
The Development and Optimization of Hot-Melt Extruded Amorphous Solid Dispersions Containing Rivaroxaban in Combination with Polymers

 , , and
, , and
Abstract
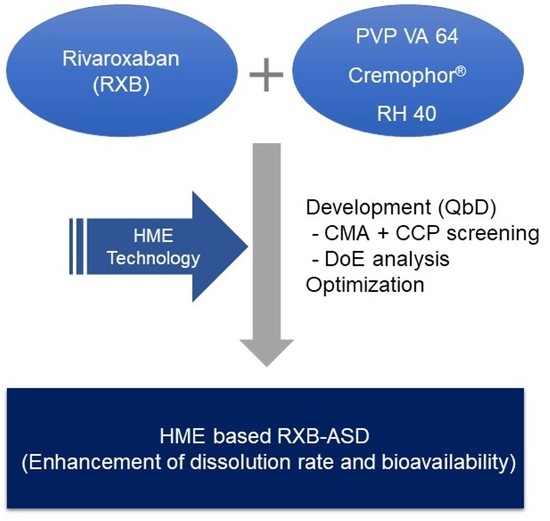
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Before the Study
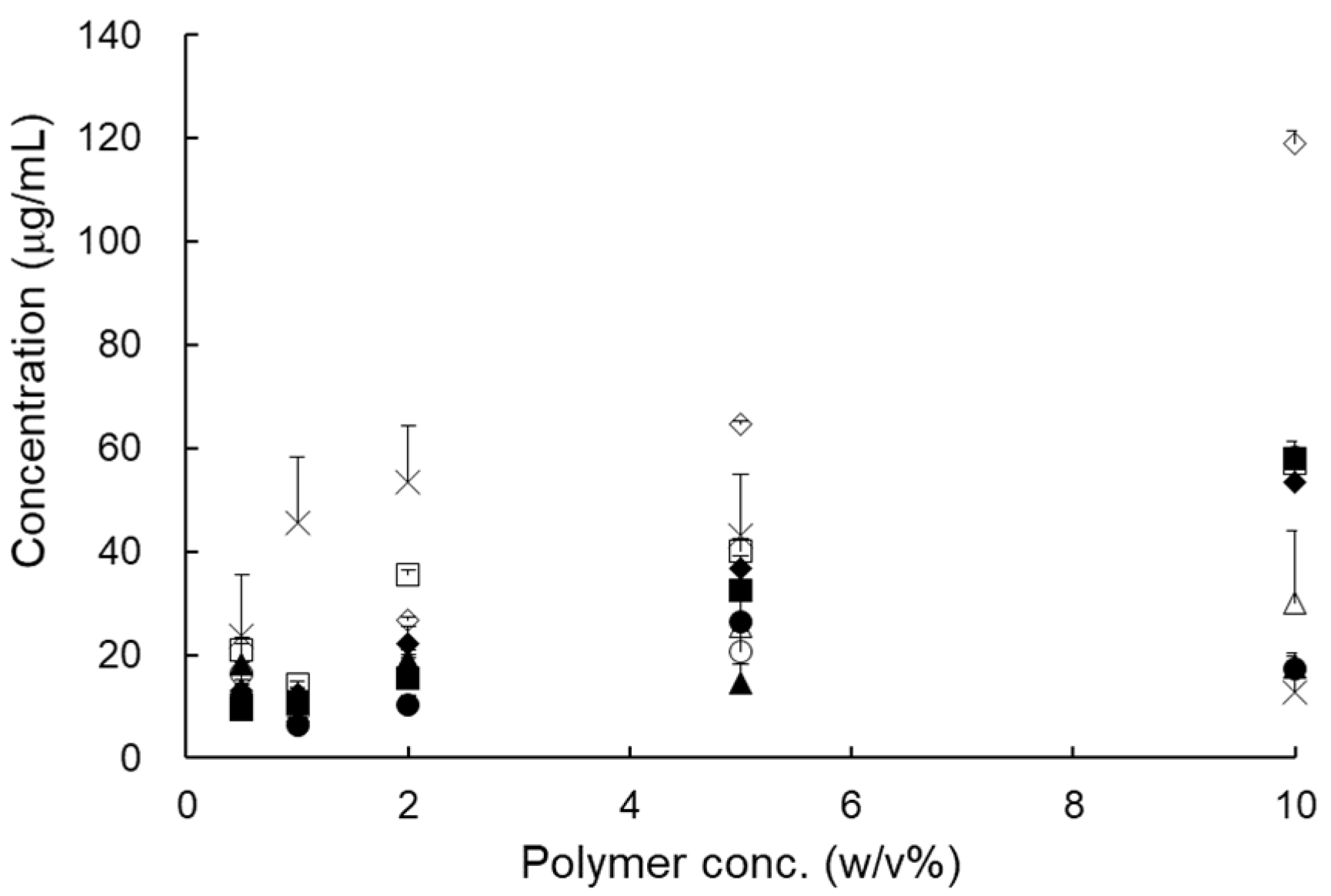
2.2.1. Selection of a Carrier
2.2.2. HME Process Condition
2.2.3. Preformulation Study of RXB-ASDs Using a Full Factorial Design (FFD)
2.3. Optimization by Central Composite Design (CCD)
2.4. Physicochemical Evaluation of RXB-ASD
2.5. Drug Content, In Vitro Dissolution Test, and Food Effect
2.6. Stability
2.7. Application to Pharmacokinetic Study
3. Results and Discussion
3.1. Before the Study
3.1.1. Selection of Carrier
3.1.2. HME Process Conditions
3.1.3. Preformulation Study of RXB-ASDs by FFD
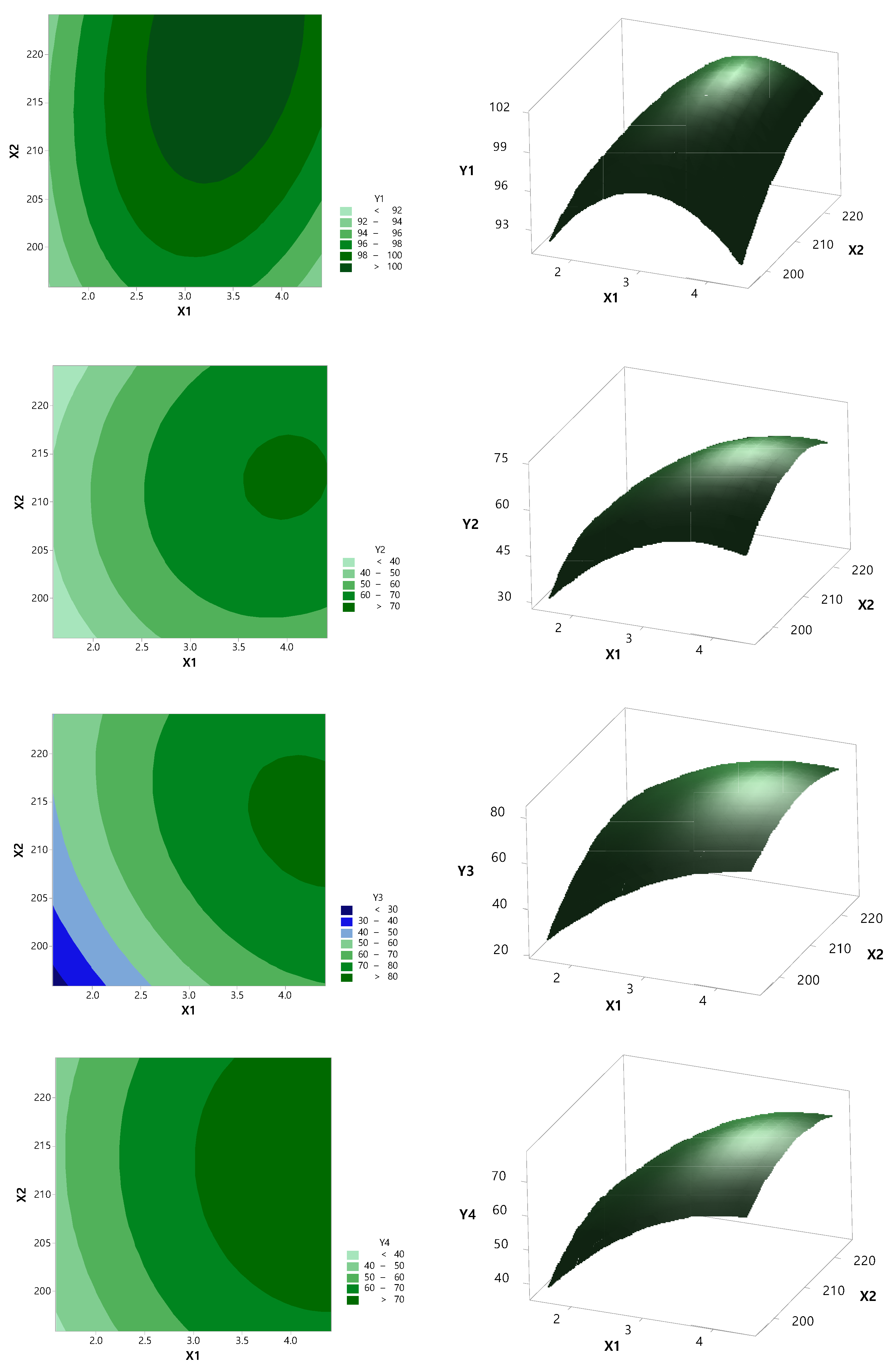
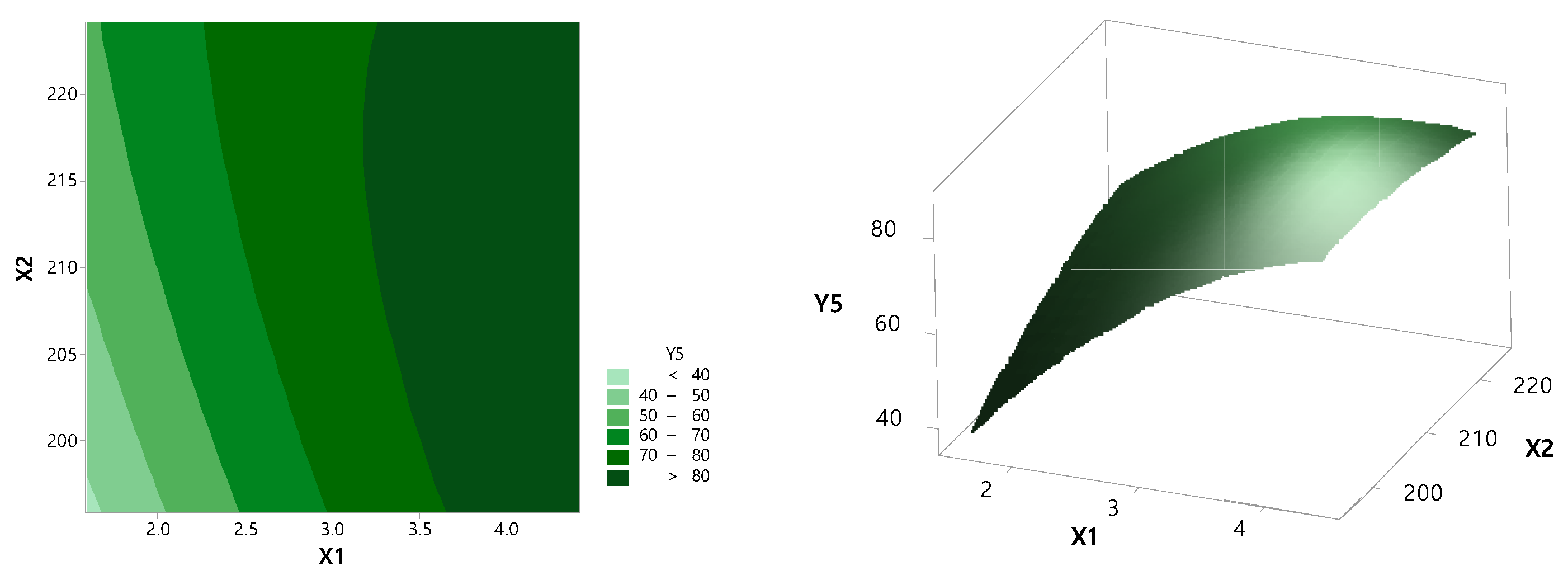
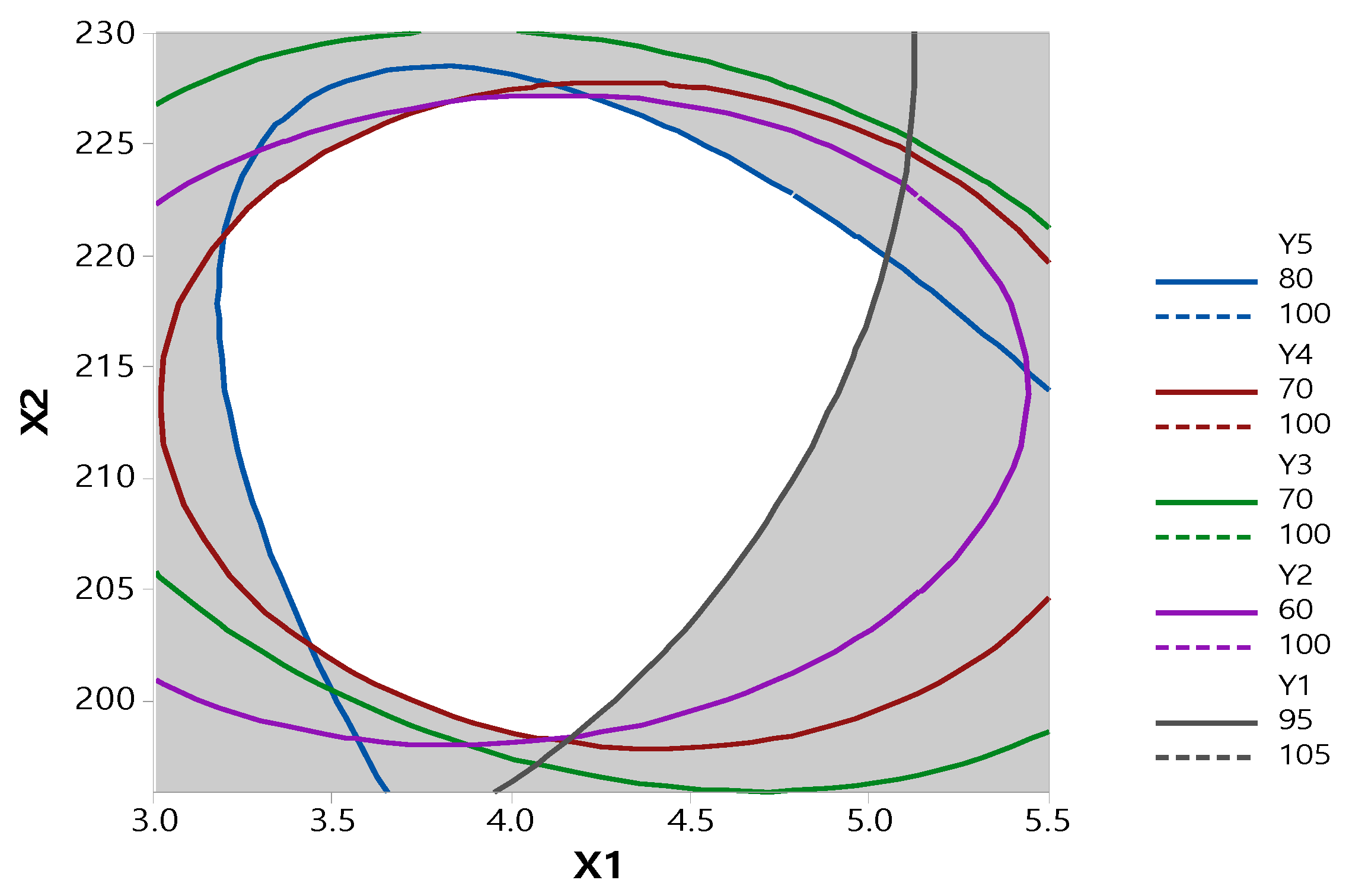
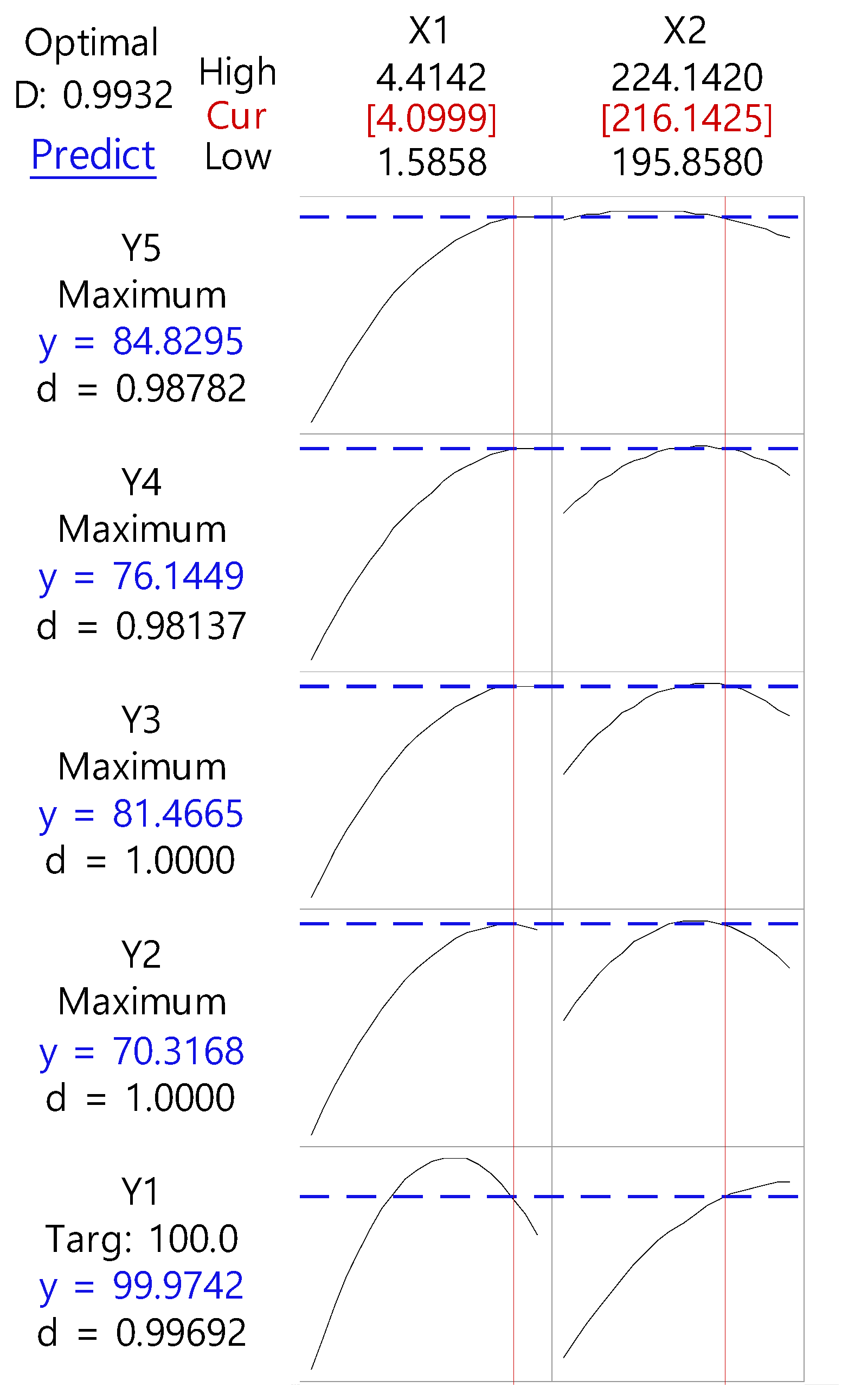
3.2. Optimization by CCD
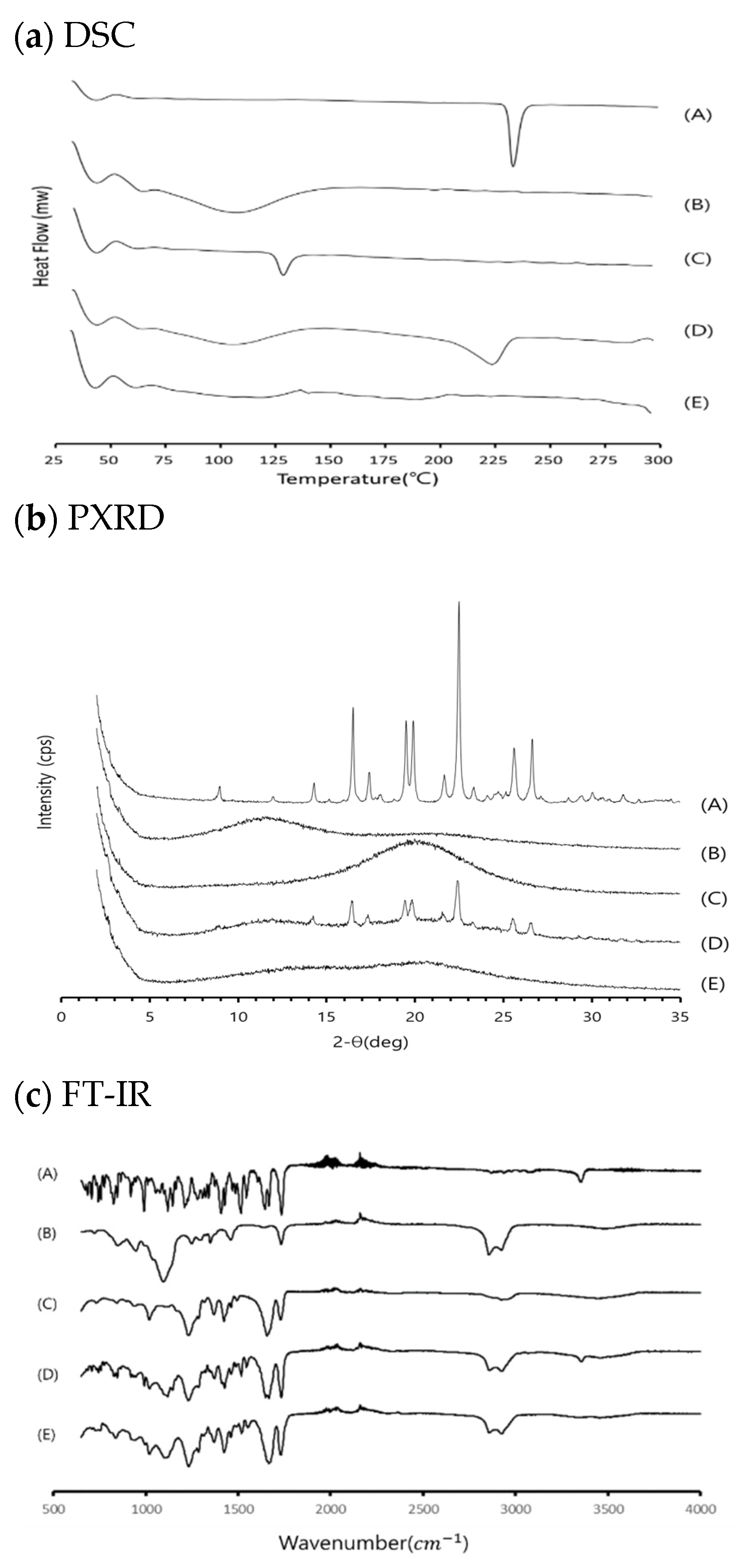
3.3. Physicochemical Evaluation of RXB-ASD
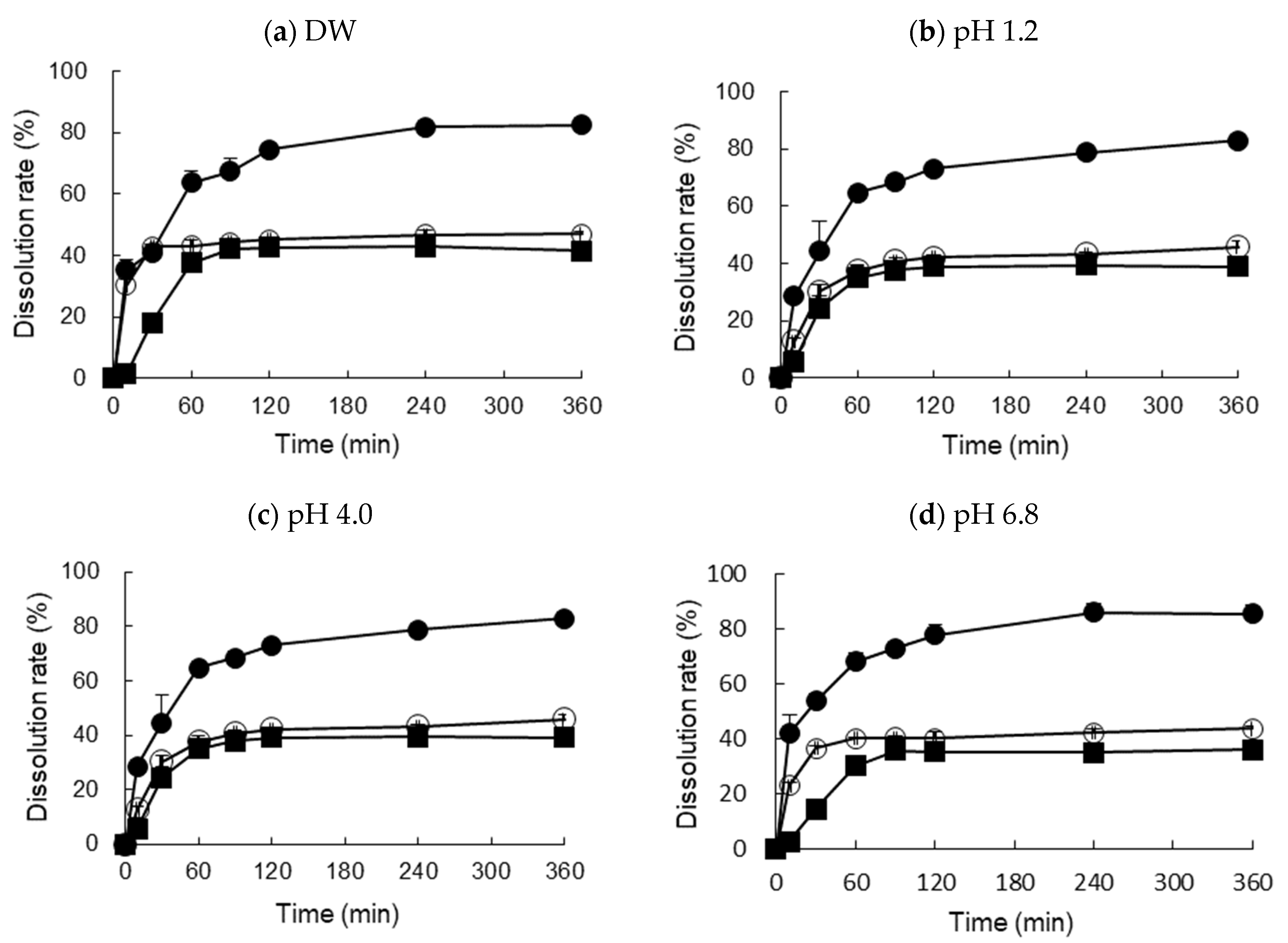
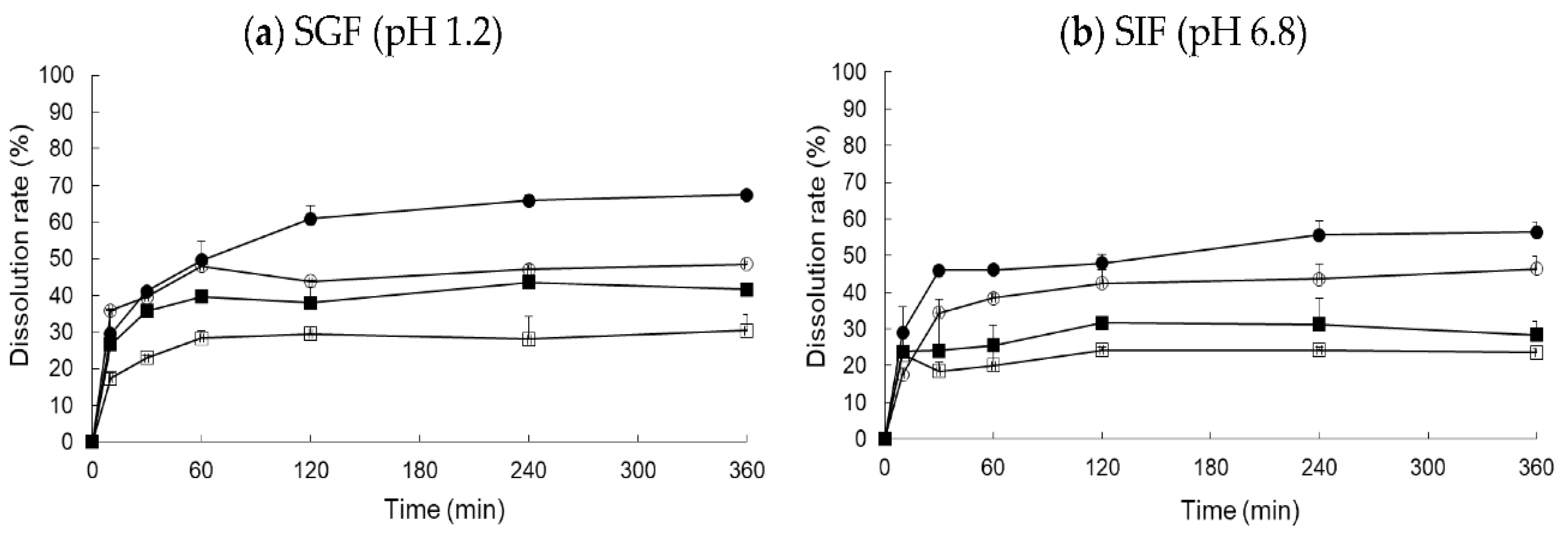
3.4. In Vitro Dissolution Test and Food Effect
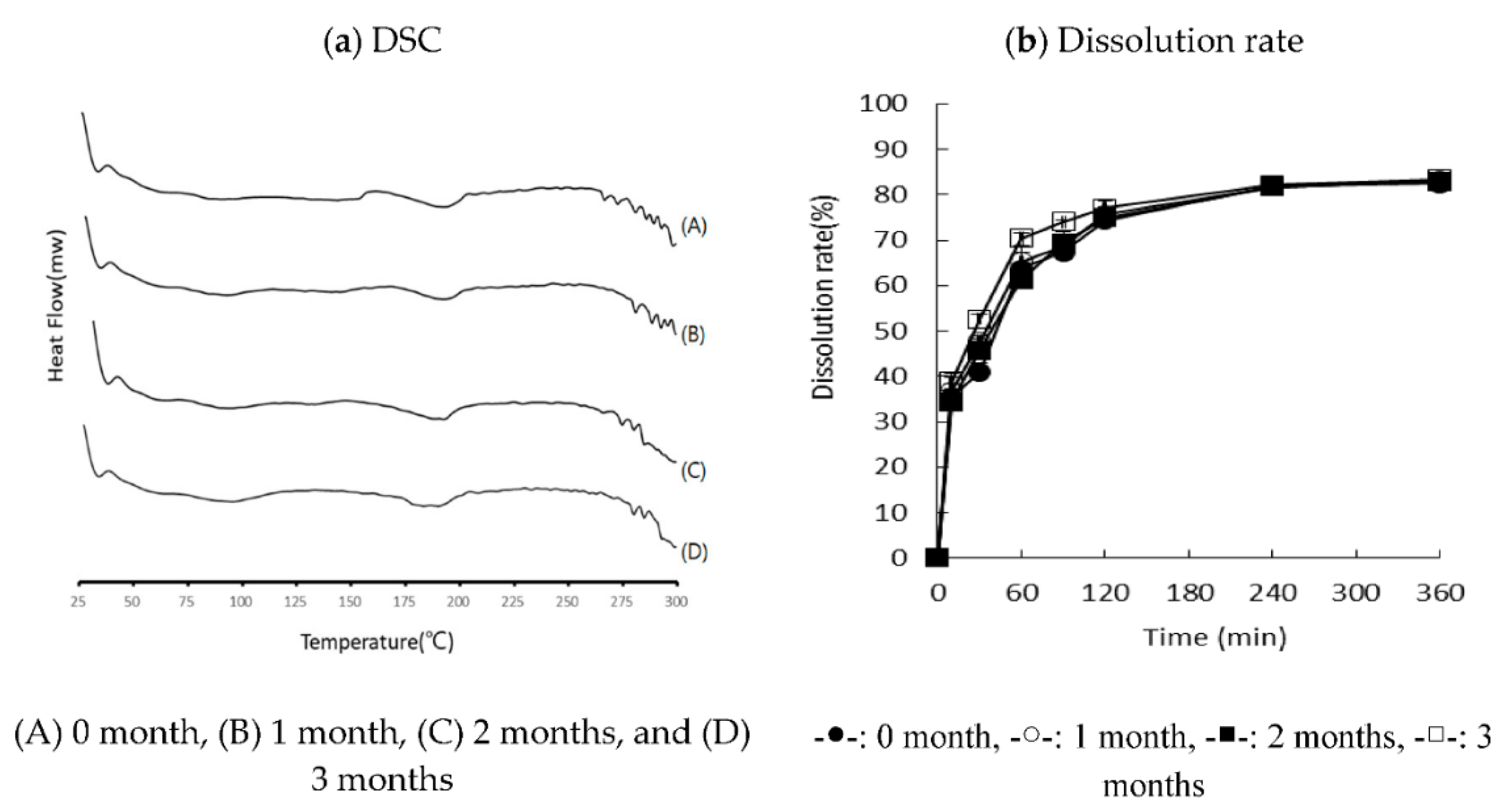
3.5. Stability
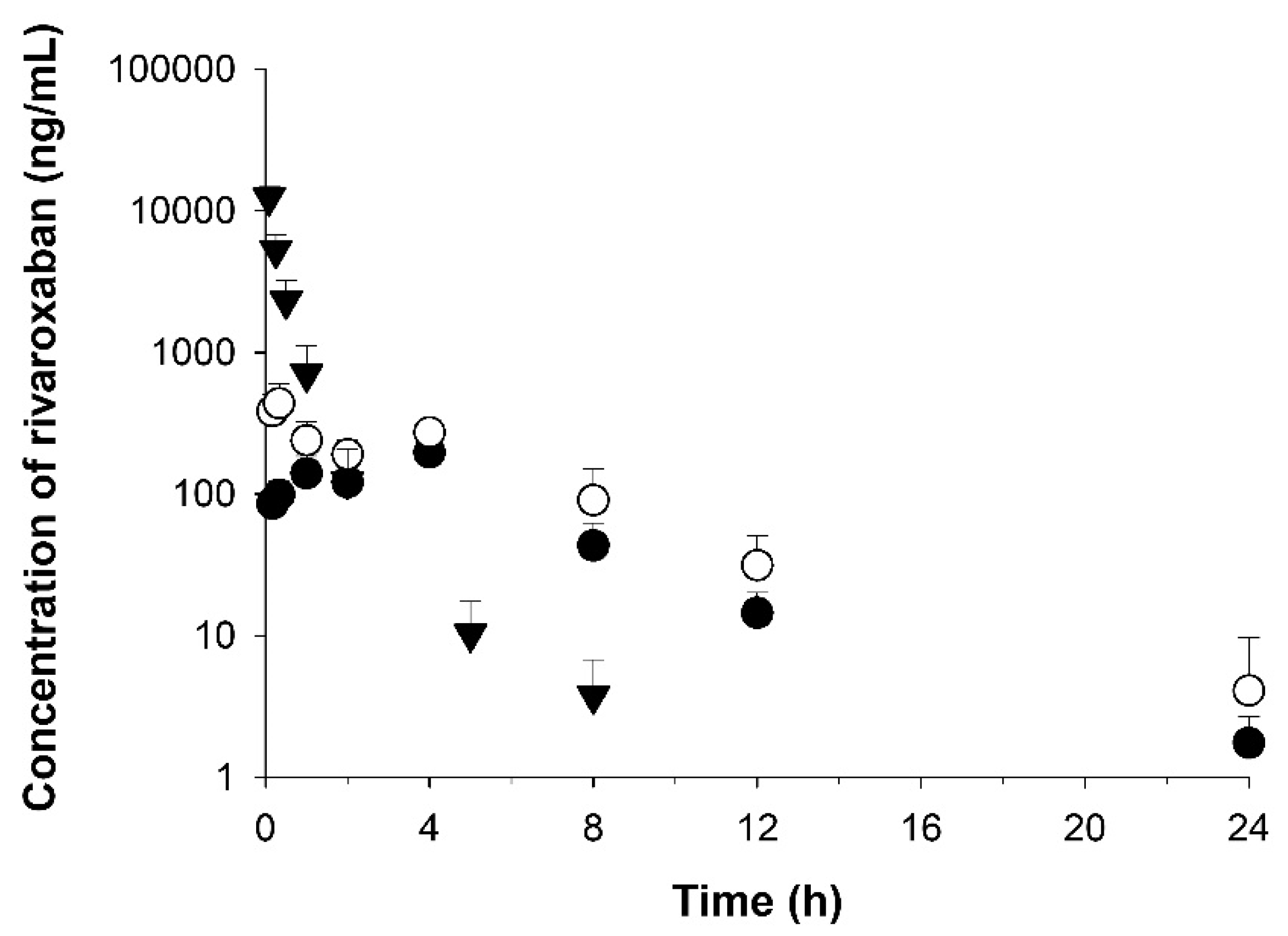
3.6. Application to Pharmacokinetic Studies
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kubitza, D.; Becka, M.; Wensing, G.; Voith, B.; Zuehlsdorf, M. Safety, pharmacodynamics, and pharmacokinetics of BAY 59-7939—An oral, direct Factor Xa inhibitor—After multiple dosing in healthy male subjects. Eur. J. Clin. Pharmacol. 2005, 61, 873–880. [Google Scholar] [CrossRef]
- Kapourani, A.; Vardaka, E.; Katopodis, K.; Kachrimanis, K.; Barmpalexis, P. Rivaroxaban polymeric amorphous solid dispersions: Moisture-induced thermodynamic phase behavior and intermolecular interactions. Eur. J. Pharm. Biopharm. 2019, 145, 98–112. [Google Scholar] [CrossRef]
- Metre, S.; Mukesh, S.; Samal, S.K.; Chand, M.; Sangamwar, A.T. Enhanced biopharmaceutical performance of rivaroxaban through polymeric amorphous solid dispersion. Mol. Pharm. 2018, 15, 652–668. [Google Scholar] [CrossRef]
- Torchilin, V.P. Recent advances with liposomes as pharmaceutical carriers. Nat. Rev. Drug Discov. 2005, 4, 145–160. [Google Scholar] [CrossRef]
- Gao, Y.; Li, Z.; Sun, M.; Li, H.; Guo, C.; Cui, J.; Li, A.; Cao, F.; Xi, Y.; Lou, H.; et al. Preparation, characterization, pharmacokinetics, and tissue distribution of curcumin nanosuspension with TPGS as stabilizer. Drug Dev. Ind. Pharm. 2010, 36, 1225–1234. [Google Scholar] [CrossRef]
- Wan, S.; Sun, Y.; Qi, X.; Tan, F. Improved bioavailability of poorly water-soluble drug curcumin in cellulose acetate solid dispersion. AAPS PharmSciTech 2011, 13, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Chen, G.; Ren, L. Preparation and characterization of the sulfobutylether-β-cyclodextrin inclusion complex of amiodarone hydrochloride with enhanced oral bioavailability in fasted state. AAPS PharmSciTech 2017, 18, 1526–1535. [Google Scholar] [CrossRef] [PubMed]
- Gurusamy, S.; Umar, V.I.K.; Ishra, D.N.M. Preparation, characterization and in vitro dissolution studies of solid dispersion. Yakugaku Zasshi 2006, 126, 657–664. [Google Scholar]
- Papageorgiou, G.Z.; Bikiaris, D.; Karavas, E.; Politis, S.; Docoslis, A.; Park, Y.; Stergiou, A.; Georgarakis, E. Effect of physical state and particle size distribution on dissolution enhancement of nimodipine/PEG solid dispersions prepared by melt mixing and solvent evaporation. AAPS J. 2006, 8, E623–E631. [Google Scholar] [CrossRef] [PubMed]
- Sethia, S.; Squillante, E. Solid dispersion of carbamazepine in PVP K30 by conventional solvent evaporation and supercritical methods. Int. J. Pharm. 2004, 272, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.-C.; Cho, C. Physicochemical characterizations of piroxicam-poloxamer solid dispersion. Pharm. Dev. Technol. 1997, 2, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Verreck, G.; Six, K.; Mooter, G.V.D.; Baert, L.; Peeters, J.; Brewster, M.E. Characterization of solid dispersions of itraconazole and hydroxypropylmethylcellulose prepared by melt extrusion—Part I. Int. J. Pharm. 2003, 251, 165–174. [Google Scholar] [CrossRef]
- Zajc, N.; Obreza, A.; Bele, M.; Srčič, S. Physical properties and dissolution behaviour of nifedipine/mannitol solid dispersions prepared by hot melt method. Int. J. Pharm. 2005, 291, 51–58. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, G.; Neilly, J.; Marsh, K.; Mawhinney, D.; Sanzgiri, Y. Enhancing the bioavailability of ABT-963 using solid dispersion containing Pluronic F-68. Int. J. Pharm. 2004, 286, 69–80. [Google Scholar] [CrossRef]
- Chokshi, R.J.; Sandhu, H.K.; Iyer, R.M.; Shah, N.H.; Malick, A.; Zia, H. Characterization of physico-mechanical properties of indomethacin and polymers to assess their suitability for hot-melt extrusion process as a means to manufacture solid dispersion/solution. J. Pharm. Sci. 2005, 94, 2463–2474. [Google Scholar] [CrossRef]
- Shah, S.; Maddineni, S.; Lu, J.; Repka, M.A. Melt extrusion with poorly soluble drugs. Int. J. Pharm. 2013, 453, 233–252. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Xie, Z.; Rao, Q.; Liamas, E.; Pan, P.; Li, Q.; Zhang, Z.J.; Lu, M.; Li, Q. Hot melt extrusion of heat-sensitive and high melting point drug: Inhibit the recrystallization of the prepared amorphous drug during extrusion to improve the bioavailability. Int. J. Pharm. 2019, 565, 316–324. [Google Scholar] [CrossRef]
- Vasoya, J.M.; Desai, H.H.; Gumaste, S.G.; Tillotson, J.; Kelemen, D.; Dalrymple, D.M.; Serajuddin, A.T.M. Development of solid dispersion by hot melt extrusion using mixtures of polyoxylglycerides with polymers as carriers for increasing dissolution rate of a poorly soluble drug model. J. Pharm. Sci. 2019, 108, 888–896. [Google Scholar] [CrossRef]
- Alshetaili, A.S.; Almutairy, B.K.; Alshahrani, S.M.; Ashour, E.A.; Tiwari, R.V.; AlShehri, S.M.; Feng, X.; Alsulays, B.B.; Majumdar, S.; Langley, N.; et al. Optimization of hot melt extrusion parameters for sphericity and hardness of polymeric face-cut pellets. Drug Dev. Ind. Pharm. 2016, 42, 1833–1841. [Google Scholar] [CrossRef]
- Alshahrani, S.M.; Lu, W.; Park, J.-B.; Morott, J.T.; Alsulays, B.B.; Majumdar, S.; Langley, N.; Kolter, K.; Gryczke, A.; Repka, M.A. Stability-enhanced hot-melt extruded amorphous solid dispersions via combinations of Soluplus® and HPMCAS-HF. AAPS PharmSciTech 2015, 16, 824–834. [Google Scholar] [CrossRef]
- Justine, T.; Pieere, L.; Chole, V.; Martine, A.; Laureanne, N.; Eric, Z.; Philippe, H.; Krier, F.; Evrard, B. Continuous production of itraconazole-based solid dispersions by hot melt extrusion: Preformulation, optimization and design space determination. Int. J. Pharm. 2016, 151, 114–124. [Google Scholar] [CrossRef]
- Fan, W.; Zhang, X.; Zhu, W.; Di, L. The preparation of curcumin sustained-release solid dispersion by hot melt extrusion-II. Optimization of preparation process and evaluation in vitro and in vivo. J. Pharm. Sci. 2020, 109, 1253–1260. [Google Scholar] [CrossRef]
- Mogal, V.; Dusane, J.; Borase, P.; Thakare, P.; Kshirsagar, S. A review on quality by design. Pharm. Biol. Eval. 2016, 3, 313–319. [Google Scholar]
- Sarwar, B.M.; Saquib, H. Pharmaceutical Quality by Design—Principles and Applications, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2019. [Google Scholar]
- Costa, P.; Sousa Lobo, J.M. Modeling and comparison of dissolution profiles. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2001, 13, 123–133. [Google Scholar] [CrossRef]
- Wang, L.; Gai, S.; Zhang, X.; Xu, X.; Gou, N.; Wang, X.; Zhou, N.; Feng, T. Simultaneous determination of RXB and TAK-438 in rat plasma by LC–MS/MS: Application to pharmacokinetic interaction study. Bioanalysis 2020, 12, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Gibaldi, M.; Perrier, D. Pharmacokinetics. In Revised and Expanded, 2nd ed.; Marcel Dekker, Inc.: New York, NY, USA, 1982; pp. 409–416. [Google Scholar]
- Patil, H.; Tiwari, R.V.; Repka, M.A. Hot-melt extrusion: From theory to application in pharmaceutical formulation. AAPS Pharm. 2016, 17, 20–42. [Google Scholar] [CrossRef]
- Pawar, J.; Tayade, A.; Gangurde, A.; Moravkar, K.; Amin, P. Solubility and dissolution enhancement of efavirenz hot melt extruded amorphous solid dispersions using combination of polymeric blends: A QbD approach. Eur. J. Pharm. Sci. 2016, 88, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Al-Zoubi, N.; Alkhatib, H.S.; Bustanji, Y.K.; Aiedeh, K.; Malamataris, S. Sustained-release of buspirone HCl by co spray-drying with aqueous polymeric dispersions. Eur. J. Pharm. Biopharm. 2008, 69, 735–742. [Google Scholar] [CrossRef]
- Godfrey, K.R.; Arundel, P.A.; Dong, Z.; Bryant, R. Modelling the double peak phenomenon in pharmacokinetics. Comput. Methods Programs Biomed. 2011, 104, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Son, H.; Noh, K.; Kim, E.; Shin, B.S.; Kang, W. Effects of verapamil and diltiazem on the pharmacokinetics and pharmacodynamics of rivaroxaban. Pharmaceutics 2019, 11, 133. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | w/w Ratio | Content (%) | Dissolution Rate (%) | |||||
|---|---|---|---|---|---|---|---|---|
| RXB | Soluplus® | PVP VA 64 | PVA | Cremophor® RH 40 | Gelucire® 50/13 | |||
| F1 | 1 | 2 | - | - | - | - | 89.63 ± 3.31 | 24.52 ± 4.13 |
| F2 | 1 | 4 | - | - | - | - | 90.56 ± 4.36 | 24.66 ±12.94 |
| F3 | 1 | - | 2 | - | - | - | 94.05 ± 3.33 | 46.57 ± 0.57 |
| F4 | 1 | - | 4 | - | - | - | 97.01 ± 1.24 | 51.89 ± 0.46 |
| F5 | 1 | 2 | 2 | - | - | - | 91.26 ± 6.77 | 18.83 ± 4.32 |
| F6 | 1 | 4 | - | 1 | - | - | 76.05 ± 2.23 | 52.72 ± 2.19 |
| F7 | 1 | 4 | - | - | 1 | - | 92.48 ± 1.33 | 35.82 ± 6.61 |
| F8 | 1 | 4 | - | - | - | 1 | 93.70 ± 1.48 | 33.44 ± 0.48 |
| F9 | 1 | - | 4 | 1 | - | - | 73.78 ± 1.77 | 80.80 ± 1.16 |
| F10 | 1 | - | 4 | - | 1 | - | 93.58 ± 1.26 | 83.29 ± 0.33 |
| F11 | 1 | - | 4 | - | - | 1 | 95.38 ± 1.26 | 73.80 ± 3.76 |
| Independent Variables | Dependent Variables | |||
|---|---|---|---|---|
| X1 | X2 | X3 | Y1 | Y2 |
| 1 | 0 | 180 | 92.32 | 48.55 |
| 1 | 0 | 200 | 101.52 | 41.17 |
| 1 | 0 | 220 | 94.75 | 33.90 |
| 1 | 10 | 180 | 97.01 | 44.46 |
| 1 | 10 | 200 | 99.49 | 43.00 |
| 1 | 10 | 220 | 96.82 | 42.30 |
| 1 | 20 | 180 | 107.36 | 40.88 |
| 1 | 20 | 200 | 97.98 | 42.71 |
| 1 | 20 | 220 | 88.12 | 45.27 |
| 2 | 0 | 180 | 88.09 | 48.96 |
| 2 | 0 | 200 | 86.67 | 51.30 |
| 2 | 0 | 220 | 91.29 | 75.32 |
| 2 | 10 | 180 | 97.71 | 44.82 |
| 2 | 10 | 200 | 101.77 | 43.91 |
| 2 | 10 | 220 | 95.68 | 77.17 |
| 2 | 20 | 180 | 94.13 | 44.41 |
| 2 | 20 | 200 | 90.45 | 49.01 |
| 2 | 20 | 220 | 101.68 | 80.51 |
| 4 | 0 | 180 | 95.40 | 60.62 |
| 4 | 0 | 200 | 97.76 | 64.29 |
| 4 | 0 | 220 | 105.12 | 73.36 |
| 4 | 10 | 180 | 94.81 | 53.18 |
| 4 | 10 | 200 | 105.81 | 68.05 |
| 4 | 10 | 220 | 97.63 | 84.40 |
| 4 | 20 | 180 | 93.35 | 50.96 |
| 4 | 20 | 200 | 98.53 | 79.89 |
| 4 | 20 | 220 | 96.31 | 87.40 |
| Polymer | Solubility (μg/mL) | Polymer | Solubility (μg/mL) |
|---|---|---|---|
| Distilled water | 0.09 ± 0.00 | HPC-L | 5.79 ± 0.02 |
| PVP VA 64 | 8.82 ± 0.16 | Poloxamer 188 | 9.67 ± 0.09 |
| PVP K90 | 6.32 ± 0.02 | Solutol® HS 15 | 12.45 ± 0.12 |
| PVA | 29.30 ± 8.76 | Soluplus® | 9.25 ± 0.04 |
| PEG 200 | 12.38 ± 1.71 | Gelucire® 44/14 | 11.00 ± 0.02 |
| PEG 4000 | 10.76 ± 0.21 | Gelucire® 50/13 | 14.23 ± 0.06 |
| PEG 10000 | 8.97 ± 0.14 | Labrasol® | 6.02 ± 0.60 |
| PEG 20000 | 9.02 ± 0.09 | Cremophor® RH 40 | 30.33 ± 5.24 |
| HPMC 4M | 6.47 ± 0.26 | Cremophor® EL | 10.44 ± 1.51 |
| HPMC 15M | 5.12 ± 0.07 | Tween 80 | 12.59 ± 1.07 |
| Independent Variable | Dependent Variable | |||||
|---|---|---|---|---|---|---|
| X1 | X2 | Y1 | Y2 | Y3 | Y4 | Y5 |
| 1.59 | 210 | 91.60 | 44.25 | 49.46 | 49.85 | 54.42 |
| 2 | 220 | 98.44 | 44.16 | 55.30 | 54.66 | 62.36 |
| 2 | 200 | 97.87 | 42.01 | 43.33 | 46.38 | 49.11 |
| 3 | 224.14 | 99.99 | 57.60 | 74.58 | 65.00 | 78.32 |
| 3 | 210 | 101.69 | 67.85 | 71.93 | 71.49 | 75.48 |
| 3 | 210 | 100.15 | 64.45 | 71.70 | 67.83 | 75.39 |
| 3 | 195.86 | 94.80 | 53.94 | 54.37 | 63.62 | 72.17 |
| 3 | 210 | 98.78 | 69.54 | 74.86 | 73.59 | 78.83 |
| 3 | 210 | 99.14 | 63.98 | 73.55 | 67.67 | 78.05 |
| 3 | 210 | 102.54 | 63.29 | 73.33 | 66.77 | 77.37 |
| 4 | 200 | 97.84 | 63.79 | 77.08 | 69.79 | 85.27 |
| 4 | 220 | 101.22 | 70.06 | 80.23 | 76.71 | 85.24 |
| 4.41 | 210 | 96.65 | 67.29 | 78.58 | 75.68 | 84.62 |
| Variable | Parameter | ||
|---|---|---|---|
| p-Value | R2 | Lack of Fit | |
| Y1: Content (%) | 0.092 | 0.6820 | 0.165 |
| X1: Polymer ratio | 0.153 | ||
| X2: Barrel Temp. | 0.110 | ||
| X1·X1 | 0.023 | ||
| X2·X2 | 0.388 | ||
| X1·X2 | 0.540 | ||
| Regression equation | |||
| Y1 = −246 + 0.9X1 + 3.13X2 − 2.397X1·X1 − 0.000762X2·X2 + 0.070X1·X2 | |||
| Y2: Dissolution rate (%)—SGF, 2 h | 0.000 | 0.9507 | 0.367 |
| X1: Polymer ratio | 0.000 | ||
| X2: Barrel Temp. | 0.146 | ||
| X1·X1 | 0.002 | ||
| X2·X2 | 0.002 | ||
| X1·X2 | 0.506 | ||
| Regression equation | |||
| Y2 = −2283 + 19.7X1 + 2177X2 − 5.22X1·X1 − 0.0522X2·X2 + 0.103X1·X2 | |||
| Y3: Dissolution rate (%)—SGF, 6 h | 0.000 | 0.9628 | 0.018 |
| X1: Polymer ratio | 0.000 | ||
| X2: Barrel Temp. | 0.002 | ||
| X1·X1 | 0.006 | ||
| X2·X2 | 0.008 | ||
| X1·X2 | 0.199 | ||
| Regression equation | |||
| Y3 = −2184 + 86.3X1 + 19.54X2 − 4.59X1·1 − 0.0437X2·X2 − 0.221X1·X2 | |||
| Y4: Dissolution rate (%)—SIF, 2 h | 0.001 | 0.9344 | 0.353 |
| X1: Polymer ratio | 0.000 | ||
| X2: Barrel Temp. | 0.100 | ||
| X1·X1 | 0.017 | ||
| X2·X2 | 0.043 | ||
| X1·X2 | 0.353 | ||
| Regression equation | |||
| Y4 = −1382 + 40.0X1 + 12.89X2 − 3.77X1·X1 − 0.0299X2·X2 − 0.034X1·X2 | |||
| Y5: Dissolution rate (%)—SIF, 6 h | 0.000 | 0.9668 | 0.057 |
| X1: Polymer ratio | 0.000 | ||
| X2: Barrel Temp. | 0.025 | ||
| X1·X1 | 0.005 | ||
| X2·X2 | 0.231 | ||
| X1·X2 | 0.046 | ||
| Regression equation | |||
| Y5 = −866 + 107.8X1 + 6.99X2 − 4.22X1·X1 − 0.0136X2·X2 − 0.332X1·X2 | |||
| Formulation (RXB-ASD) | Reference (RXB Powder) | ||
|---|---|---|---|
| P.O. | P.O. | I.V. | |
| T1/2 (h) | 3.10 ± 0.98 | 3.18 ± 0.98 | 1.21 ± 0.19 |
| AUClast (h·ng/mL) | 2180 ± 455 | 1240 ± 170 | 5130 ± 1530 |
| AUC0–4 h (h·ng/mL) | 1000 ± 257 | 548 ± 104 | - |
| AUC4–24 h (h·ng/mL) | 1190 ± 295 | 696 ± 101 | - |
| AUCinf (h·ng/mL) | 2210 ± 459 | 1250 ± 172 | 5140 ± 1530 |
| MRT (h) | 4.78 ± 0.74 | 4.73 ± 0.32 | 0.381 ± 0.093 |
| Tmax (h) | 3.25 ± 1.50 | 0.33 ± 0.00 | - |
| Cmax (ng/mL) | 436 ± 168 | 206 ± 26 | - |
| Vd (mL/kg) | 22,700 ± 6300 | 36,700 ± 2400 | 740 ± 267 |
| CL (mL/h/kg) | 4690 ± 1030 | 8130 ± 1100 | 420 ± 140 |
| Kel (1/h) | 0.237 ± 0.059 | 0.223 ± 0.039 | 0.583 ± 0.100 |
| Bioavailability (%) | 8.6 | 4.9 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.-H.; Jeong, H.S.; Jeong, J.-W.; Koo, T.-S.; Kim, D.-K.; Cho, Y.H.; Lee, G.W. The Development and Optimization of Hot-Melt Extruded Amorphous Solid Dispersions Containing Rivaroxaban in Combination with Polymers. Pharmaceutics 2021, 13, 344. https://doi.org/10.3390/pharmaceutics13030344
Lee J-H, Jeong HS, Jeong J-W, Koo T-S, Kim D-K, Cho YH, Lee GW. The Development and Optimization of Hot-Melt Extruded Amorphous Solid Dispersions Containing Rivaroxaban in Combination with Polymers. Pharmaceutics. 2021; 13(3):344. https://doi.org/10.3390/pharmaceutics13030344
Chicago/Turabian StyleLee, Jong-Hwa, Hyeong Sik Jeong, Jong-Woo Jeong, Tae-Sung Koo, Do-Kyun Kim, Young Ho Cho, and Gye Won Lee. 2021. "The Development and Optimization of Hot-Melt Extruded Amorphous Solid Dispersions Containing Rivaroxaban in Combination with Polymers" Pharmaceutics 13, no. 3: 344. https://doi.org/10.3390/pharmaceutics13030344
APA StyleLee, J.-H., Jeong, H. S., Jeong, J.-W., Koo, T.-S., Kim, D.-K., Cho, Y. H., & Lee, G. W. (2021). The Development and Optimization of Hot-Melt Extruded Amorphous Solid Dispersions Containing Rivaroxaban in Combination with Polymers. Pharmaceutics, 13(3), 344. https://doi.org/10.3390/pharmaceutics13030344