
The Ins and Outs of Messenger RNA Electroporation for Physical Gene Delivery in Immune Cell-Based Therapy

 , , , ,
, , , ,
Abstract
1. Introduction
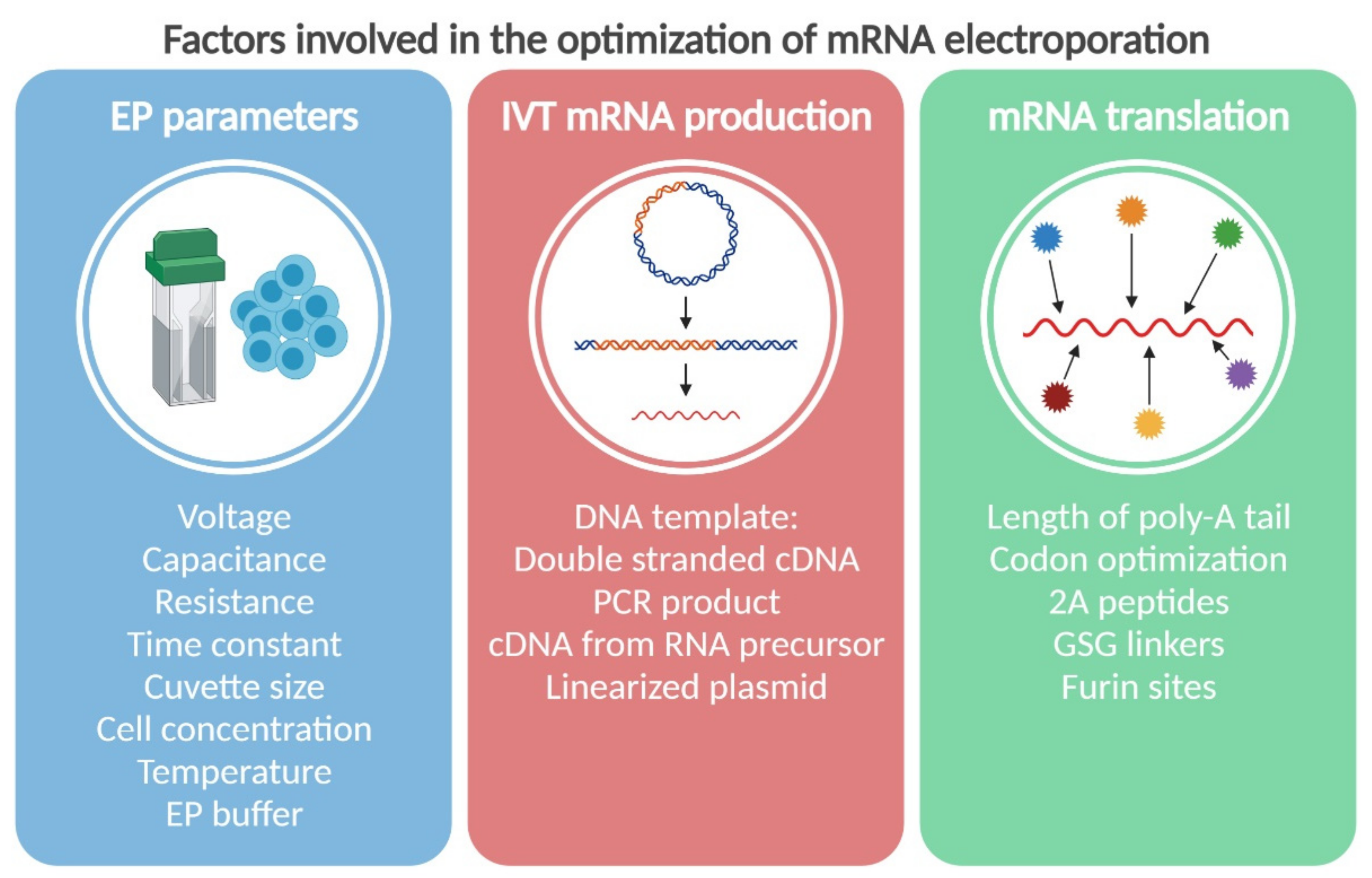
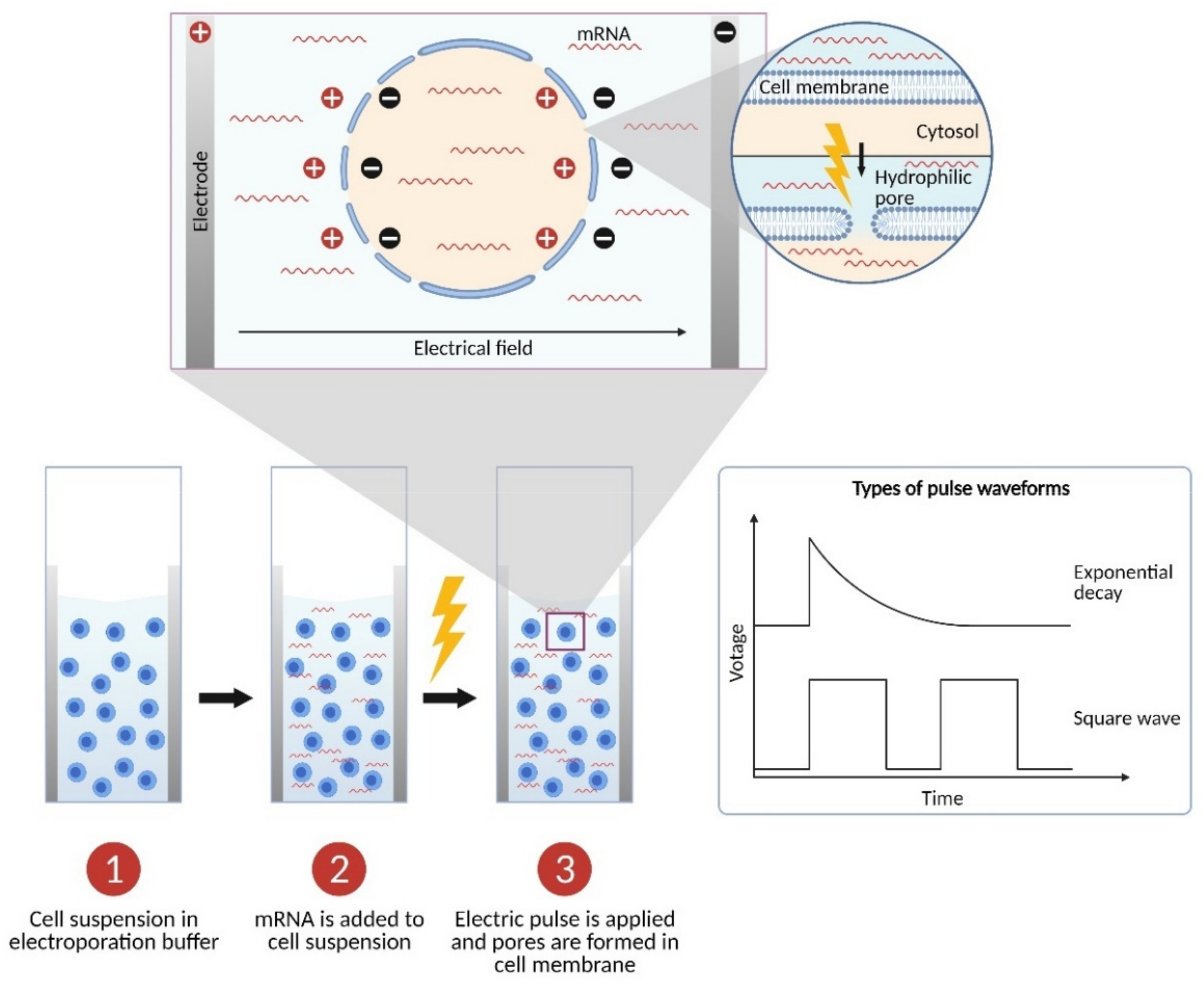
2. The Physics: Parameters of Electroporation
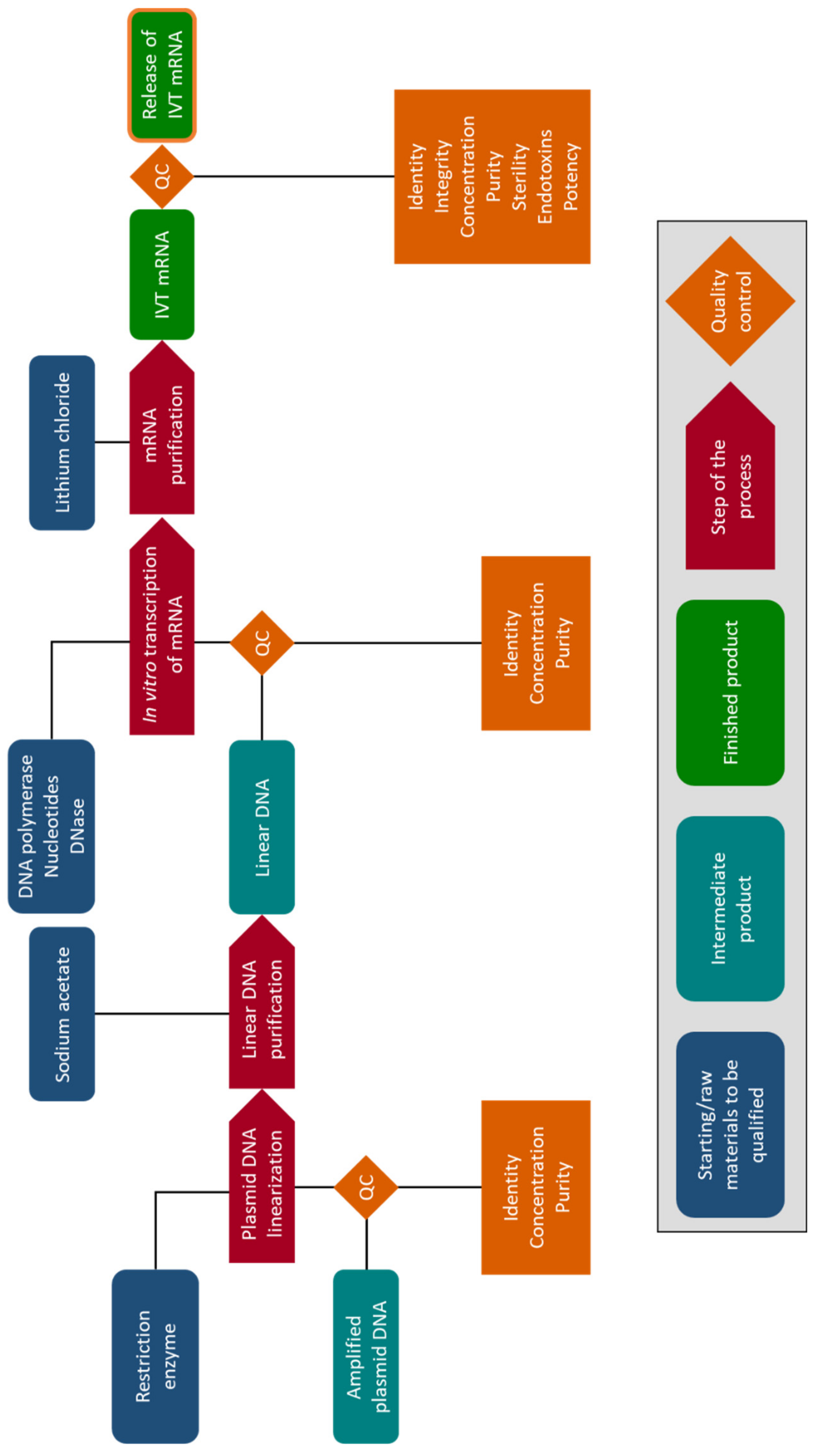
3. The Chemistry: In Vitro Synthesis of mRNA for Electroporation
4. The Biology: How to Improve mRNAs for Better Stability and Translation
5. Clinical Production of mRNA for Electroporation
6. Clinical Application of mRNA Electroporation in Cell-Based Immunotherapies
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rolong, A.; Davalos, R.V.; Rubinsky, B. History of Electroporation. In Irreversible Electroporation in Clinical Practice; Meijerink, M.R., Scheffer, H.J., Narayanan, G., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 13–37. [Google Scholar] [CrossRef]
- Stampfli, R.; Willi, M. Membrane potential of a Ranvier node measured after electrical destruction of its membrane. Experientia 1957, 13, 297–298. [Google Scholar] [CrossRef] [PubMed]
- Neumann, E.; Schaefer-Ridder, M.; Wang, Y.; Hofschneider, P.H. Gene transfer into mouse lyoma cells by electroporation in high electric fields. EMBO J. 1982, 1, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Dullaers, M.; Breckpot, K.; Van Meirvenne, S.; Bonehill, A.; Tuyaerts, S.; Michiels, A.; Straetman, L.; Heirman, C.; De Greef, C.; Van Der Bruggen, P.; et al. Side-by-side comparison of lentivirally transduced and mRNA-electroporated dendritic cells: Implications for cancer immunotherapy protocols. Mol. Ther. 2004, 10, 768–779. [Google Scholar] [CrossRef]
- Devoldere, J.; Dewitte, H.; De Smedt, S.C.; Remaut, K. Evading innate immunity in nonviral mRNA delivery: don’t shoot the messenger. Drug Discov. Today 2016, 21, 11–25. [Google Scholar] [CrossRef]
- Van Tendeloo, V.F.; Ponsaerts, P.; Lardon, F.; Nijs, G.; Lenjou, M.; Van Broeckhoven, C.; Van Bockstaele, D.R.; Berneman, Z.N. Highly efficient gene delivery by mRNA electroporation in human hematopoietic cells: Superiority to lipofection and passive pulsing of mRNA and to electroporation of plasmid cDNA for tumor antigen loading of dendritic cells. Blood 2001, 98, 49–56. [Google Scholar] [CrossRef]
- Smits, E.; Ponsaerts, P.; Lenjou, M.; Nijs, G.; Van Bockstaele, D.R.; Berneman, Z.N.; Van Tendeloo, V.F. RNA-based gene transfer for adult stem cells and T cells. Leukemia 2004, 18, 1898–1902. [Google Scholar] [CrossRef]
- Gehl, J. Electroporation: Theory and methods, perspectives for drug delivery, gene therapy and research. Acta Physiol. Scand. 2003, 177, 437–447. [Google Scholar] [CrossRef]
- Chang, D. Electroporation and Electrofusion. Rev. Cell Biol. Mol. Med. 2006, 2, 198–206. [Google Scholar]
- Luft, C.; Ketteler, R. Electroporation Knows no Boundaries: The Use of Electrostimulation for siRNA Delivery in Cells and Tissues. J. Biomol. Screen. 2015, 20, 932–942. [Google Scholar] [CrossRef]
- Rosazza, C.; Meglic, S.H.; Zumbusch, A.; Rols, M.P.; Miklavcic, D. Gene Electrotransfer: A Mechanistic Perspective. Curr. Gene Ther. 2016, 16, 98–129. [Google Scholar] [CrossRef]
- Heiser, W.C. Optimizing electroporation conditions for the transformation of mammalian cells. Methods Mol. Biol. 2000, 130, 117–134. [Google Scholar] [CrossRef]
- Weaver, J.C.; Smith, K.C.; Esser, A.T.; Son, R.S.; Gowrishankar, T.R. A brief overview of electroporation pulse strength-duration space: A region where additional intracellular effects are expected. Bioelectrochemistry 2012, 87, 236–243. [Google Scholar] [CrossRef]
- Teissié, J. Cell Membrane Electropermeabilization. In Bioelectrochemistry of Membranes; Walz, D., Teissié, J., Milazzo, G., Eds.; Birkhäuser Basel: Basel, Switzerland, 2004; pp. 205–235. [Google Scholar] [CrossRef]
- Sherba, J.J.; Hogquist, S.; Lin, H.; Shan, J.W.; Shreiber, D.I.; Zahn, J.D. The effects of electroporation buffer composition on cell viability and electro-transfection efficiency. Sci. Rep. 2020, 10, 3053. [Google Scholar] [CrossRef]
- Silve, A.; Leray, I.; Poignard, C.; Mir, L.M. Impact of external medium conductivity on cell membrane electropermeabilization by microsecond and nanosecond electric pulses. Sci. Rep. 2016, 6, 19957. [Google Scholar] [CrossRef]
- Pucihar, G.; Kotnik, T.; Kanduser, M.; Miklavcic, D. The influence of medium conductivity on electropermeabilization and survival of cells in vitro. Bioelectrochemistry 2001, 54, 107–115. [Google Scholar] [CrossRef]
- Schlaak, C.; Hoffmann, P.; May, K.; Weimann, A. Desalting minimal amounts of DNA for electroporation in E. coli: A comparison of different physical methods. Biotechnol. Lett. 2005, 27, 1003–1005. [Google Scholar] [CrossRef]
- Bahnson, A.B.; Boggs, S.S. Addition of serum to electroporated cells enhances survival and transfection efficiency. Biochem. Biophys. Res. Commun. 1990, 171, 752–757. [Google Scholar] [CrossRef]
- Welter, J.F.; Solchaga, L.A.; Stewart, M.C. High-efficiency nonviral transfection of primary chondrocytes. Methods Mol. Med. 2004, 100, 129–146. [Google Scholar] [CrossRef]
- Jordan, E.T.; Collins, M.; Terefe, J.; Ugozzoli, L.; Rubio, T. Optimizing electroporation conditions in primary and other difficult-to-transfect cells. J. Biomol. Tech. 2008, 19, 328–334. [Google Scholar]
- Ramanathan, A.; Robb, G.B.; Chan, S.H. mRNA capping: Biological functions and applications. Nucleic Acids Res. 2016, 44, 7511–7526. [Google Scholar] [CrossRef]
- Shi, Y.; Manley, J.L. The end of the message: Multiple protein-RNA interactions define the mRNA polyadenylation site. Genes Dev. 2015, 29, 889–897. [Google Scholar] [CrossRef]
- Moore, M.J. From birth to death: The complex lives of eukaryotic mRNAs. Science 2005, 309, 1514–1518. [Google Scholar] [CrossRef]
- Kuhn, U.; Gundel, M.; Knoth, A.; Kerwitz, Y.; Rudel, S.; Wahle, E. Poly(A) tail length is controlled by the nuclear poly(A)-binding protein regulating the interaction between poly(A) polymerase and the cleavage and polyadenylation specificity factor. J. Biol. Chem. 2009, 284, 22803–22814. [Google Scholar] [CrossRef]
- Wiederhold, K.; Passmore, L.A. Cytoplasmic deadenylation: Regulation of mRNA fate. Biochem. Soc. Trans. 2010, 38, 1531–1536. [Google Scholar] [CrossRef]
- Weill, L.; Belloc, E.; Bava, F.A.; Mendez, R. Translational control by changes in poly(A) tail length: Recycling mRNAs. Nat. Struct. Mol. Biol. 2012, 19, 577–585. [Google Scholar] [CrossRef]
- Russell, J.E.; Liebhaber, S.A. The stability of human beta-globin mRNA is dependent on structural determinants positioned within its 3′ untranslated region. Blood 1996, 87, 5314–5323. [Google Scholar] [CrossRef]
- Holtkamp, S.; Kreiter, S.; Selmi, A.; Simon, P.; Koslowski, M.; Huber, C.; Tureci, O.; Sahin, U. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood 2006, 108, 4009–4017. [Google Scholar] [CrossRef] [PubMed]
- Ferizi, M.; Aneja, M.K.; Balmayor, E.R.; Badieyan, Z.S.; Mykhaylyk, O.; Rudolph, C.; Plank, C. Human cellular CYBA UTR sequences increase mRNA translation without affecting the half-life of recombinant RNA transcripts. Sci. Rep. 2016, 6, 39149. [Google Scholar] [CrossRef] [PubMed]
- Kozak, M. An analysis of 5′-noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res. 1987, 15, 8125–8148. [Google Scholar] [CrossRef]
- Yisraeli, J.K.; Melton, D.A. Synthesis of long, capped transcripts in vitro by SP6 and T7 RNA polymerases. Methods Enzym. 1989, 180, 42–50. [Google Scholar]
- Krieg, P.A.; Melton, D.A. Functional messenger RNAs are produced by SP6 in vitro transcription of cloned cDNAs. Nucleic Acids Res. 1984, 12, 7057–7070. [Google Scholar] [CrossRef] [PubMed]
- Milligan, J.F.; Groebe, D.R.; Witherell, G.W.; Uhlenbeck, O.C. Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res. 1987, 15, 8783–8798. [Google Scholar] [CrossRef]
- Gurevich, V.V.; Pokrovskaya, I.D.; Obukhova, T.A.; Zozulya, S.A. Preparative in vitro mRNA synthesis using SP6 and T7 RNA polymerases. Anal. Biochem. 1991, 195, 207–213. [Google Scholar] [CrossRef]
- Kozak, M. Pushing the limits of the scanning mechanism for initiation of translation. Gene 2002, 299, 1–34. [Google Scholar] [CrossRef]
- Brunelle, J.L.; Green, R. In vitro transcription from plasmid or PCR-amplified DNA. Methods Enzym. 2013, 530, 101–114. [Google Scholar] [CrossRef]
- Avci-Adali, M.; Behring, A.; Steinle, H.; Keller, T.; Krajeweski, S.; Schlensak, C.; Wendel, H.P. In vitro synthesis of modified mRNA for induction of protein expression in human cells. J. Vis. Exp. 2014, e51943. [Google Scholar] [CrossRef]
- Muttach, F.; Muthmann, N.; Rentmeister, A. Synthetic mRNA capping. Beilstein J. Org. Chem. 2017, 13, 2819–2832. [Google Scholar] [CrossRef]
- Grier, A.E.; Burleigh, S.; Sahni, J.; Clough, C.A.; Cardot, V.; Choe, D.C.; Krutein, M.C.; Rawlings, D.J.; Jensen, M.C.; Scharenberg, A.M.; et al. pEVL: A Linear Plasmid for Generating mRNA IVT Templates with Extended Encoded Poly(A) Sequences. Mol. Ther. Nucleic Acids 2016, 5, e306. [Google Scholar] [CrossRef]
- Meador, J.W., 3rd; McElroy, H.E.; Pasloske, B.L.; Milburn, S.C.; Winkler, M.M. pTRIPLEscript: A novel cloning vector for generating in vitro transcripts from tandem promoters for SP6, T7 and T3 RNA polymerase. Biotechniques 1995, 18, 152–157. [Google Scholar]
- Mauro, V.P.; Chappell, S.A. A critical analysis of codon optimization in human therapeutics. Trends Mol. Med. 2014, 20, 604–613. [Google Scholar] [CrossRef]
- Scholten, K.B.; Kramer, D.; Kueter, E.W.; Graf, M.; Schoedl, T.; Meijer, C.J.; Schreurs, M.W.; Hooijberg, E. Codon modification of T cell receptors allows enhanced functional expression in transgenic human T cells. Clin. Immunol. 2006, 119, 135–145. [Google Scholar] [CrossRef]
- De Felipe, P. Polycistronic viral vectors. Curr. Gene Ther. 2002, 2, 355–378. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, Z. IRES-mediated capindependent translation, a path leading to hidden proteome. J. Mol. Cell Biol. 2019, 11, 911–919. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, S.R.; Li, L.H.; Park, H.J.; Park, J.H.; Lee, K.Y.; Kim, M.K.; Shin, B.A.; Choi, S.Y. High cleavage efficiency of a 2A peptide derived from porcine teschovirus-1 in human cell lines, zebrafish and mice. PLoS ONE 2011, 6, e18556. [Google Scholar] [CrossRef]
- Leisegang, M.; Engels, B.; Meyerhuber, P.; Kieback, E.; Sommermeyer, D.; Xue, S.A.; Reuss, S.; Stauss, H.; Uckert, W. Enhanced functionality of T cell receptor-redirected T cells is defined by the transgene cassette. J. Mol. Med. 2008, 86, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Chen, O.; Wall, J.B.J.; Zheng, M.; Zhou, Y.; Wang, L.; Ruth Vaseghi, H.; Qian, L.; Liu, J. Systematic comparison of 2A peptides for cloning multi-genes in a polycistronic vector. Sci. Rep. 2017, 7, 2193. [Google Scholar] [CrossRef]
- De Felipe, P.; Luke, G.A.; Brown, J.D.; Ryan, M.D. Inhibition of 2A-mediated ‘cleavage’ of certain artificial polyproteins bearing N-terminal signal sequences. Biotechnol. J. 2010, 5, 213–223. [Google Scholar] [CrossRef]
- Minskaia, E.; Ryan, M.D. Protein coexpression using FMDV 2A: Effect of “linker” residues. Biomed. Res. Int. 2013, 2013, 291730. [Google Scholar] [CrossRef]
- Yang, S.; Cohen, C.J.; Peng, P.D.; Zhao, Y.; Cassard, L.; Yu, Z.; Zheng, Z.; Jones, S.; Restifo, N.P.; Rosenberg, S.A.; et al. Development of optimal bicistronic lentiviral vectors facilitates high-level TCR gene expression and robust tumor cell recognition. Gene Ther. 2008, 15, 1411–1423. [Google Scholar] [CrossRef]
- Chng, J.; Wang, T.; Nian, R.; Lau, A.; Hoi, K.M.; Ho, S.C.; Gagnon, P.; Bi, X.; Yang, Y. Cleavage efficient 2A peptides for high level monoclonal antibody expression in CHO cells. MAbs 2015, 7, 403–412. [Google Scholar] [CrossRef]
- Fang, J.; Qian, J.J.; Yi, S.; Harding, T.C.; Tu, G.H.; VanRoey, M.; Jooss, K. Stable antibody expression at therapeutic levels using the 2A peptide. Nat. Biotechnol. 2005, 23, 584–590. [Google Scholar] [CrossRef] [PubMed]
- Van Driessche, A.; Van de Velde, A.L.; Nijs, G.; Braeckman, T.; Stein, B.; De Vries, J.M.; Berneman, Z.N.; Van Tendeloo, V.F. Clinical-grade manufacturing of autologous mature mRNA-electroporated dendritic cells and safety testing in acute myeloid leukemia patients in a phase I dose-escalation clinical trial. Cytotherapy 2009, 11, 653–668. [Google Scholar] [CrossRef]
- European Commission. Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the Community Code Relating to Medicinal Products for Human Use. 2001. EU law (EUR-Lex), ELI. 2001. Available online: http://data.europa.eu/eli/dir/2001/83/2012-11-16 (accessed on 19 November 2020).
- European Commission. Commission Directive 2005/28/EC of 8 April 2005 Laying Down Principles and Detailed Guidelines for Good Clinical Practice as Regards Investigational Medicinal Products for Human Use, as Well as the Requirements for Authorisation of the Manufacturing or Importation of Such Products. EU law (EUR-Lex), ELI. 2005. Available online: http://data.europa.eu/eli/dir/2005/28/oj (accessed on 19 November 2020).
- European Commission. Commission Directive 2003/94/EC of 8 October 2003 Laying down the Principles and Guidelines of Good Manufacturing Practice in Respect of Medicinal Products for Human Use and Investigational Medicinal Products for Human Use. Retrieved from EU Law (EUR-Lex), ELI. 2003. Available online: http://data.europa.eu/eli/dir/2003/94/oj (accessed on 19 November 2020).
- EMA. CPMP/ICH/381/95—ICH Harmonised Tripartite Guideline—Validation of Analytical Procedures: Text and Methodology Q2(R1). Available online: https://www.ema.europa.eu/en/ich-q2-r1-validation-analytical-procedures-text-methodology (accessed on 4 November 2020).
- Annunziata, C.M.; Ghobadi, A.; Pennella, E.J.; Vanas, J.; Powell, C.; Pavelova, M.; Wagner, C.; Kuo, M.; Ullmann, C.D.; Hassan, R.; et al. Feasibility and preliminary safety and efficacy of first-in-human intraperitoneal delivery of MCY-M11, anti-human-mesothelin CAR mRNA transfected into peripheral blood mononuclear cells, for ovarian cancer and malignant peritoneal mesothelioma. J. Clin. Oncol. 2020, 38, 3014. [Google Scholar] [CrossRef]
- Derdelinckx, J.; Mansilla, M.J.; De Laere, M.; Lee, W.P.; Navarro-Barriuso, J.; Wens, I.; Nkansah, I.; Daans, J.; De Reu, H.; Jolanta Keliris, A.; et al. Clinical and immunological control of experimental autoimmune encephalomyelitis by tolerogenic dendritic cells loaded with MOG-encoding mRNA. J. Neuroinflamm. 2019, 16, 167. [Google Scholar] [CrossRef]
- Chung, D.J.; Carvajal, R.D.; Postow, M.A.; Sharma, S.; Pronschinske, K.B.; Shyer, J.A.; Singh-Kandah, S.; Dickson, M.A.; D’Angelo, S.P.; Wolchok, J.D.; et al. Langerhans-type dendritic cells electroporated with TRP-2 mRNA stimulate cellular immunity against melanoma: Results of a phase I vaccine trial. Oncoimmunology 2017, 7, e1372081. [Google Scholar] [CrossRef]
- Aarntzen, E.H.; Schreibelt, G.; Bol, K.; Lesterhuis, W.J.; Croockewit, A.J.; de Wilt, J.H.; van Rossum, M.M.; Blokx, W.A.; Jacobs, J.F.; Duiveman-de Boer, T.; et al. Vaccination with mRNA-electroporated dendritic cells induces robust tumor antigen-specific CD4+ and CD8+ T cells responses in stage III and IV melanoma patients. Clin. Cancer Res. 2012, 18, 5460–5470. [Google Scholar] [CrossRef] [PubMed]
- Vik-Mo, E.O.; Nyakas, M.; Mikkelsen, B.V.; Moe, M.C.; Due-Tonnesen, P.; Suso, E.M.; Saeboe-Larssen, S.; Sandberg, C.; Brinchmann, J.E.; Helseth, E.; et al. Therapeutic vaccination against autologous cancer stem cells with mRNA-transfected dendritic cells in patients with glioblastoma. Cancer Immunol. Immunother. 2013, 62, 1499–1509. [Google Scholar] [CrossRef]
- Suso, E.M.; Dueland, S.; Rasmussen, A.M.; Vetrhus, T.; Aamdal, S.; Kvalheim, G.; Gaudernack, G. hTERT mRNA dendritic cell vaccination: Complete response in a pancreatic cancer patient associated with response against several hTERT epitopes. Cancer Immunol. Immunother. 2011, 60, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Saeboe-Larssen, S.; Fossberg, E.; Gaudernack, G. mRNA-based electrotransfection of human dendritic cells and induction of cytotoxic T lymphocyte responses against the telomerase catalytic subunit (hTERT). J. Immunol. Methods 2002, 259, 191–203. [Google Scholar] [CrossRef]
- Bol, K.F.; Aarntzen, E.H.; Pots, J.M.; Olde Nordkamp, M.A.; van de Rakt, M.W.; Scharenborg, N.M.; de Boer, A.J.; van Oorschot, T.G.; Croockewit, S.A.; Blokx, W.A.; et al. Prophylactic vaccines are potent activators of monocyte-derived dendritic cells and drive effective anti-tumor responses in melanoma patients at the cost of toxicity. Cancer Immunol. Immunother. 2016, 65, 327–339. [Google Scholar] [CrossRef]
- Wilgenhof, S.; Van Nuffel, A.M.T.; Benteyn, D.; Corthals, J.; Aerts, C.; Heirman, C.; Van Riet, I.; Bonehill, A.; Thielemans, K.; Neyns, B. A phase IB study on intravenous synthetic mRNA electroporated dendritic cell immunotherapy in pretreated advanced melanoma patients. Ann. Oncol. 2013, 24, 2686–2693. [Google Scholar] [CrossRef]
- Wilgenhof, S.; Corthals, J.; Heirman, C.; Neyns, B.; Thielemans, K. Clinical trials with MRNA electroporated dendritic cells for stage III/IV melanoma patients. J. Immunother. Cancer 2015, 3, P211. [Google Scholar] [CrossRef]
- De Keersmaecker, B.; Claerhout, S.; Carrasco, J.; Bar, I.; Corthals, J.; Wilgenhof, S.; Neyns, B.; Thielemans, K. TriMix and tumor antigen mRNA electroporated dendritic cell vaccination plus ipilimumab: Link between T-cell activation and clinical responses in advanced melanoma. J. Immunother. Cancer 2020, 8, e000329. [Google Scholar] [CrossRef]
- Borch, T.H.; Engell-Noerregaard, L.; Zeeberg Iversen, T.; Ellebaek, E.; Met, O.; Hansen, M.; Andersen, M.H.; Thor Straten, P.; Svane, I.M. mRNA-transfected dendritic cell vaccine in combination with metronomic cyclophosphamide as treatment for patients with advanced malignant melanoma. Oncoimmunology 2016, 5, e1207842. [Google Scholar] [CrossRef]
- Bol, K.; van den Bosch, T.; Schreibelt, G.; Punt, C.; Figdor, C.; Paridaens, D.; de Vries, J. Adjuvant dendritic cell vaccination in high-risk uveal melanoma patients. J. Immunother. Cancer 2015, 3, P127. [Google Scholar] [CrossRef]
- Amin, A.; Dudek, A.; Logan, T.; Lance, R.S.; Holzbeierlein, J.M.; Williams, W.L.; Jain, R.; Chew, T.G.; Nicolette, C.A.; Figlin, R.A.; et al. A phase II study testing the safety and activity of AGS-003 as an immunotherapeutic in subjects with newly diagnosed advanced stage renal cell carcinoma (RCC) in combination with sunitinib. J. Clin. Oncol. 2010, 28, 4588. [Google Scholar] [CrossRef]
- Amin, A.; Dudek, A.Z.; Logan, T.F.; Lance, R.S.; Holzbeierlein, J.M.; Knox, J.J.; Master, V.A.; Pal, S.K.; Miller, W.H., Jr.; Karsh, L.I.; et al. Survival with AGS-003, an autologous dendritic cell-based immunotherapy, in combination with sunitinib in unfavorable risk patients with advanced renal cell carcinoma (RCC): Phase 2 study results. J. Immunother. Cancer 2015, 3, 14. [Google Scholar] [CrossRef]
- Figlin, R.A. Personalized immunotherapy (AGS-003) when combined with sunitinib for the treatment of metastatic renal cell carcinoma. Expert Opin. Biol. Ther. 2015, 15, 1241–1248. [Google Scholar] [CrossRef] [PubMed]
- Figlin, R.A.; Tannir, N.M.; Uzzo, R.G.; Tykodi, S.S.; Chen, D.Y.T.; Master, V.; Kapoor, A.; Vaena, D.; Lowrance, W.; Bratslavsky, G.; et al. Results of the ADAPT Phase 3 Study of Rocapuldencel-T in Combination with Sunitinib as First-Line Therapy in Patients with Metastatic Renal Cell Carcinoma. Clin. Cancer Res. 2020, 26, 2327–2336. [Google Scholar] [CrossRef]
- Batich, K.A.; Reap, E.A.; Archer, G.E.; Sanchez-Perez, L.; Nair, S.K.; Schmittling, R.J.; Norberg, P.; Xie, W.; Herndon, J.E., 2nd; Healy, P.; et al. Long-term Survival in Glioblastoma with Cytomegalovirus pp65-Targeted Vaccination. Clin. Cancer Res. 2017, 23, 1898–1909. [Google Scholar] [CrossRef]
- Mitchell, D.A.; Batich, K.A.; Gunn, M.D.; Huang, M.N.; Sanchez-Perez, L.; Nair, S.K.; Congdon, K.L.; Reap, E.A.; Archer, G.E.; Desjardins, A.; et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature 2015, 519, 366–369. [Google Scholar] [CrossRef]
- Lesterhuis, W.J.; De Vries, I.J.; Schreibelt, G.; Schuurhuis, D.H.; Aarntzen, E.H.; De Boer, A.; Scharenborg, N.M.; Van De Rakt, M.; Hesselink, E.J.; Figdor, C.G.; et al. Immunogenicity of dendritic cells pulsed with CEA peptide or transfected with CEA mRNA for vaccination of colorectal cancer patients. Anticancer Res. 2010, 30, 5091–5097. [Google Scholar]
- Berneman, Z.N.; Germonpre, P.; Huizing, M.T.; Velde, A.V.D.; Nijs, G.; Stein, B.; Tendeloo, V.F.V.; Lion, E.; Smits, E.L.; Anguille, S. Dendritic cell vaccination in malignant pleural mesothelioma: A phase I/II study. J. Clin. Oncol. 2014, 32, 7583. [Google Scholar] [CrossRef]
- Kongsted, P.; Ellebæk, E.; Borch, T.H.; Iversen, T.Z.; Andersen, R.; Met, Ö.; Hansen, M.; Sengeløv, L.; Svane, I.M. Dendritic cell vaccination in combination with docetaxel for patients with prostate cancer – a randomized phase II study. Ann. Oncol. 2016, 27, vi371. [Google Scholar] [CrossRef]
- Van Tendeloo, V.F.; Van de Velde, A.; Van Driessche, A.; Cools, N.; Anguille, S.; Ladell, K.; Gostick, E.; Vermeulen, K.; Pieters, K.; Nijs, G.; et al. Induction of complete and molecular remissions in acute myeloid leukemia by Wilms’ tumor 1 antigen-targeted dendritic cell vaccination. Proc. Natl. Acad. Sci. USA 2010, 107, 13824–13829. [Google Scholar] [CrossRef] [PubMed]
- Anguille, S.; Van de Velde, A.L.; Smits, E.L.; Van Tendeloo, V.F.; Juliusson, G.; Cools, N.; Nijs, G.; Stein, B.; Lion, E.; Van Driessche, A.; et al. Dendritic cell vaccination as postremission treatment to prevent or delay relapse in acute myeloid leukemia. Blood 2017, 130, 1713–1721. [Google Scholar] [CrossRef] [PubMed]
- Khoury, H.J.; Collins, R.H., Jr.; Blum, W.; Stiff, P.S.; Elias, L.; Lebkowski, J.S.; Reddy, A.; Nishimoto, K.P.; Sen, D.; Wirth, E.D., 3rd; et al. Immune responses and long-term disease recurrence status after telomerase-based dendritic cell immunotherapy in patients with acute myeloid leukemia. Cancer 2017, 123, 3061–3072. [Google Scholar] [CrossRef] [PubMed]
- Khoury, H.; Collins, R.; Blum, W.; Stiff, P.; Lebkowski, J.; Wirth, E.D.; Nishimoto, K.; DiPersio, J. Long-term follow-up of patients with acute myelogenous leukemia receiving an autologous telomerase-based dendritic cell vaccine. J. Clin. Oncol. 2015, 33, 7007. [Google Scholar] [CrossRef]
- Lichtenegger, F.S.; Schnorfeil, F.M.; Rothe, M.; Deiser, K.; Altmann, T.; Bucklein, V.L.; Kohnke, T.; Augsberger, C.; Konstandin, N.P.; Spiekermann, K.; et al. Toll-like receptor 7/8-matured RNA-transduced dendritic cells as post-remission therapy in acute myeloid leukaemia: Results of a phase I trial. Clin. Transl. Immunol. 2020, 9, e1117. [Google Scholar] [CrossRef]
- Frauke, S.; Felix, L.; Christiane, G.; Reinhard, H.; Beate, W.; Iris, B.; Gunnar, K.; Dolores, S.; Wolfgang, H.; Marion, S. ITOC2—Vaccination with next-generation dendritic cells for AML postremission therapy induces antigen-specific T cell responses. Eur. J. Cancer 2015, 51, S8. [Google Scholar] [CrossRef]
- Schnorfeil, F.; Lichtenegger, F.; Geiger, C.; Köhnke, T.; Bücklein, V.; Altmann, T.; Wagner, B.; Henschler, R.; Bigalke, I.; Kvalheim, G.; et al. Next-generation dendritic cells for immunotherapy of acute myeloid leukemia. J. Immunother. Cancer 2014, 2, P84. [Google Scholar] [CrossRef]
- Routy, J.P.; Nicolette, C. Arcelis AGS-004 dendritic cell-based immunotherapy for HIV infection. Immunotherapy 2010, 2, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Gay, C.L.; DeBenedette, M.A.; Tcherepanova, I.Y.; Gamble, A.; Lewis, W.E.; Cope, A.B.; Kuruc, J.D.; McGee, K.S.; Kearney, M.F.; Coffin, J.M.; et al. Immunogenicity of AGS-004 Dendritic Cell Therapy in Patients Treated During Acute HIV Infection. AIDS Res. Hum. Retrovir. 2018, 34, 111–122. [Google Scholar] [CrossRef]
- Gay, C.L.; Kuruc, J.D.; Falcinelli, S.D.; Warren, J.A.; Reifeis, S.A.; Kirchherr, J.L.; James, K.S.; Dewey, M.G.; Helms, A.; Allard, B.; et al. Assessing the impact of AGS-004, a dendritic cell-based immunotherapy, and vorinostat on persistent HIV-1 Infection. Sci. Rep. 2020, 10, 5134. [Google Scholar] [CrossRef]
- Routy, J.P.; Boulassel, M.R.; Yassine-Diab, B.; Nicolette, C.; Healey, D.; Jain, R.; Landry, C.; Yegorov, O.; Tcherepanova, I.; Monesmith, T.; et al. Immunologic activity and safety of autologous HIV RNA-electroporated dendritic cells in HIV-1 infected patients receiving antiretroviral therapy. Clin. Immunol. 2010, 134, 140–147. [Google Scholar] [CrossRef]
- Jacobson, J.M.; Routy, J.P.; Welles, S.; DeBenedette, M.; Tcherepanova, I.; Angel, J.B.; Asmuth, D.M.; Stein, D.K.; Baril, J.G.; McKellar, M.; et al. Dendritic Cell Immunotherapy for HIV-1 Infection Using Autologous HIV-1 RNA: A Randomized, Double-Blind, Placebo-Controlled Clinical Trial. J. Acquir. Immune Defic. Syndr. 2016, 72, 31–38. [Google Scholar] [CrossRef]
- Gandhi, R.T.; Kwon, D.S.; Macklin, E.A.; Shopis, J.R.; McLean, A.P.; McBrine, N.; Flynn, T.; Peter, L.; Sbrolla, A.; Kaufmann, D.E.; et al. Immunization of HIV-1-Infected Persons With Autologous Dendritic Cells Transfected With mRNA Encoding HIV-1 Gag and Nef: Results of a Randomized, Placebo-Controlled Clinical Trial. J. Acquir. Immune Defic. Syndr. 2016, 71, 246–253. [Google Scholar] [CrossRef]
- Allard, S.D.; De Keersmaecker, B.; de Goede, A.L.; Verschuren, E.J.; Koetsveld, J.; Reedijk, M.L.; Wylock, C.; De Bel, A.V.; Vandeloo, J.; Pistoor, F.; et al. A phase I/IIa immunotherapy trial of HIV-1-infected patients with Tat, Rev and Nef expressing dendritic cells followed by treatment interruption. Clin. Immunol. 2012, 142, 252–268. [Google Scholar] [CrossRef] [PubMed]
- Van Craenenbroeck, A.H.; Smits, E.L.; Anguille, S.; Van de Velde, A.; Stein, B.; Braeckman, T.; Van Camp, K.; Nijs, G.; Ieven, M.; Goossens, H.; et al. Induction of cytomegalovirus-specific T cell responses in healthy volunteers and allogeneic stem cell recipients using vaccination with messenger RNA-transfected dendritic cells. Transplantation 2015, 99, 120–127. [Google Scholar] [CrossRef]
- Maus, M.V.; Haas, A.R.; Beatty, G.L.; Albelda, S.M.; Levine, B.L.; Liu, X.; Zhao, Y.; Kalos, M.; June, C.H. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol. Res. 2013, 1, 26–31. [Google Scholar] [CrossRef]
- Zhao, Y.; Moon, E.; Carpenito, C.; Paulos, C.M.; Liu, X.; Brennan, A.L.; Chew, A.; Carroll, R.G.; Scholler, J.; Levine, B.L.; et al. Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res. 2010, 70, 9053–9061. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Haas, A.R.; Maus, M.V.; Torigian, D.A.; Soulen, M.C.; Plesa, G.; Chew, A.; Zhao, Y.; Levine, B.L.; Albelda, S.M.; et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol. Res. 2014, 2, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; O’Hara, M.H.; Lacey, S.F.; Torigian, D.A.; Nazimuddin, F.; Chen, F.; Kulikovskaya, I.M.; Soulen, M.C.; McGarvey, M.; Nelson, A.M.; et al. Activity of Mesothelin-Specific Chimeric Antigen Receptor T Cells Against Pancreatic Carcinoma Metastases in a Phase 1 Trial. Gastroenterology 2018, 155, 29–32. [Google Scholar] [CrossRef]
- Beatty, G.L.; O’Hara, M.H.; Nelson, A.M.; McGarvey, M.; Torigian, D.A.; Lacey, S.F.; Melenhorst, J.J.; Levine, B.; Plesa, G.; June, C.H. Safety and antitumor activity of chimeric antigen receptor modified T cells in patients with chemotherapy refractory metastatic pancreatic cancer. J. Clin. Oncol. 2015, 33, 3007. [Google Scholar] [CrossRef]
- Tchou, J.; Zhao, Y.; Levine, B.L.; Zhang, P.J.; Davis, M.M.; Melenhorst, J.J.; Kulikovskaya, I.; Brennan, A.L.; Liu, X.; Lacey, S.F.; et al. Safety and Efficacy of Intratumoral Injections of Chimeric Antigen Receptor (CAR) T Cells in Metastatic Breast Cancer. Cancer Immunol. Res. 2017, 5, 1152–1161. [Google Scholar] [CrossRef]
- Shah, P.D.; Huang, A.C.C.; Xu, X.; Zhang, P.J.; Orlowski, R.; Matlawski, T.; Shea, J.; Cervini, A.; Amaravadi, R.K.; Tchou, J.C.; et al. Phase I trial of autologous cMET-directed CAR-t cells administered intravenously in patients with melanoma & breast carcinoma. J. Clin. Oncol. 2020, 38, 10035. [Google Scholar] [CrossRef]
- Tan, A.T.; Yang, N.; Lee Krishnamoorthy, T.; Oei, V.; Chua, A.; Zhao, X.; Tan, H.S.; Chia, A.; Le Bert, N.; Low, D.; et al. Use of Expression Profiles of HBV-DNA Integrated Into Genomes of Hepatocellular Carcinoma Cells to Select T Cells for Immunotherapy. Gastroenterology 2019, 156, 1862–1876.E9. [Google Scholar] [CrossRef] [PubMed]
- Koh, S.; Shimasaki, N.; Suwanarusk, R.; Ho, Z.Z.; Chia, A.; Banu, N.; Howland, S.W.; Ong, A.S.; Gehring, A.J.; Stauss, H.; et al. A practical approach to immunotherapy of hepatocellular carcinoma using T cells redirected against hepatitis B virus. Mol. Ther. Nucleic Acids 2013, 2, e114. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Cheng, J.; Zheng, X.; Yang, F.; Fam, R.; Koh, S.; Wai, L.-E.; Wang, T.; Bertoletti, A.; Zhang, Q. 273 Phase i study of LioCyx-M, autologous hepatitis B virus (HBV)-specific T cell receptor (TCR) T-cells, in recurrent HBV-related hepatocellular carcinoma (HCC) post-liver transplantation. J. Immunother. Cancer 2020, 8, A167. [Google Scholar] [CrossRef]
- Wang, F.-S.; Meng, F.; Jin, J.; Li, Y.; Wong, R.W.; Tan, A.T.; Wang, T.; Bertoletti, A. 272 Use of LioCyx-M, autologous hepatitis B virus (HBV)-Specific T cell receptor (TCR) T-cells, in advanced HBV-related hepatocellular carcinoma (HCC). J. Immunother. Cancer 2020, 8, A166–A167. [Google Scholar] [CrossRef]
- Inderberg, E.M.; Walchli, S. Long-term surviving cancer patients as a source of therapeutic TCR. Cancer Immunol Immunother 2020, 69, 859–865. [Google Scholar] [CrossRef]
- Inderberg, E.M.; Walchli, S.; Myhre, M.R.; Trachsel, S.; Almasbak, H.; Kvalheim, G.; Gaudernack, G. T cell therapy targeting a public neoantigen in microsatellite instable colon cancer reduces in vivo tumor growth. Oncoimmunology 2017, 6, e1302631. [Google Scholar] [CrossRef]
- Svoboda, J.; Rheingold, S.R.; Gill, S.I.; Grupp, S.A.; Lacey, S.F.; Kulikovskaya, I.; Suhoski, M.M.; Melenhorst, J.J.; Loudon, B.; Mato, A.R.; et al. Nonviral RNA chimeric antigen receptor-modified T cells in patients with Hodgkin lymphoma. Blood 2018, 132, 1022–1026. [Google Scholar] [CrossRef]
- Riet, T.; Holzinger, A.; Dorrie, J.; Schaft, N.; Schuler, G.; Abken, H. Nonviral RNA transfection to transiently modify T cells with chimeric antigen receptors for adoptive therapy. Methods Mol. Biol. 2013, 969, 187–201. [Google Scholar] [CrossRef]
- Svoboda, J.; Rheingold, S.R.; Gill, S.I.; Grupp, S.A.; Lacey, S.F.; Melenhorst, J.J.; Kulikovskaya, I.; Loudon, B.; Kerr, N.; Marcucci, K.T.; et al. Pilot Study of Non-Viral, RNA-Redirected Autologous Anti-CD19 Chimeric Antigen Receptor Modified T-Cells in Patients with Refractory/Relapsed Hodgkin Lymphoma (HL). Blood 2017, 130, 653. [Google Scholar] [CrossRef]
- Poon, M.; Linn, Y.C.; Shimasaki, N.; Tan, L.K.; Koh, L.P.; Coustan-Smith, E.; Campana, D. A First-in-Human Study of Autologous T Lymphocytes with Antibody-Dependent Cell Cytotoxicity (ADCC) in Patients with B-Cell Non-Hodgkin Lymphoma (NHL). Blood 2016, 128, 3031. [Google Scholar] [CrossRef]
- Tasian, S.K.; Kenderian, S.S.; Shen, F.; Ruella, M.; Shestova, O.; Kozlowski, M.; Li, Y.; Schrank-Hacker, A.; Morrissette, J.J.D.; Carroll, M.; et al. Optimized depletion of chimeric antigen receptor T cells in murine xenograft models of human acute myeloid leukemia. Blood 2017, 129, 2395–2407. [Google Scholar] [CrossRef]
- Barrett, D.M.; Liu, X.; Jiang, S.; June, C.H.; Grupp, S.A.; Zhao, Y. Regimen-specific effects of RNA-modified chimeric antigen receptor T cells in mice with advanced leukemia. Hum. Gene Ther. 2013, 24, 717–727. [Google Scholar] [CrossRef]
- Cummins, K.D.; Frey, N.; Nelson, A.M.; Schmidt, A.; Luger, S.; Isaacs, R.E.; Lacey, S.F.; Hexner, E.; Melenhorst, J.J.; June, C.H.; et al. Treating Relapsed / Refractory (RR) AML with Biodegradable Anti-CD123 CAR Modified T Cells. Blood 2017, 130, 1359. [Google Scholar] [CrossRef]
- Lin, L.; Cho, S.F.; Xing, L.; Wen, K.; Li, Y.; Yu, T.; Hsieh, P.A.; Chen, H.; Kurtoglu, M.; Zhang, Y.; et al. Preclinical evaluation of CD8+ anti-BCMA mRNA CAR T cells for treatment of multiple myeloma. Leukemia 2020. [Google Scholar] [CrossRef]
- Lin, L.; Xing, L.; Cho, S.-F.; Wen, K.; Hsieh, P.A.; Kurtoglu, M.; Zhang, Y.; Stewart, C.A.; Anderson, K.C.; Tai, Y.-T. Preclinical Evaluation of CD8+ Anti-Bcma mRNA CAR T-Cells for the Control of Human Multiple Myeloma. Blood 2019, 134, 1811. [Google Scholar] [CrossRef]
- Tebas, P.; Jadlowsky, J.K.; Shaw, P.A.; Tian, L.; Esparza, E.; Brennan, A.; Kim, S.; Naing, S.Y.; Richardson, M.W.; Vogel, A.N.; et al. CCR5-edited CD4 T cells augment HIV-specific immunity to enable post rebound control of HIV replication. J. Clin. Investig. 2021. [Google Scholar] [CrossRef]
- Xiao, L.; Cen, D.; Gan, H.; Sun, Y.; Huang, N.; Xiong, H.; Jin, Q.; Su, L.; Liu, X.; Wang, K.; et al. Adoptive Transfer of NKG2D CAR mRNA-Engineered Natural Killer Cells in Colorectal Cancer Patients. Mol. Ther. 2019, 27, 1114–1125. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Gene(s) | mRNA Synthesis | EP Conditions | Clinical Trial Identifier and References | ||
|---|---|---|---|---|---|---|
| Template | Production | Device | Settings | |||
| Solid malignancies | ||||||
| Melanoma | TAA (murine TRP2) | Linearized pING vector | mMessage mMachine T7 kit | BTX ECM 830 square wave electroporator | 700 V (two pulses) 2-mm cuvette | NCT01456104 [61] |
| Melanoma | TAA (gp100, tyrosinase) | Linearized pGEM4Z/hgp100/A64 pGEM4Z/tyrosinase/A64 vectors | Produced by CureVac GmbH Purified by PUREmessengerTM (chromatography) | Gene Pulser Xcell (Bio-Rad) | Exponential decay pulse (300 V, 150 μF) 4-mm cuvette | NCT00243529 [62] |
| Melanoma | TAA (h-TERT, survivin) + tumor cell mRNA | ND | T7 mMESSAGE mMACHINE large-scale transcription kit (Ambion) Purified with MEGAclear column (Ambion) | BTX ECM 830 square wave electroporator | Square wave pulse | NCT00961844 [63,64,65] |
| Melanoma | TAA (gp100 and tyrosinase) + immune modulating molecules (active TLR4, CD70) | Linearized pGEM4Z/hgp100/A64 pGEM4Z/tyrosinase/A64 vectors | Produced by CureVac GmbH Purified by PUREmessenger technology (chromatography) | Gene Pulser Xcell (Bio-Rad) | Exponential decay pulse (300 V, 150 μF) 4-mm cuvette | NCT01530698 NCT00940004 [62,66] |
| Melanoma | TAA (MAGE-A3, MAGE-C2, tyrosinase, gp100) + immune modulating molecules (CD40L, CD70, caTLR4) (TriMixDC-MEL product) | Linearized pGEM-CD40L pGEM-CD70 pGEM-caTLR4 pGEM-sig-MageA3-DCLamp pGEM-sig-MageC2-DCLamp pGEM-sig-gp100-Lamp pGEM-sig-tyrosinase-Lamp vectors | mMESSAGE mMACHINE Ultra T7 Kit Length, concentration and purity evaluated with Agilent 2100 Bioanalyzer (Agilent Technologies) using RNA 6000 Nano LabChip Kit (Agilent Technologies) | EQUIBIO Easyject Plus | 300 V, 450 μF, 99 Ω (pulse time ~5 ms) | NCT01066390 [67,68] NCT01676779 [68] NCT01302496 [68,69] |
| Breast cancer Melanoma | TAA (hTERT, survivin, p53) | Linearized pCI/hTERT/A102 pSP73/p53/A64 pSP73/survivin/A64 vectors | mMESSAGE mMACHINE T7 Ultra kit (Life Technologies) Purified with MEGAclear kit (Ambion) Length, concentration, and purity evaluated with Agilent 2100 Bioanalyzer (Agilent Technologies) using RNA 6000 Nano LabChip Kit (Agilent Technologies) | BTX ECM 830 square wave electroporator | Square wave pulse (500 V, 2 ms) 4-mm cuvette (placed for 5 min on ice) | NCT00978913 [70] |
| Uveal melanoma | TAA (gp100, tyrosinase) | ND | ND | ND | ND | NCT00929019 [71] |
| Renal cell carcinoma | huCD40L + autologous tumor cell mRNA (AGS-003 product) | Linearized pCR2.1/CD40L wt vector from pCR2.1 (Invitrogen) | mMessage mMachine T7 Ultra kits (Ambion) Purified using RNeasy column (QIAGEN) | Bio-Rad | 4-mm cuvette | NCT00272649 [72] NCT00678119 [73,74] NCT01582672 [74,75] |
| Renal cell carcinoma | huCD40L + autologous tumor cell mRNA (AGS-003 product) | ND | ND | ND | ND | NCT02170389 NCT01482949 NCT04203901 |
| Bladder urothelial carcinoma | huCD40L + autologous tumor cell mRNA (AGS-003-BLD product) | ND | ND | ND | ND | NCT02944357 |
| Non-small cell lung cancer | huCD40L + autologous tumor cell mRNA (AGS-003-LNG product) | ND | ND | ND | ND | NCT02662634 |
| Lung cancer | TAA (MIDRIX4-LUNG product) | ND | ND | ND | ND | NCT04082182 |
| Glioblastoma multiforme | CMV pp65-LAMP | pp65-LAMP/A64 | ND | ND | ND | NCT00626483 |
| Glioblastoma multiforme | CMV pp65-LAMP | pp65-LAMP/A64 | ND | ND | ND | NCT00639639 [76,77] |
| Colorectal cancer | TAA (CEA) | ND | ND | Gene Pulser Xcell (Bio-Rad) | Exponential decay pulse (300 V, 150 μF) 4-mm cuvette | NCT00228189 [78] |
| Solid tumors (malignant pleural mesothelioma) | TAA (WT1) | Linearized pGEM/WT1 pST1/sig-WT1-DC-LAMP pST1/sig-WT1-DC-LAMP-OPT (codon-optimized version of pST1/sig-WT1-DC-LAMP) vectors | Produced by CureVac GmbH | Gene Pulser Xcell (Bio-Rad) | Exponential decay pulse (300 V, 7 ms) 4-mm cuvette | NCT01291420 [79] |
| Prostate cancer | TAA (PSA, PAP, survivin, hTERT) | ND | ND | ND | ND | NCT01446731 [80] |
| Hematological malignancies | ||||||
| Hematological malignancies | TAA | ND | ND | ND | ND | NCT02528682 |
| Acute myeloid leukemia | TAA | ND | ND | ND | ND | NCT01686334 |
| Acute myeloid leukemia | TAA (WT1) | ND | Produced by CureVac GmbH | Gene Pulser Xcell (Bio-Rad) | Exponential decay pulse (300 V, 7 ms) 4-mm cuvette | NCT00834002 [81] |
| Acute myeloid leukemia Chronic myeloid leukemia Multiple myeloma | TAA (WT1) | Linearized pGEM/WT1 pST1/sig-WT1-DC-LAMP pST1/sig-WT1-DC-LAMP-OPT (codon-optimized version of pST1/sig-WT1-DC-LAMP) vectors | Produced by CureVac GmbH | Gene Pulser Xcell (Bio-Rad) | Exponential decay pulse (300 V, 7 ms) 4-mm cuvette | NCT00965224 [82] |
| Acute myeloid leukemia | TAA (hTERT-LAMP-1) | Linearized pGEM4Z/hTERT/LAMP/A64 vector | mMESSAGE mMACHINE high yield capped RNA transcription kit (Ambion) Purified with RNeasy kit (Qiagen) | Gene Pulser II (Bio-Rad) | Cells + mRNA for 5 min on ice Exponential decay pulse (300 V, 150 μF) 4-mm cuvette | NCT00510133 [83,84] |
| Acute myeloid leukemia | TAA (WT1 isoform A, PRAME, CMV pp65) | Codon-optimized mRNA | Produced at Oslo University Hospital | ND | ND | NCT01734304 [85,86,87] |
| Myelodysplastic syndromes Acute myeloid leukemia | TAA | ND | ND | ND | ND | NCT03083054 |
| Multiple myeloma | TAA | ND | ND | ND | ND | NCT01995708 |
| Infectious diseases | ||||||
| HIV | HIV antigen (Gag, Nef, Vpr, Rev (GNVR)) + immune modulating molecules (hCD40L) (AGS-004 product) | HIV antigens: PCR fragments hCD40L: Linearized pCR2.1 vector | mMessage mMachine T7 Ultra kit (Life Technologies) Purified with RNeasy columns (QIAGEN) | ND | ND | NCT02042248 [88,89] NCT02707900 [90] NCT00381212 [91] NCT01069809 NCT00672191 [92] |
| HIV | HIV antigen (HIV-1 Gag, Nef) | Codon-optimized coding sequence including endoplasmic reticulum translocation signal peptide, antigen polypeptide, and human lysosome-associated membrane protein-1 targeting sequence | Produced by Asuragen | Gene Pulser II (Bio-Rad) | Square wave pulse (900 V, 0.75 ms) | NCT00833781 [93] |
| HIV | HIV antigen (Tat, Rev, Nef, Gag, NP1) | Linearized pGEM-sig-Tat-DC-LAMP pGEM-sig-Rev-DC-LAMP pGEM-sig-Nef-DC-LAMP pST1-sig-Gag-DC-LAMP pGEM-Sig-Flu-NP1-DC-LAMP vectors | mMESSAGE mMACHINE™ kit (Life Technologies) | EQUIBIO EasyjecT Plus® (EQUIBIO) | 12 × 106 DC: 300 V, 150 μF, 99 Ω (pulse time 5–6 ms) 50 × 106 DC: 300 V, 450 μF, 99 Ω 4-mm cuvette | VUB-05-001 MEC-2005-227 [94] |
| CMV | CMV pp65 | ND | Produced by Curevac GmbH | ND | ND | EudraCT 2008-006074-15 EudraCT 2008-000430-45 [95] |
| Condition | Gene | mRNA Synthesis | EP Conditions | Clinical Trial Identifier and References | ||
|---|---|---|---|---|---|---|
| Template | Production | Device | Settings | |||
| Solid malignancies | ||||||
| Malignant peritoneal mesothelioma | CAR | Linearized pDrive vector (Qiagen) (GOI + two repeats of 3′-UTR from beta globulin (2bgUTR) with or without 150 poly(A) tail) | mMESSAGE mMACHINE T7 kit (including regular cap analog; Life Technologies) mMESSAGE mMACHINE T7 Ultra kit (including anti-reverse cap analog; Life Technologies) mScript™ RNA System (including capping enzyme and 2′-O-Methyltransferase capping enzyme to generate Cap 1 IVT RNA; Epicentre) | BTX ECM 830 square wave electroporator/Maxcyte | 2-mm cuvette (BTX) /OC-400 (Maxcyte) | NCT01355965 [96,97,98] |
| Pancreatic ductal adenocarcinoma Breast cancer | CAR | Linearized pDrive vector (Qiagen) (GOI + two repeats of 3′-UTR from beta globulin (2bgUTR) with or without 150 poly(A) tail) | mMESSAGE mMACHINE T7 kit (including regular cap analog; Life Technologies) mMESSAGE mMACHINE T7 Ultra kit (including anti-reverse cap analog; Life Technologies) mScript™ RNA System (including capping enzyme and 2′-O-Methyltransferase capping enzyme to generate Cap 1 IVT RNA; Epicentre) | Maxcyte | ND | NCT01897415 [98,99,100] NCT01837602 [98,99,100,101] |
| Breast cancer | CAR | ND | ND | ND | ND | NCT03060356 [102] |
| Hepatocellular carcinoma | TCR | Linearized pVAX1 vector | mMESSAGE mMACHINE T7 Ultra kit (including anti-reverse cap analog; Life Technologies) Concentrated by lithium chloride precipitation Dissolved in T4 buffer (BTX) | AgilePulse Max system (BTX) | Manufacturer’s recommended protocol | NCT02719782 [103,104,105] NCT03634683 [103,104] NCT03899415 [103,104,106] |
| Hepatocellular carcinoma | TCR | ND | ND | ND | ND | NCT04745403 |
| Colorectal cancer | TCR | mRNA expression vector Sequence containing 2A construct | Capping: Anti-Reverse Cap Analog (TriLink Biotechnologies Inc.) | BTX ECM 830 square wave electroporator | Square Wave pulse (500 V, 2 ms) 4-mm cuvette | NCT03431311 [107,108] |
| Hematological malignancies | ||||||
| Hodgkin lymphoma | CAR | Linearized pGEM4-Z/A64 vector | mMESSAGE mMACHINE T7 Ultra kit (including anti-reverse cap analog and in vitro poly(A) tailing (“E-PAP”); Life Technologies) | Gene Pulser Xcell (BioRad) | Square wave pulse (500 V, 5 ms) | NCT02277522 NCT02624258 [109,110,111] |
| B-cell non-Hodgkin’s lymphoma B-cell chronic lymphocytic leukemia | CAR | ND | ND | ND | ND | NCT02315118 [112] |
| Acute myeloid leukemia | CAR | Linearized pDA vector | mMESSAGE mMACHINE T7 Ultra kit (including anti-reverse cap analog; Life Technologies) mRNA purified by RNeasy Mini Kit (Qiagen) | BTX ECM 830 square wave electroporator | 2-mm cuvette | NCT02623582 [113,114,115] |
| Multiple myeloma | CAR | Linearized DNA plasmid Codon-optimized nucleotide sequence containing 3′-UTR, mouse alpha globin 5′-UTR, and poly(A) tail | ND | ND | ND | NCT03448978 [116,117] |
| Autoimmune diseases | ||||||
| Type 1 diabetes | Peptide-MHC-CD3-zeta construct | ND | ND | ND | ND | NCT02117518 |
| Infectious diseases | ||||||
| HIV | ZFN | Linearized pDA-A.2bg.150A vector | mMESSAGE mMACHINE T7 Ultra kit (including anti-reverse cap analog; Life Technologies) mRNA purified by RNeasy Maxi kit (Qiagen) | MaxCyte GTTM Flow Transfection System | ND | NCT02388594 [118] |
| Condition | Gene | mRNA Synthesis | EP Conditions | Clinical Trial Identifier and Reference | ||
|---|---|---|---|---|---|---|
| Template | Production | Device | Settings | |||
| Colorectal cancer | CAR | PCR product from pFBCMV-T7 vector GOI + 5′-UTR with Kozak sequence, and ClaI, the GM-CSF signal peptide encoding sequence (SP) and the alpha-globin 3′-UTR sequence | mMESSAGE mMACHINE T7 Ultra kit (including anti-reverse cap analog (ARCA); Life Technologies) mScript™ RNA system (Epicentre) | NEPA21 electroporator (Nepagene) BTX electroporator (AgilePulse) | 2 or 4-mm cuvette | NCT03415100 [119] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campillo-Davo, D.; De Laere, M.; Roex, G.; Versteven, M.; Flumens, D.; Berneman, Z.N.; Van Tendeloo, V.F.I.; Anguille, S.; Lion, E. The Ins and Outs of Messenger RNA Electroporation for Physical Gene Delivery in Immune Cell-Based Therapy. Pharmaceutics 2021, 13, 396. https://doi.org/10.3390/pharmaceutics13030396
Campillo-Davo D, De Laere M, Roex G, Versteven M, Flumens D, Berneman ZN, Van Tendeloo VFI, Anguille S, Lion E. The Ins and Outs of Messenger RNA Electroporation for Physical Gene Delivery in Immune Cell-Based Therapy. Pharmaceutics. 2021; 13(3):396. https://doi.org/10.3390/pharmaceutics13030396
Chicago/Turabian StyleCampillo-Davo, Diana, Maxime De Laere, Gils Roex, Maarten Versteven, Donovan Flumens, Zwi N. Berneman, Viggo F. I. Van Tendeloo, Sébastien Anguille, and Eva Lion. 2021. "The Ins and Outs of Messenger RNA Electroporation for Physical Gene Delivery in Immune Cell-Based Therapy" Pharmaceutics 13, no. 3: 396. https://doi.org/10.3390/pharmaceutics13030396
APA StyleCampillo-Davo, D., De Laere, M., Roex, G., Versteven, M., Flumens, D., Berneman, Z. N., Van Tendeloo, V. F. I., Anguille, S., & Lion, E. (2021). The Ins and Outs of Messenger RNA Electroporation for Physical Gene Delivery in Immune Cell-Based Therapy. Pharmaceutics, 13(3), 396. https://doi.org/10.3390/pharmaceutics13030396