV vaccine development. Proc. Natl. Acad. Sci. USA 2014, 111, 15614. [Google Scholar] [CrossRef]
- Joyce, M.G.; Zhang, B.; Ou, L.; Chen, M.; Chuang, G.-Y.; Druz, A.; Kong, W.-P.; Lai, Y.-T.; Rundlet, E.J.; Tsybovsky, Y.; et al. Iterative structure-based improvement of a fusion-glycoprotein vaccine against RSV. Nat. Struct. Mol. Biol. 2016, 23, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Ngwuta, J.O.; Chen, M.; Modjarrad, K.; Joyce, M.G.; Kanekiyo, M.; Kumar, A.; Yassine, H.M.; Moin, S.M.; Killikelly, A.M.; Chuang, G.-Y.; et al. Prefusion F–specific antibodies determine the magnitude of RSV neutralizing activity in human sera. Sci. Transl. Med. 2015, 7, 309ra162. [Google Scholar] [CrossRef]
- Hollingshead, S.; Jongerius, I.; Exley, R.M.; Johnson, S.; Lea, S.M.; Tang, C.M. Structure-based design of chimeric antigens for multivalent protein vaccines. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Geldmacher, A.; Freier, A.; Losch, F.O.; Walden, P. Therapeutic vaccination for cancer immunotherapy: Antigen selection and clinical response. Human Vaccines 2011, 7, 115. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, L.H. Lessons learned from cancer vaccine trials and target antigen choice. Cancer Immunol. Immunother. 2016, 65, 805–812. [Google Scholar] [CrossRef]
- Xu, Z.; Kulp, D.W. Protein engineering and particulate display of B-cell epitopes to facilitate development of novel vaccines. Curr. Opin. Immunol. 2019, 59, 49. [Google Scholar] [CrossRef]
- Cheever, M.A.; Allison, J.P.; Ferris, A.S.; Finn, O.J.; Hastings, B.M.; Hecht, T.T.; Mellman, I.; Prindiville, S.A.; Steinman, R.M.; Viner, J.L.; et al. The prioritization of cancer an-tigens: A national cancer institute pilot project for the acceleration of translational research Clin. Cancer Res 2009, 15, 5323. [Google Scholar]
- Radford, K.J.; Caminschi, I. New generation of dendritic cell vaccines. Hum. Vaccines Immunother. 2013, 9, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Sharma, S.K.; Srivastava, V.; Kumar, A. Proteomic Exploration of Listeria monocytogenes for the Purpose of Vaccine Designing Using a Reverse Vaccinology Approach. Int. J. Pept. Res. Ther. 2021, 27, 779–799. [Google Scholar] [CrossRef]
- Calderon-Gonzalez, R.; Tobes, R.; Pareja, E.; Frande-Cabanes, E.; Petrovsky, N.; Alvarez-Dominguez, C. Identification and characterisation of T cell epitopes for incorporation into dendritic cell-delivered Listeria vaccines. J. Immunol. Methods 2015, 424, 111–119. [Google Scholar] [CrossRef]
- Kono, M.; Nakamura, Y.; Suda, T.; Uchijima, M.; Tsujimura, K.; Nagata, T.; Giermasz, A.S.; Kalinski, P.; Nakamura, H.; Chida, K. En-hancement of protective immunity against intracellular bacteria using type-1 polarized dendritic cell (DC) vaccine. Vaccine 2012, 30, 2633–2639. [Google Scholar] [CrossRef] [PubMed]
- Calderon-Gonzalez, R.; Frande-Cabanes EBronchalo-Vicente, L.; Lecea-Cuello, M.J.; Bosch-Martinez, A.; Fanarraga, M.L.; Yañez-Diaz, S.; Carrasco-Marin, E.; Alvarez-Dominguez, C. Cellular vaccines in listeriosis: Role of the Listeria antigen GAPDH. Front. Cell. Infect. Microbiol. 2014, 4, 22. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Dominguez, C.; Salcines-Cuevas, D.; Teran-Navarro, H.; Calderon-Gonzalez, R.; Tober, R.; Garcia, I.; Grijalvo, S.; Paradela, A.; Seoane, A.; Sangari, F.J.; et al. Epitopes for multivalent vaccines against Listeria, Mycobacterium and Streptococcus spp: A novel role for glyceraldehyde-3-phosphate dehydrogenase. Front. Cell. Infect. Microbiol. 2020, 10, 573348. [Google Scholar] [CrossRef] [PubMed]
- Brown, F.; Dougan GMoey, E.M. A short history of vaccination. In Vaccine Design; Brown, F., Dougan, G., Moey, E.M., Eds.; John Wiley and Sons: Chichester, UK, 1993; pp. 1–6. [Google Scholar]
- Schijns, V.E. Immunological concepts of vaccine adjuvant activity. Curr. Opin. Immunol. 2020, 12, 456–463. [Google Scholar] [CrossRef]
- Christensen, D. Vaccine adjuvants: Why and how. Hum. Vaccines Immunother. 2016, 12, 2709–2711. [Google Scholar] [CrossRef] [PubMed]
- Del Giudice, G.; Rappuoli, R.; Didierlaurent, A.M. Correlates of adjuvanticity: A review on adjuvants in licensed vaccines. Semin. Immunol. 2018, 39, 14–21. [Google Scholar] [CrossRef]
- O’Hagan, D.T.; Fox, C.B. New generation adjuvants—From empiricism to rational design. Vaccine 2015, 33, B14–B20. [Google Scholar] [CrossRef]
- Ribeiro, C.M.; Schijns, V.E. Immunology of Vaccine Adjuvants. In Methods in Molecular Biology; Springer International Publishing: New York, NY, USA, 2009; Volume 626, pp. 1–14. [Google Scholar]
- Shi, S.; Zhu, H.; Xia, X.; Liang, Z.; Ma, X.; Sun, B. Vaccine adjuvants: Understanding the structure and mechanism of adjuvanticity. Vaccine 2019, 37, 3167–3178. [Google Scholar] [CrossRef]
- Bonam, S.R.; Partidos, C.D.; Halmuthur, S.K.M.; Muller, S. An overview of novel adjuvants designed for improving vaccine efficacy. Trends Pharmacol. Sci. 2017, 38, 771–793. [Google Scholar] [CrossRef]
- Rueckert, C.; Guzmán, C.A. Vaccines: From empirical development to rational design. PLoS Pathog. 2012, 8, e1003001. [Google Scholar] [CrossRef]
- Garçon, N.; Chomez, P.; Van Mechelen, M. GlaxoSmithKline Adjuvant Systems in vaccines: Concepts, achievements and perspectives. Expert Rev. Vaccines 2007, 6, 723–739. [Google Scholar] [CrossRef]
- Pedersen, G.K.; Andersen, P.; Christensen, D. Immunocorrelates of CAF family adjuvants. Semin. Immunol. 2018, 39, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Molinaro, A.; Holst, O.; Di Lorenzo, F.; Callaghan, M.; Nurisso, A.; D’Errico, G.; Zamyatina, A.; Peri, F.; Berisio, R.; Jerala, R.; et al. Chemistry of Lipid A: At the Heart of Innate Immunity. Chem. Eur. J. 2015, 21, 500–519. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A.; Medzhitov, R. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 2004, 5, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Schromm, A.B.; Brandenburg, K.; Loppnow, H.; Moran, A.P.; Koch, M.H.J.; Rietschel, E.T.; Seydel, U. Biological activities of lipopolysaccharides are determined by the shape of their lipid A portion. FEBS J. 2000, 267, 2008–2013. [Google Scholar] [CrossRef] [PubMed]
- Kong, Q.; Six, D.A.; Liu, Q.; Gu, L.; Wang, S.; Alamuri, P.; Raetz, C.R.H.; Curtiss, R. Phosphate groups of lipid A are essential for Salmonella enterica serovar Typhimurium virulence and affect innate and adaptive immunity. Infect. Immun. 2012, 80, 3215–3224. [Google Scholar] [CrossRef] [PubMed]
- Bhat, U.R.; Forsberg, L.S.; Carlson, R.W. Structure of lipid A component of Rhizobium leguminosarum bv. phaseoli lipopolysaccharide. Unique nonphosphorylated lipid A containing 2-amino- 2-deoxygluconate, galacturonate, and glu-cosamine. J. Biol. Chem. 1994, 269, 14402–14410. [Google Scholar] [CrossRef]
- Raetz, C.R.H.; Reynolds, C.M.; Trent, M.S.; Bishop, R.E. Lipid A modification systems in Gram-negative bacteria. Annu. Rev. Biochem 2007, 76, 295–329. [Google Scholar] [CrossRef]
- Plotz, B.M.; Lindner, B.; Stetter, K.O.; Holst, O. Characterization of a Novel Lipid A Containing D-Galacturonic Acid That Replaces Phosphate Residues. J. Biol. Chem. 2000, 275, 11222–11228. [Google Scholar] [CrossRef]
- Silipo, A.; Vitiello, G.; Gully, D.; Sturiale, L.; Chaintreuil, C.; Fardoux, J.; Gargani, D.; Lee, H.I.; Kulkarni, G.; Busset, N.; et al. Covalently linked hopanoid-lipid A improves outer-membrane resistance of a Bradyrhizobium symbiont of legumes. Nat. Commun. 2014, 5, 5106. [Google Scholar] [CrossRef]
- Di Lorenzo, F.; Pither, M.D.; Martufi, M.; Scarinci, I.; Guzmán-Caldentey, J.; Łakomiec, E.; Jachymek, W.; Bruijns, S.C.M.; Santamaría, S.M.; Frick, J.-S.; et al. Pairing Bacteroides vulgatus LPS Structure with Its Immunomodulatory Effects on Human Cellular Models. ACS Cent. Sci. 2020, 6, 1602–1616. [Google Scholar] [CrossRef] [PubMed]
- Schwudke, D.; Linscheid, M.; Strauch, E.; Appel, B.; Zahringer, U.; Moll, H.; Muller, M.; Brecker, L.; Gronow, S.; Lindner, B. The Obligate Predatory Bdellovibrio bacteriovorus Possesses a Neutral Lipid A Containing α-D-Mannoses That Replace Phosphate Residues. J. Biol. Chem. 2003, 278, 27502–27512. [Google Scholar] [CrossRef]
- Zhou, Z.; Ribeiro, A.A.; Lin, S.; Cotter, R.J.; Miller, S.I.; Raetz, C.R.H. Lipid A Modifications in Polymyxin-resistant Salmonella typhimurium. PRMA dependent 4-amino-4-deoxy-L-arabinose, and phosphoethanolamine incorporation. J. Biol. Chem. 2001, 276, 43111–43121. [Google Scholar] [CrossRef] [PubMed]
- Silipo, A.; Molinaro, A.; Cescutti, P.; Bedini, E.; Rizzo, R.; Parrilli, M.; Lanzetta, R. Complete structural characterization of the lipid A fraction of a clinical strain of B. cepacia genomovar I lipopolysaccharide. Glycobiology 2005, 15, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Casabuono, A.C.; Czibener, C.; del Giudice, M.G.; Valguarnera, E.; Ugalde, J.E.; Couto, A.S. New Features in the Lipid A Structure of Brucella suis and Brucella abortus Lipopolysaccharide. J. Am. Chem. Soc. Mass. Spec. 2017, 28, 2716–2723. [Google Scholar] [CrossRef]
- Zahringer, U.; Lindner, B.; Knirel, Y.A.; van den Akker, W.M.R.; Hiestand, R.; Heine, H.; Dehio, C. Structure and Biological Activity of the Short-chain Lipopolysaccharide from Bartonella henselae ATCC 49882T. J. Biol. Chem. 2004, 279, 21046–21054. [Google Scholar] [CrossRef] [PubMed]
- Di Lorenzo, F.; de Castro, C.; Silipo, A.; Molinaro, A. Lipopolysaccharide structures of Gram-negative populations in the gut microbiota and effects on host interactions. FEMS Microbiol. Rev. 2019, 43, 257–272. [Google Scholar] [CrossRef]
- Van Vliet, S.J.; Steeghs, L.; Bruijns, S.C.M.; Vaezirad, M.M.; Blok, C.S.; Busto, J.A.A.; Deken, M.; van Putten, J.P.M.; van Kooyk, Y. Variation of Neisseria gonorrhoeae Lipooligosaccharide Directs Dendritic Cell–Induced T Helper Responses. PLoS Pathog. 2009, 5, e1000625. [Google Scholar] [CrossRef]
- Hagar, J.A.; Powell, D.A.; Aachoui, Y.; Ernst, R.K.; Miao, E.A. Cytoplasmic LPS activates caspase-11: Implications in TLR4-independent endotoxic shock. Science 2013, 341, 1250–1253. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Wong, M.T.; Stowe, I.B.; Ramani, S.R.; Gonzalez, L.C.; Akashi-Takamura, S.; Miyake, K.; Zhang, J.; Lee, W.P.; Muszýnski, A.; et al. Noncanonical inflammasome activation by intracellular LPS in-dependent of TLR4. Science 2013, 341, 1246–1249. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014, 514, 187–192. [Google Scholar] [CrossRef]
- Viganò, E.; Diamond, C.E.; Spreafico, R.; Balachander, A.; Sobota, R.M.; Mortellaro, A. Human caspase-4 and caspase-5 regulate the one-step non-canonical inflammasome activation in monocytes. Nat. Commun. 2015, 6, 8761. [Google Scholar] [CrossRef]
- Chavarría-Velázquez, C.O.; Torres-Martínez, A.C.; Montaño, L.F.; Rendón-Huerta, E.P. TLR2 activation induced by H. pylori LPS promotes the differential expression of claudin-4, -6, -7 and -9 via either STAT3 and ERK1/2 in AGS cells. Immunobiology 2018, 223, 38–48. [Google Scholar] [CrossRef]
- Jamalan, M.; Ardestani, S.K.; Zeinali, M.; Mosaveri, N.; Taheri, M.M. Effectiveness of Brucella abortus lipopol-ysaccharide as an adjuvant for tuberculin PPD. Biologicals 2011, 39, 23–28. [Google Scholar] [CrossRef]
- Kianmehr, Z.; Soleimanjahi, H.; Ardestani, S.K.; Fotouhi, F.; Abdoli, A. Influence of Brucella abortus lipopolysaccharide as an adjuvant on the immunogenicity of HPV-16 L1VLP vaccine in mice. Med. Microbiol. Immunol. 2015, 204, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Chilton, P.M.; Hadel, D.M.; To, T.T.; Mitchell, T.C.; Darveau, R.P. Adjuvant activity of naturally occurring monophos-phoryl lipopolysaccharide preparations from mucosa-associated bacteria. Infect. Immun. 2013, 81, 3317–3325. [Google Scholar] [CrossRef] [PubMed]
- Ko, A.; Wui, S.R.; Ryu, J.I.; Do, H.T.T.; Lee, Y.J.; Lim, S.J.; Rhee, I.; Jung, D.I.; Park, J.-A.; Choi, J.-A.; et al. Comparison of the adjuvanticity of two adjuvant formulations containing de-O-acylated lipooligosaccharide on Japanese encephalitis vaccine in mice. Arch. Pharmacal Res. 2018, 41, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Casella, C.; Mitchell, T. Putting endotoxin to work for us: Monophosphoryl lipid A as a safe and effective vaccine adjuvant. Cell. Mol. Life Sci. 2008, 65, 3231–3240. [Google Scholar] [CrossRef]
- Embry, C.A.; Franchi, L.; Nunez, G.; Mitchell, T.C. Mechanism of Impaired NLRP3 Inflammasome Priming by Mono-phosphoryl Lipid A. Sci. Signal. 2011, 4, ra28. [Google Scholar] [CrossRef] [PubMed]
- Casella, C.R.; Mitchell, T.C. Inefficient TLR4/MD-2 Heterotetramerization by Monophosphoryl Lipid A. PLoS ONE 2013, 8, e62622. [Google Scholar] [CrossRef] [PubMed]
- Tanimura, N.; Saitoh, S.-I.; Ohto, U.; Akashi-Takamura, S.; Fujimoto, Y.; Fukase, K.; Shimizu, T.; Miyake, K. The attenuated inflammation of MPL is due to the lack of CD14-dependent tight dimerization of the TLR4/MD2 complex at the plasma membrane. Int. Immunol. 2014, 26, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Pantel, A.; Cheong, C.; Dandamudi, D.; Shrestha, E.; Mehandru, S.; Brane, L.; Ruane, D.; Teixeira, A.; Bozzacco, L.; Steinman, R.M.; et al. A new synthetic TLR4 agonist, GLA, allows dendritic cells targeted with antigen to elicit Th1 T cell immunity in vivo. Eur. J. Immunol. 2012, 42, 101–109. [Google Scholar] [CrossRef]
- Carter, D.; Fox, C.B.; Day, T.A.; Guderian, J.A.; Liang, H.; Rolf, T.; Vergara, J.; Sagawa, Z.K.; Ireton, G.; Orr, M.T.; et al. A structure-function approach to optimizing TLR4 ligands for human vaccines. Clin. Transl. Immunol. 2016, 5, e108. [Google Scholar] [CrossRef]
- Gregg, K.A.; Harberts, E.; Gardner, F.M.; Pelletier, M.R.; Cayatte, C.; Yu, L.; McCarthy, M.P.; Marshall, J.D.; Ernst, R.K. Rationally designed TLR4 ligands for vaccine adjuvant discovery. mBio 2017, 8, e00492-17. [Google Scholar] [CrossRef]
- Johnson, D. Synthetic TLR4-active Glycolipids as Vaccine Adjuvants and Stand-alone Immunotherapeutics. Curr. Top. Med. Chem. 2008, 8, 64–79. [Google Scholar] [CrossRef]
- Khalaf, J.K.; Bowen, W.S.; Bazin, H.G.; Ryter, K.T.; Livesay, M.T.; Ward, J.R.; Evans, J.T.; Johnson, D.A. Characterization of TRIF selectivity in the AGP class of lipid A mimetics: Role of secondary lipid chains. Bioorg. Med. Chem. Lett 2015, 25, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.H.; Budzynski, W.A.; Skeels, L.N.; Krantz, M.J.; Koganty, R.R. Novel lipid A mimetics derived from pentae-rythritol: Synthesis and their potent agonistic activity. Tetrahedron 2002, 58, 8833–8842. [Google Scholar] [CrossRef]
- Akamatsu, M.; Fujimoto, Y.; Kataoka, M.; Suda, Y.; Kusumoto, S.; Fukase, K. Synthesis of lipid A monosaccharide ana-logues containing acidic amino acid: Exploring the structural basis for the endotoxic and antagonistic activities. Bioorg. Med. Chem. 2006, 14, 6759–6777. [Google Scholar] [CrossRef] [PubMed]
- Ishizaka, S.T.; Hawkins, L.D. E6020: A synthetic Toll-like receptor 4 agonist as a vaccine adjuvant. Expert Rev. Vaccines 2007, 6, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Morefield, G.L.; Hawkins, L.D.; Ishizaka, S.T.; Kissner, T.L.; Ulrich, R.G. Synthetic Toll-like receptor 4 agonist enhances vaccine efficacy in an experimental model of toxic shock syndrome. Clin. Vaccine Immunol. 2007, 14, 1499–1504. [Google Scholar] [CrossRef] [PubMed]
- Adanitsch, F.; Ittig, S.; Stöckl, J.; Oblak, A.; Haegman, M.; Jerala, R.; Beyaert, R.; Kosma, P.; Zamyatina, A. Development of aGlcN(1⇿1)aMan-based Lipid A mimetics as a novel class of potent Toll-like Receptor 4 agonists. J. Med. Chem. 2014, 57, 8056–8071. [Google Scholar] [CrossRef] [PubMed]
- Adanitsch, F.; Shi, J.; Shao, F.; Beyaert, R.; Heine, H.; Zamyatina, A. Synthetic glycan-based TLR4 agonists targeting caspa-se-4/11 for the development of adjuvants and immunotherapeutics. Chem. Sci. 2018, 9, 3957–3963. [Google Scholar] [CrossRef]
- Gordya, N.; Yakovlev, A.; Kruglikova, A.; Tulin, D.; Potolitsina, E.; Suborova, T.; Bordo, D.; Rosano, C.; Chernysh, S. Natural antimicrobial peptide complexes in the fighting of antibiotic resistant biofilms: Calliphora vicina medicinal maggots. PLoS ONE 2017, 12, e0173559. [Google Scholar] [CrossRef]
- Chernysh, S.; Kozuharova, I. Anti-tumor activity of a peptide combining patterns of insect alloferons and mammalian immunoglobulins in naïve and tumor antigen vaccinated mice. Int. Immunopharmacol. 2013, 17, 1090–1093. [Google Scholar] [CrossRef]
- Kim, Y.; Lee, S.K.; Bae, S.; Kim, H.; Park, Y.; Chu, N.K.; Kim, S.G.; Kim, H.-R.; Hwang, Y.-I.; Kang, J.S.; et al. The anti-inflammatory effect of alloferon on UVB-induced skin inflammation through the down-regulation of pro-inflammatory cytokines. Immunol. Lett. 2013, 149, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Griffin, M.E.; Hespen, C.W.; Wang, Y.; Hang, H.C. Translation of peptidoglycan metabolites into immunotherapeutics. Clin. Transl. Immunol. 2019, 8, e1095. [Google Scholar] [CrossRef] [PubMed]
- Magalhaes, J.G.; Fritz, J.H.; Le Bourhis, L.; Sellge, G.; Travassos, L.H.; Selvanantham, T.; Girardin, S.E.; Gommerman, J.L.; Philpott, D.J. Nod2-Dependent Th2 Polarization of Antigen-Specific Immunity. J. Immunol. 2008, 181, 7925–7935. [Google Scholar] [CrossRef]
- Rubino, S.J.; Magalhaes, J.G.; Philpott, D.; Bahr, G.M.; Blanot, D.; Girardin, S.E. Identification of a synthetic muramyl pep-tide derivative with enhanced Nod2 stimulatory capacity. Innate. Immun. 2013, 19, 493–503. [Google Scholar] [CrossRef]
- Maisonneuve, C.; Bertholet, S.; Philpott, D.J.; De Gregorio, E. Unleashing the potential of NOD- and Toll-like agonists as vaccine adjuvants. Proc. Natl. Acad. Sci. USA 2014, 111, 12294–12299. [Google Scholar] [CrossRef]
- Bumgardner, S.A.; Zhang, L.; LaVoy, A.S.; Andre, B.; Frank, C.B.; Kajikawa, A.; Klaenhammer, T.R.; Dean, G.A. Nod2 is required for antigen-specific humoral responses against antigens orally delivered using a recombinant Lactobacillus vaccine platform. PLoS ONE 2018, 13, e0196950. [Google Scholar] [CrossRef] [PubMed]
- Jackson, E.M.; Herbst-Kralovetz, M.M. Intranasal Vaccination with Murabutide Enhances Humoral and Mucosal Immune Responses to a Virus-Like Particle Vaccine. PLoS ONE 2012, 7, e41529. [Google Scholar] [CrossRef]
- Nabergoj, S.; Mlinarič-Raščan, I.; Jakopin, Ž. Harnessing the untapped potential of nucleotide-binding oligomerization domain ligands for cancer immunotherapy. Med. Res. Rev. 2019, 39, 1447–1484. [Google Scholar] [CrossRef]
- Jakopin, Z. Murabutide Revisited: A Review of its Pleiotropic Biological Effects. Curr. Med. Chem. 2013, 20, 2068–2079. [Google Scholar] [CrossRef]
- Telzak, E.; Wolff, S.M.; Dinarello, C.A.; Conlon, T.; El Kholy, A.; Bahr, G.M.; Choay, J.P.; Morin, A.; Chedid, L. Clinical Evaluation of the Immunoadjuvant Murabutide, a Derivative of MDP, Administered with a Tetanus Toxoid Vaccine. J. Infect. Dis. 1986, 153, 628–633. [Google Scholar] [CrossRef]
- Przewlocki, G.; Audibert, F.; Jolivet, M.; Chedid, L.; Kent, S.B.; Neurath, A.R. Production of antibodies recognizing a hepa-titis B virus (HBV) surface antigen by administration of murabutide associated to a synthetic pre-S HBV peptide conjugated to a toxoid carrier. Biochem. Biophys. Res. Commun. 1986, 140, 557–564. [Google Scholar] [CrossRef]
- Byars, N.E.; Nakano, G.; Welch, M.; Lehman, D.; Allison, A.C. Improvement of hepatitis B vaccine by the use of a new adju-vant. Vaccine 1991, 9, 309–318. [Google Scholar] [CrossRef]
- Keefer, M.C.; Graham, B.S.; McElrath, M.J.; Matthews, T.J.; Stablein, D.M.; Corey, L.; Wright, P.F.; Lawrence, D.; Fast, P.E.; Weinhold, K.; et al. Safety and immunogenicity of Env 2-3, a human immunode-ficiency virus type 1 candidate vaccine, in combination with a novel adjuvant, MTP-PE/MF59. AIDS Res. Hum. Retrov. 1996, 12, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Tamura, M.; Yoo, Y.C.; Yoshimatsu, K.; Yoshida, R.; Oka, T.; Ohkuma, K.; Arikawa, J.; Azuma, I. Effects of muramyl dipep-tide derivatives as adjuvants on the induction of antibody response to recombinant hepatitis B surface antigen. Vaccine 1995, 13, 77–82. [Google Scholar] [CrossRef]
- Yoo, Y.C.; Yoshimatsu, K.; Koike, Y.; Hatsuse, R.; Yamanishi, K.; Tanishita, O.; Arikawa, J.; Azuma, I. Adjuvant activity of muramyl dipeptide derivatives to enhance immunogenicity of a hantavirus-inactivated vaccine. Vaccine 1998, 16, 216–224. [Google Scholar] [CrossRef]
- Effenberg, R.; Turánek Knötigová, P.; Zyka, D.; Célechovská, H.; Mašek, J.; Bartheldyová, E.; Hubatka, F.; Koudelka, S.; Lukáč, R.; Kovalová, A.; et al. Nonpyrogenic molecular adjuvants based on norAbu-muramyldipeptide and norAbu-glucosaminyl muramyldipeptide: Synthesis, molecular mechanisms of action, and biological activities in vitro and in vivo. J. Med. Chem. 2017, 60, 7745–7763. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.Z.; Xu, S.; Liao, X.Y.; Zhang, S.D.; Liang, Z.L.; Liu, B.H.; Bai, J.Y.; Jiang, C.; Ding, J.; Cheng, G.F.; et al. A Novel Immunostimulator, N2-[α-O-Benzyl-N-(acetylmuramyl)-l-alanyl-d- isoglutaminyl]-N6-trans-(m-nitrocinnamoyl)-l-lysine, and Its Adjuvancy on the Hepatitis B Surface Antigen. J. Med. Chem. 2005, 48, 5112–5122. [Google Scholar] [CrossRef] [PubMed]
- Gobec, M.; Tomašič, T.; Štimac, A.; Frkanec, R.; Trontelj, J.; Anderluh, M.; Mlinarič-Raščan, I.; Jakopin, Ž. Discovery of na-nomolar desmuramylpeptide agonists of the innate immune receptor nucleotide-binding oligomerization domain-containing protein 2 (NOD2) possessing immunostimulatory properties. J. Med. Chem. 2018, 61, 2707–2724. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Tejada, A.; Tan, D.S.; Gin, D.Y. Development of Improved Vaccine Adjuvants Based on the Saponin Natural Product QS-21 through Chemical Synthesis. Acc. Chem. Res. 2016, 49, 1741–1756. [Google Scholar] [CrossRef]
- Pifferi, C.; Fuentes, R.; Fernández-Tejada, A. Natural and synthetic carbohydrate-based vaccine adjuvants and their mechanisms of action. Nat. Rev. Chem. 2021, 5, 197–216. [Google Scholar] [CrossRef]
- Fernández-Tejada, A. Design, synthesis and evaluation of optimized saponin variants derived from the vaccine adjuvant QS-21. Pure Appl. Chem. 2017, 89, 1359–1378. [Google Scholar] [CrossRef]
- Fuentes, R.; Ruiz-de-Angulo, A.; Sacristán, N.; Navo, C.D.; Jiménez-Osés, G.; Anguita, J.; Fernández-Tejada, A. Replacing the Rhamnose-Xylose Moiety of QS-21 with Simpler Terminal Disaccharide Units Attenuates Adjuvant Activity in Truncated Saponin Variants. Chem. Eur. J. 2021, 27, 4731–4737. [Google Scholar] [CrossRef]
- Fernández-Tejada, A.; Chea, E.K.; George, C.; Pillarsetty, N.V.K.; Gardner, J.R.; Livingston, P.O.; Ragupathi, G.; Lewis, J.S.; Tan, D.S.; Gin, D.Y. Development of a minimal saponin vaccine adjuvant based on QS-21. Nat. Chem. 2014, 6, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Ghirardello, M.; Ruiz-de-Angulo, A.; Sacristan, N.; Barriales, D.; Jiménez-Barbero, J.; Poveda, A.; Corzana, F.; Anguita, J.; Fernández-Tejada, A. Exploting structure–activity relationships of QS-21 in the design and synthesis of streamlined saponin vaccine adjuvants. Chem. Commun. 2020, 56, 719–722. [Google Scholar] [CrossRef]
- Le, T.T.; Cramer, J.P.; Chen, R.; Mayhew, S. Evolution of the COVID-19 vaccine development landscape. Nat. Rev. Drug Discov. 2020, 19, 667–668. [Google Scholar] [CrossRef] [PubMed]
- Jeyanathan, M.; Afkhami, S.; Smaill, F.; Miller, M.S.; Lichty, B.D.; Xing, Z. Immunological considerations for COVID-19 vaccine strategies. Nat. Rev. Immunol. 2020, 20, 1–18. [Google Scholar] [CrossRef]
- Huang, X.; Wang, X.; Zhang, J.; Xia, N.; Zhao, Q. Escherichia coli-derived virus-like particles in vaccine development. Npj Vaccines 2017, 2, 1–9. [Google Scholar] [CrossRef]
- Donaldson, B.; Lateef, Z.; Walker, G.F.; Young, S.L.; Ward, V.K. Virus-like particle vaccines: Immunology and formula-tion for clinical translation. Expert. Rev. Vaccines 2018, 17, 833–849. [Google Scholar] [CrossRef]
- Ward, B.J.; Gobeil, P.; Séguin, A.; Atkins, J.; Boulay, I.; Charbonneau, P.Y.; Couture, M.; D’Aoust, M.-A.; Dhaliwall, J.; Finkle, C.; et al. Phase 1 trial of a Candidate Recombinant Virus-Like Particle Vaccine for Covid-19 Disease Produced in Plants. medRxiv 2020. [Google Scholar] [CrossRef]
- Gursel, M.; Gursel, I. Development of CpG ODN Based Vaccine Adjuvant Formulations. Adv. Struct. Saf. Stud. 2016, 1404, 289–298. [Google Scholar] [CrossRef]
- Bode, C.; Zhao, G.; Steinhagen, F.; Kinjo, T.; Klinman, D.M. CpG DNA as a vaccine adjuvant. Expert Rev. Vaccines 2011, 10, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Klimek, L.; Willers, J.; Hammann-Haenni, A.; Pfaar, O.; Stocker, H.; Mueller, P.; Renner, W.A.; Bachmann, M.F. Assessment of clinical efficacy of CYT003-QbG10 in patients with allergic rhinoconjunctivitis: A phase IIb study. Clin. Exp. Allergy 2011, 41, 1305–1312. [Google Scholar] [CrossRef]
- Park, Y.; Casey, D.; Joshi, I.; Zhu, J.; Cheng, F. Emergence of New Disease: How Can Artificial Intelligence Help? Trends Mol. Med. 2020, 26, 627–629. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pharmaceutical function (affecting vaccine antigen delivery) |
| Prolong antigen residence time at the site of administration |
| Protect the nature of the antigen |
| Prevent the antigen from degradation (improve stability) |
| Protect the 3D structure of the exposed antigen epitope(s) |
| Induce an environment that mimics the infection |
| Secure absorption into the lymphatics over systemic circulation |
| Decrease the number of boosters required for successful immune response |
| Have a good safety and toxicological profile |
| Immunological function (impact on immune function) |
| Attract antigen presenting cells to the site of administration |
| Augment immune response type: e.g., Th1, Th2, Th3 or Th17 |
| Augment the generation of memory cells |
| Augment mucosal, or systemic responses |
| Improve the generation of neutralizing antibodies and /or effector T cells |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schijns, V.; Majhen, D.; van der Ley, P.; Thakur, A.; Summerfield, A.; Berisio, R.; Nativi, C.; Fernández-Tejada, A.; Alvarez-Dominguez, C.; Gizurarson, S.; et al. Rational Vaccine Design in Times of Emerging Diseases: The Critical Choices of Immunological Correlates of Protection, Vaccine Antigen and Immunomodulation. Pharmaceutics 2021, 13, 501. https://doi.org/10.3390/pharmaceutics13040501
Schijns V, Majhen D, van der Ley P, Thakur A, Summerfield A, Berisio R, Nativi C, Fernández-Tejada A, Alvarez-Dominguez C, Gizurarson S, et al. Rational Vaccine Design in Times of Emerging Diseases: The Critical Choices of Immunological Correlates of Protection, Vaccine Antigen and Immunomodulation. Pharmaceutics. 2021; 13(4):501. https://doi.org/10.3390/pharmaceutics13040501
Chicago/Turabian StyleSchijns, Virgil, Dragomira Majhen, Peter van der Ley, Aneesh Thakur, Artur Summerfield, Rita Berisio, Cristina Nativi, Alberto Fernández-Tejada, Carmen Alvarez-Dominguez, Sveinbjörn Gizurarson, and et al. 2021. "Rational Vaccine Design in Times of Emerging Diseases: The Critical Choices of Immunological Correlates of Protection, Vaccine Antigen and Immunomodulation" Pharmaceutics 13, no. 4: 501. https://doi.org/10.3390/pharmaceutics13040501
APA StyleSchijns, V., Majhen, D., van der Ley, P., Thakur, A., Summerfield, A., Berisio, R., Nativi, C., Fernández-Tejada, A., Alvarez-Dominguez, C., Gizurarson, S., Zamyatina, A., Molinaro, A., Rosano, C., Jakopin, Ž., Gursel, I., & McClean, S. (2021). Rational Vaccine Design in Times of Emerging Diseases: The Critical Choices of Immunological Correlates of Protection, Vaccine Antigen and Immunomodulation. Pharmaceutics, 13(4), 501. https://doi.org/10.3390/pharmaceutics13040501