
Improved Dissolution Rate and Intestinal Absorption of Fexofenadine Hydrochloride by the Preparation of Solid Dispersions: In Vitro and In Situ Evaluation


Abstract
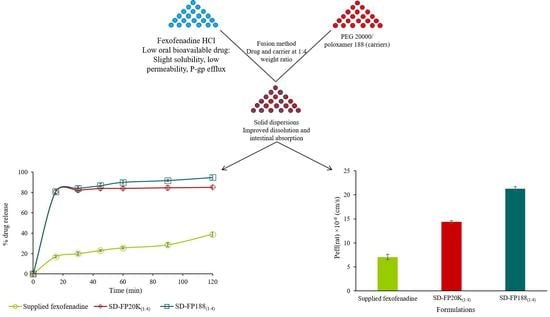
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Animals
2.3. Drug Quantification by HPLC
2.4. Phase Solubility Study
2.5. Preparation of Solid Dispersions and Physical Mixtures
2.6. Drug Content Estimation
2.7. In Vitro Dissolution Studies
2.8. Characterization of the Physicochemical Properties
2.8.1. Scanning Electron Microscopy (SEM)
2.8.2. Differential Scanning Calorimetry (DSC)
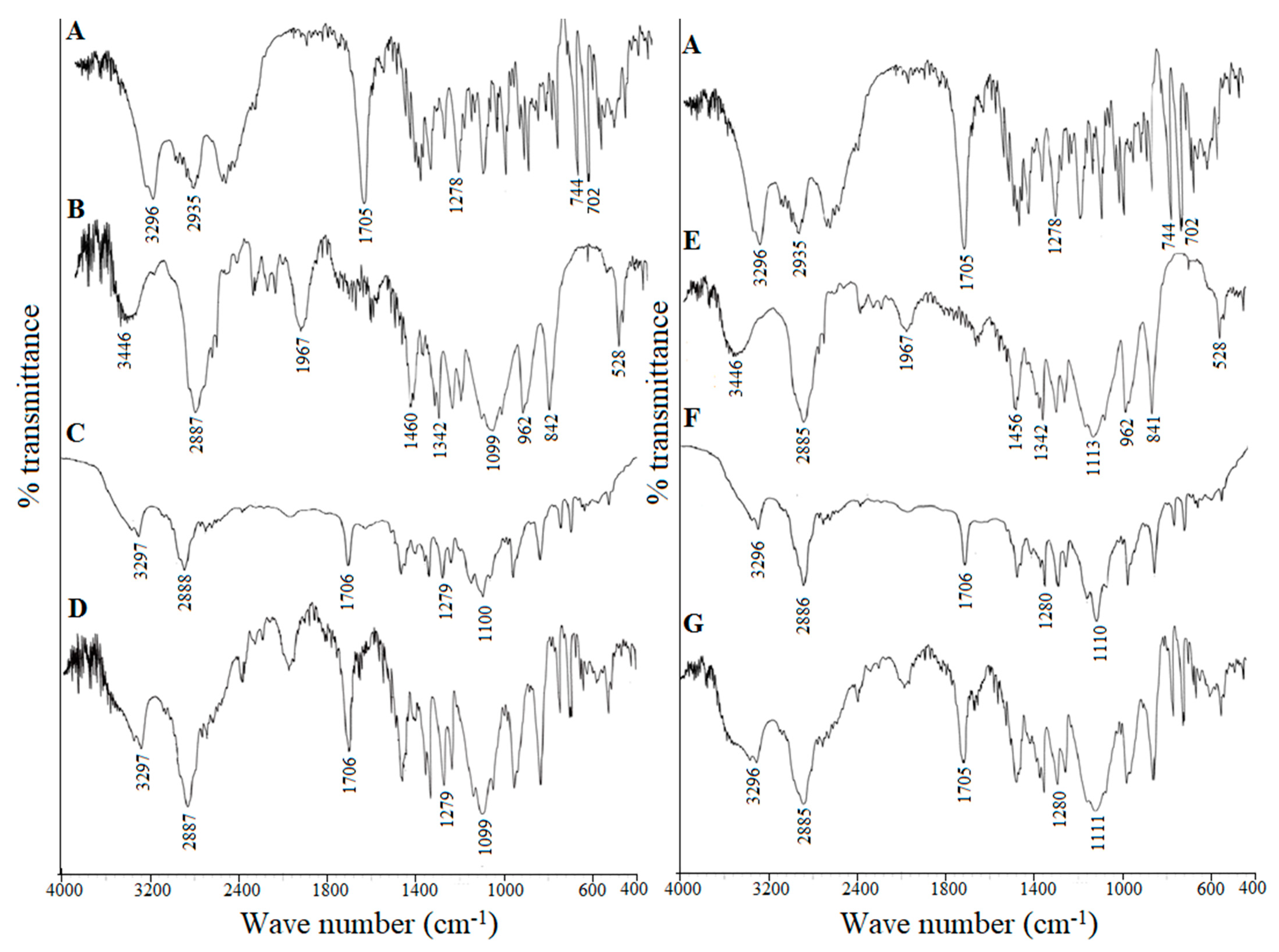
2.8.3. Fourier Transform Infrared Spectroscopy (FTIR)
2.9. In Situ Intestinal Absorption Study
2.10. Statistical Analysis
3. Results
3.1. Phase Solubility Study
3.2. Drug Content
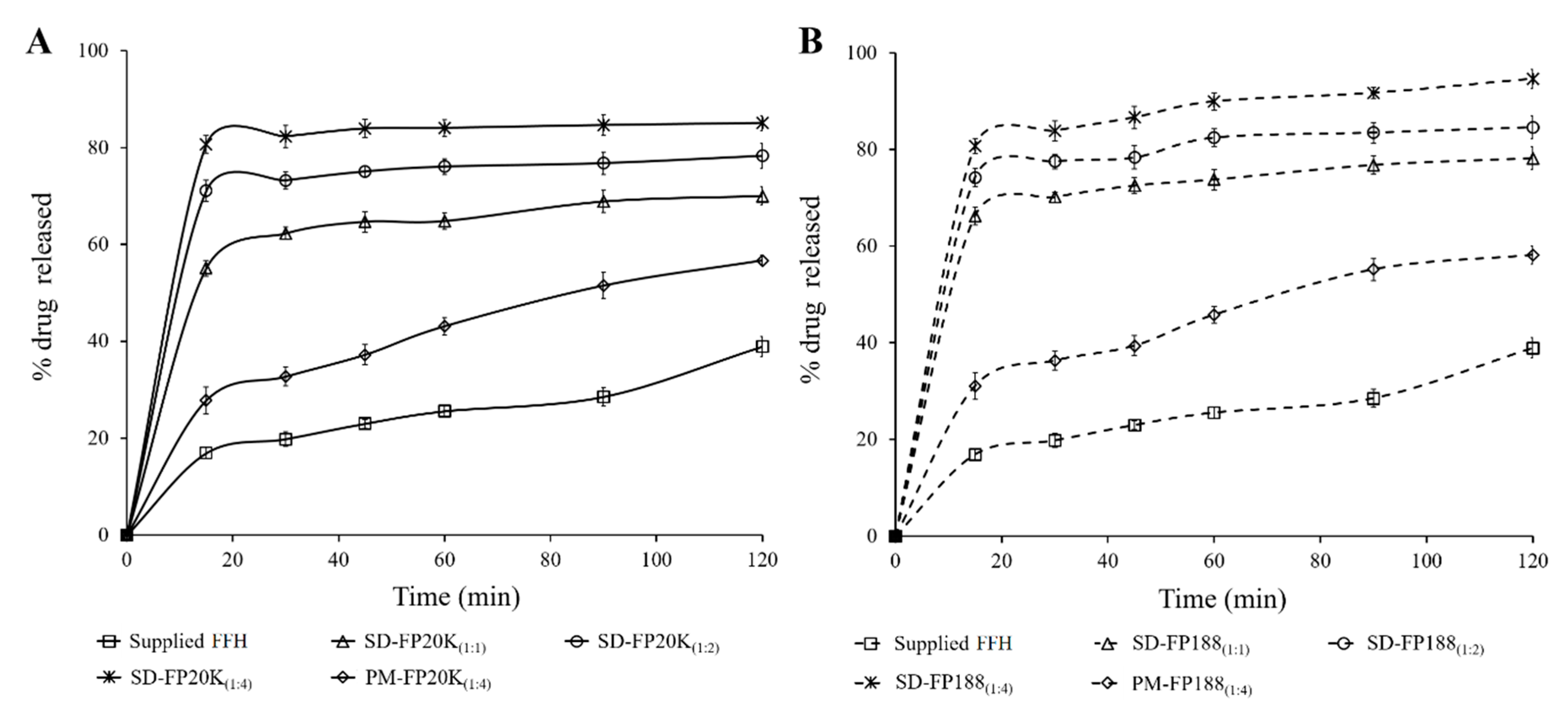
3.3. In Vitro Dissolution Study
3.4. Solid State Characterization
3.4.1. Scanning Electron Microscopy
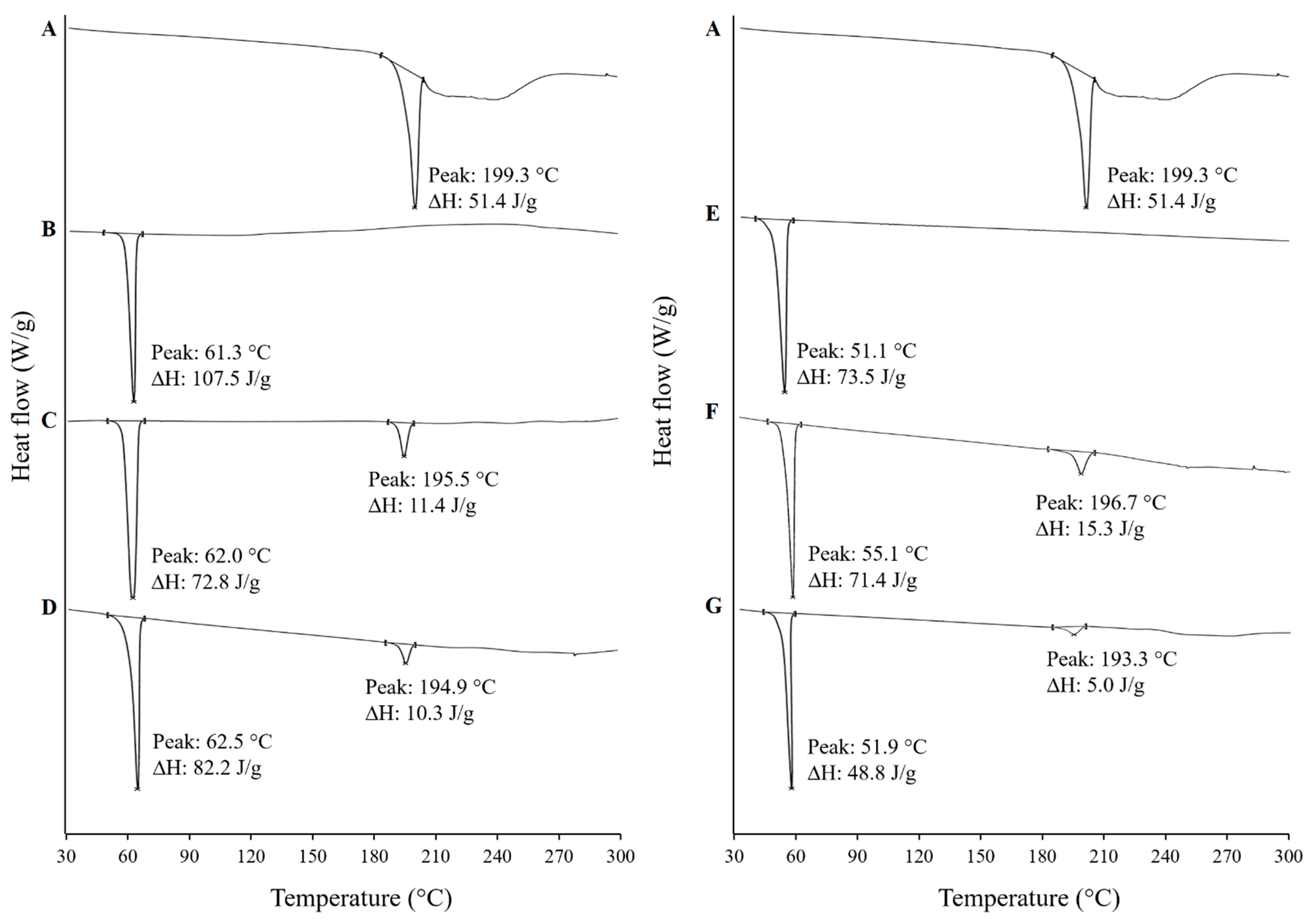
3.4.2. Differential Scanning Calorimetry (DSC)
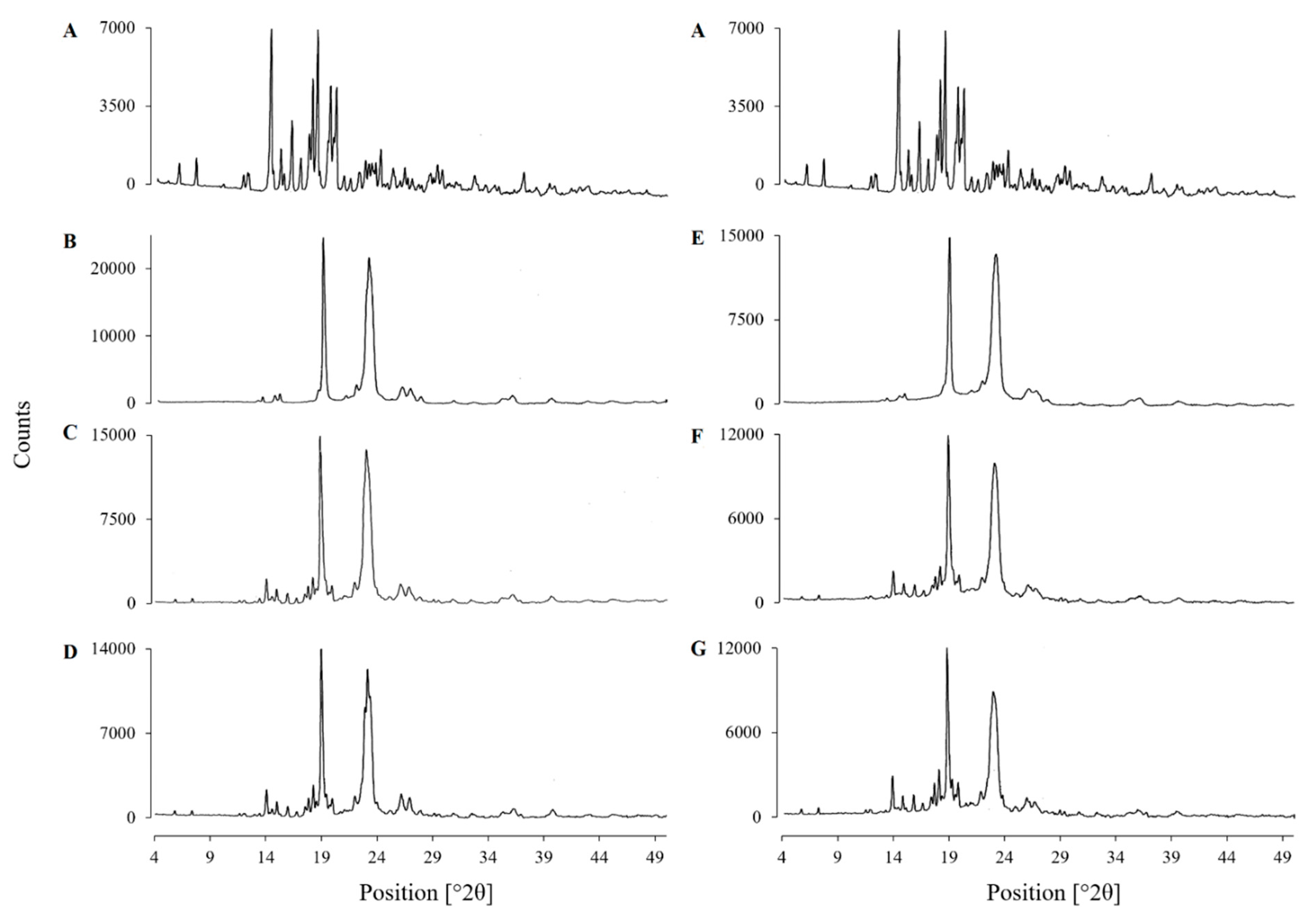
3.4.3. X-Ray Powder Diffraction (XRPD)
3.4.4. Fourier Transform Infrared Spectroscopy (FTIR)
3.5. In Situ Intestinal Absorption Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Pawankar, R.; Baena-Cagnani, C.E.; Bousquet, J.; Canonica, G.W.; Cruz, A.A.; Kaliner, M.A.; Lanier, B.Q.; Henley, K. State of world allergy report 2008: Allergy and chronic respiratory diseases. World Allergy Organ. J. 2008, 1, S4–S17. [Google Scholar] [CrossRef]
- Howarth, P.; Wilson, S.; Lau, L.; Raj Akulasingam, K. The nasal mast cell and rhinitis. Clin. Exp. Allergy 1991, 21, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Axelrod, D.; Bielory, L. Fexofenadine hydrochloride in the treatment of allergic disease: A review. J. Asthma Allergy 2008, 1, 19. [Google Scholar]
- Dresser, G.K.; Bailey, D.G.; Leake, B.F.; Schwarz, U.I.; Dawson, P.A.; Freeman, D.J.; Kim, R.B. Fruit juices inhibit organic anion transporting polypeptide–mediated drug uptake to decrease the oral availability of fexofenadine. Clin. Pharm. Ther. 2002, 71, 11–20. [Google Scholar] [CrossRef]
- Drescher, S.; Schaeffeler, E.; Hitzl, M.; Hofmann, U.; Schwab, M.; Brinkmann, U.; Eichelbaum, M.; Fromm, M.F. MDR1 gene polymorphisms and disposition of the P-glycoprotein substrate fexofenadine. Br. J. Clin. Pharm. 2002, 53, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Clough, S.R. Fexofenadine. In Encyclopedia of Toxicology, 3rd ed.; Academic Press: Cambridge, MA, USA, 2014. [Google Scholar]
- Carrier, R.L.; Miller, L.A.; Ahmed, I. The utility of cyclodextrins for enhancing oral bioavailability. J. Control. Release 2007, 123, 78–99. [Google Scholar] [CrossRef]
- Bandari, S.; Jadav, S.; Eedara, B.B.; Jukanti, R.; Veerareddy, P.R. Physicochemical characterization and dissolution enhancement of loratadine by solid dispersion technique. Korean J. Chem. Eng. 2013, 30, 238–244. [Google Scholar] [CrossRef]
- Eedara, B.B.; Kankane, M.; Jukanti, R.; Nagabandi, V.K.; Bandari, S. Enhanced solubility and permeability of exemestane solid dispersion powders for improved oral delivery. J. Pharm. Investig. 2013, 43, 229–242. [Google Scholar] [CrossRef]
- Eedara, B.B.; Kallakunta, V.R.; Bandari, S. Self-nanoemulsifying powders for improved oral delivery of poorly water-soluble drugs. Ther. Deliv. 2015, 6, 899–901. [Google Scholar] [CrossRef]
- Gangishetty, H.; Eedara, B.B.; Bandari, S. Development of ketoprofen loaded proliposomal powders for improved gastric absorption and gastric tolerance: In Vitro and in situ evaluation. Pharm. Dev. Technol. 2015, 20, 641–651. [Google Scholar] [CrossRef]
- Sunkavalli, S.; Eedara, B.B.; Janga, K.Y.; Velpula, A.; Jukanti, R.; Bandari, S. Preparation and characterization of docetaxel self-nanoemulsifying powders (SNEPs): A strategy for improved oral delivery. Korean J. Chem. Eng. 2016, 33, 1115–1124. [Google Scholar] [CrossRef]
- Bandari, S.; Dronam, V.R.; Eedara, B.B. Development and preliminary characterization of levofloxacin pharmaceutical cocrystals for dissolution rate enhancement. J. Pharm. Investig. 2017, 47, 583–591. [Google Scholar] [CrossRef]
- Eedara, B.B.; Bandari, S. Lipid-based dispersions of exemestane for improved dissolution rate and intestinal permeability: In Vitro and ex vivo characterization. Artif. Cells Nanomed. Biotechnol. 2017, 45, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Nalla, P.; Bagam, S.; Eedara, B.B.; Dhurke, R. Formulation and evaluation of domperidone oral proliposomal powders. Int. J. Pharm. Technol. Res. 2015, 7, 108–118. [Google Scholar]
- Ramasahayam, B.; Eedara, B.B.; Kandadi, P.; Jukanti, R.; Bandari, S. Development of isradipine loaded self-nano emulsifying powders for improved oral delivery: In Vitro and in vivo evaluation. Drug Dev. Ind. Pharm. 2015, 41, 753–763. [Google Scholar] [CrossRef]
- Shimizu, M.; Uno, T.; Sugawara, K.; Tateishi, T. Effects of itraconazole and diltiazem on the pharmacokinetics of fexofenadine, a substrate of P-glycoprotein. Br. J. Clin. Pharmacol. 2006, 61, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Türkmen, Ö.; Ay Şenyiğit, Z.; Baloğlu, E. Formulation and evaluation of fexofenadine hydrochloride orally disintegrating tablets for pediatric use. J. Drug Deliv. Sci. Technol. 2018, 43, 201–210. [Google Scholar] [CrossRef]
- Gundogdu, E.; Alvarez, I.G.; Karasulu, E. Improvement of effect of water-in-oil microemulsion as an oral delivery system for fexofenadine: In Vitro and in vivo studies. Int. J. Nanomed. 2011, 6, 1631. [Google Scholar] [CrossRef]
- Mandeep; Kaur, S.; Samal, S.K.; Roy, S.; Sangamwar, A.T. Successful oral delivery of fexofenadine hydrochloride by improving permeability via phospholipid complexation. Eur. J. Pharm. Sci. 2020, 149, 105338. [Google Scholar] [CrossRef]
- Eedara, B.B.; Veerareddy, P.R.; Jukanti, R.; Bandari, S. Improved oral bioavailability of fexofenadine hydrochloride using lipid surfactants: Ex Vivo, in situ and in vivo studies. Drug Dev. Ind. Pharm. 2014, 40, 1030–1043. [Google Scholar] [CrossRef]
- Parray, Z.A.; Hassan, M.I.; Ahmad, F.; Islam, A. Amphiphilic nature of polyethylene glycols and their role in medical research. Polym. Test. 2020, 82, 106316. [Google Scholar] [CrossRef]
- Hugger, E.D.; Audus, K.L.; Borchardt, R.T. Effects of poly (ethylene glycol) on efflux transporter activity in Caco-2 cell monolayers. J. Pharm. Sci. 2002, 91, 1980–1990. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.M.; Charman, W.N.; Porter, C.J. An in vitro examination of the impact of polyethylene glycol 400, pluronic P85, and vitamin E da-tocopheryl polyethylene glycol 1000 succinate on P-glycoprotein efflux and enterocyte-based metabolism in excised rat intestine. AAPS Pharmsci. 2002, 4, 193–205. [Google Scholar] [CrossRef]
- Shen, Q.; Li, W.; Lin, Y.; Katsumi, H.; Okada, N.; Sakane, T.; Fujita, T.; Yamamoto, A. Modulating effect of polyethylene glycol on the intestinal transport and absorption of prednisolone, methylprednisolone and quinidine in rats by in-vitro and in-situ absorption studies. J. Pharm. Pharmacol. 2008, 60, 1633–1641. [Google Scholar] [CrossRef]
- Kabanov, A.V.; Batrakova, E.V.; Alakhov, V.Y. Pluronic® block copolymers as novel polymer therapeutics for drug and gene delivery. J. Control. Release 2002, 82, 189–212. [Google Scholar] [CrossRef]
- Tambe, A.; Pandita, N. Enhanced solubility and drug release profile of boswellic acid using a poloxamer-based solid dispersion technique. J. Drug Deliv. Sci. Technol. 2018, 44, 172–180. [Google Scholar] [CrossRef]
- Newa, M.; Bhandari, K.H.; Li, D.X.; Kwon, T.-H.; Kim, J.A.; Yoo, B.K.; Woo, J.S.; Lyoo, W.S.; Yong, C.S.; Choi, H.G. Preparation, characterization and in vivo evaluation of ibuprofen binary solid dispersions with poloxamer 188. Int. J. Pharm. 2007, 343, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Ghareeb, M.M.; Abdulrasool, A.A.; Hussein, A.A.; Noordin, M.I. Kneading technique for preparation of binary solid dispersion of meloxicam with poloxamer 188. AAPS PharmSciTech 2009, 10, 1206–1215. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Si, L.; Jiang, L.; Fan, Z.; Qiu, J.; Li, G. Effect of pluronic F68 block copolymer on P-glycoprotein transport and CYP3A4 metabolism. Int. J. Pharm. 2008, 356, 351–353. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, T. A phase solubility technique. Adv. Anal. Chem. Instrum. 1965, 4, 117–211. [Google Scholar]
- Ni, J.; Tian, F.; Dahmani, F.Z.; Yang, H.; Yue, D.; He, S.; Zhou, J.; Yao, J. Curcumin-carboxymethyl chitosan (CNC) conjugate and CNC/LHR mixed polymeric micelles as new approaches to improve the oral absorption of P-gp substrate drugs. Drug Deliv. 2016, 23, 3424–3435. [Google Scholar] [CrossRef] [PubMed]
- Varma, M.V.; Panchagnula, R. Enhanced oral paclitaxel absorption with vitamin E-TPGS: Effect on solubility and permeability in vitro, in situ and in vivo. Eur. J. Pharm. Sci. 2005, 25, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, G.; Neilly, J.; Marsh, K.; Mawhinney, D.; Sanzgiri, Y. Enhancing the bioavailability of ABT-963 using solid dispersion containing Pluronic F-68. Int. J. Pharm. 2004, 286, 69–80. [Google Scholar] [CrossRef]
- Kolašinac, N.; Kachrimanis, K.; Homšek, I.; Grujić, B.; Đurić, Z.; Ibrić, S. Solubility enhancement of desloratadine by solid dispersion in poloxamers. Int. J. Pharm. 2012, 436, 161–170. [Google Scholar] [CrossRef]
- Baghel, S.; Cathcart, H.; O’Reilly, N.J. Polymeric amorphous solid dispersions: A review of amorphization, crystallization, stabilization, solid-state characterization, and aqueous solubilization of biopharmaceutical classification system class II drugs. J. Pharm. Sci. 2016, 105, 2527–2544. [Google Scholar] [CrossRef] [PubMed]
- Damian, F.; Blaton, N.; Naesens, L.; Balzarini, J.; Kinget, R.; Augustijns, P.; Van den Mooter, G. Physicochemical characterization of solid dispersions of the antiviral agent UC-781 with polyethylene glycol 6000 and Gelucire 44/14. Eur. J. Pharm. Sci. 2000, 10, 311–322. [Google Scholar] [CrossRef]
- Mehanna, M.M.; Motawaa, A.M.; Samaha, M.W. In sight into tadalafil–block copolymer binary solid dispersion: Mechanistic investigation of dissolution enhancement. Int. J. Pharm. 2010, 402, 78–88. [Google Scholar] [CrossRef]
- Vo, C.L.-N.; Park, C.; Lee, B.-J. Current trends and future perspectives of solid dispersions containing poorly water-soluble drugs. Eur. J. Pharm. Biopharm. 2013, 85, 799–813. [Google Scholar] [CrossRef]
- Weerapol, Y.; Limmatvapirat, S.; Nunthanid, J.; Konthong, S.; Suttiruengwong, S.; Sriamornsak, P. Development and characterization of nifedipine-amino methacrylate copolymer solid dispersion powders with various adsorbents. Asian J. Pharm. Sci. 2017, 12, 335–343. [Google Scholar] [CrossRef]
- Kumar, L.; Alam, M.S.; Meena, C.L.; Jain, R.; Bansal, A.K. Fexofenadine hydrochloride. In Profiles of Drug Substances, Excipients and Related Methodology; Elsevier: Amsterdam, The Netherlands, 2009; Volume 34, pp. 153–192. [Google Scholar]
- Passerini, N.; Albertini, B.; González-Rodríguez, M.L.; Cavallari, C.; Rodriguez, L. Preparation and characterisation of ibuprofen–poloxamer 188 granules obtained by melt granulation. Eur. J. Pharm. Sci. 2002, 15, 71–78. [Google Scholar] [CrossRef]
- Windbergs, M.; Strachan, C.J.; Kleinebudde, P. Influence of structural variations on drug release from lipid/polyethylene glycol matrices. Eur. J. Pharm. Sci. 2009, 37, 555–562. [Google Scholar] [CrossRef]
- Pardhi, V.P.; Jain, K. Impact of binary/ternary solid dispersion utilizing poloxamer 188 and TPGS to improve pharmaceutical attributes of bedaquiline fumarate. J. Drug Deliv. Sci. Technol. 2021, 62, 102349. [Google Scholar] [CrossRef]
- Ige, P.P.; Baria, R.K.; Gattani, S.G. Fabrication of fenofibrate nanocrystals by probe sonication method for enhancement of dissolution rate and oral bioavailability. Colloids Surf. B Biointerfaces 2013, 108, 366–373. [Google Scholar] [CrossRef]
- Bandari, S.; Jadav, S.; Eedara, B.B.; Dhurke, R.; Jukanti, R. Enhancement of solubility and dissolution rate of Loratadine with Gelucire 50/13. J. Pharm. Innov. 2014, 9, 141–149. [Google Scholar] [CrossRef]
- Strelevitz, T.J.; Foti, R.S.; Fisher, M.B. In Vivo use of the P450 inactivator 1-aminobenzotriazole in the rat: Varied dosing route to elucidate gut and liver contributions to first-pass and systemic clearance. J. Pharm. Sci. 2006, 95, 1334–1341. [Google Scholar] [CrossRef]
- Lappin, G.; Shishikura, Y.; Jochemsen, R.; Weaver, R.J.; Gesson, C.; Houston, B.; Oosterhuis, B.; Bjerrum, O.J.; Rowland, M.; Garner, C. Pharmacokinetics of fexofenadine: Evaluation of a microdose and assessment of absolute oral bioavailability. Eur. J. Pharm. Sci. 2010, 40, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Piao, H.-M.; Balakrishnan, P.; Cho, H.-J.; Kim, H.; Kim, Y.-S.; Chung, S.-J.; Shim, C.-K.; Kim, D.-D. Preparation and evaluation of fexofenadine microemulsions for intranasal delivery. Int. J. Pharm. 2010, 395, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Bansal, T.; Akhtar, N.; Jaggi, M.; Khar, R.K.; Talegaonkar, S. Novel formulation approaches for optimising delivery of anticancer drugs based on P-glycoprotein modulation. Drug Discov. Today 2009, 14, 1067–1074. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Concentration of Carrier (% w/v) | PEG 20,000 | Poloxamer 188 | ||
|---|---|---|---|---|
| Drug Solubility (mg/mL) | ΔG°tr (J/mole) | Drug Solubility (mg/mL) | ΔG°tr (J/mole) | |
| 0 | 1.45 ± 0.15 | - | 1.45 ± 0.15 | - |
| 5 | 3.14 ± 0.19 | −1.99 | 4.06 ± 0.21 | −2.66 |
| 10 | 5.06 ± 0.23 | −3.22 | 5.73 ± 0.24 | −3.55 |
| 15 | 7.16 ± 0.18 | −4.12 | 6.90± 0.19 | −4.03 |
| 20 | 7.99 ± 0.26 | −4.40 | 9.32 ± 0.14 | −4.80 |
| 25 | 10.02 ± 0.17 | −4.99 | 10.65 ± 0.28 | −5.14 |
| 30 | 11.78 ± 0.18 | −5.40 | 12.27 ± 0.22 | −5.51 |
| Parameters | Q15 | Q120 | DE15 | DE120 | t50% |
|---|---|---|---|---|---|
| Supplied FFH | 16.9 ± 1.2 | 38.9 ± 2.1 | 8.5 ± 0.6 | 24.3 ± 1.4 | ˃ 120 |
| SD-FP20K(1:1) * | 55.1 ± 1.6 | 70.0 ± 1.9 | 27.5 ± 0.8 | 60.9 ± 1.8 | ˃ 120 |
| SD-FP20K(1:2) * | 71.1 ± 2.2 | 78.3 ± 2.6 | 35.5 ± 1.1 | 70.7 ± 1.8 | 60 |
| SD-FP20K(1:4) * | 80.6 ± 1.8 | 85.1 ± 1.5 | 40.3 ± 0.9 | 78.4 ± 1.8 | 15 |
| PM-FP20K(1:4) * | 27.8 ± 2.8 | 56.7 ± 1.1 | 13.9 ± 1.4 | 40.3 ± 2.0 | ˃ 120 |
| SD-FP188(1:1) * | 66.3 ± 1.8 | 78.2 ± 2.3 | 27.5 ± 0.9 | 68.9 ± 1.7 | ˃ 120 |
| SD-FP188(1:2) * | 74.2 ± 1.9 | 84.7 ± 2.3 | 35.5 ±1.0 | 75.7 ± 1.9 | 60 |
| SD-FP188(1:4) * | 80.7 ± 1.6 | 94.7 ± 1.9 | 40.3 ± 0.8 | 83.1 ± 1.6 | 15 |
| PM-FP188(1:4) * | 31.1 ± 2.8 | 58.2 ± 1.8 | 13.9 ± 0.6 | 43.0 ± 1.4 | ˃ 120 |
| Formulation | Peff(rat) × 10−6 (cm/s) | Peff (human) × 10−5 (cm/s) | ka (min−1) | ER |
|---|---|---|---|---|
| Supplied FFH | 7.04 ± 0.56 | 2.61 ± 0.18 | 0.016 ± 0.001 | - |
| SD-FP20K(1:4) | 14.40 ± 0.27 † | 5.00 ± 0.08 † | 0.030 ± 0.001 † | 2.04 † |
| SD-FP188(1:4) | 21.30 ± 0.45 † | 7.26 ± 0.14 † | 0.044 ± 0.001 † | 3.03 † |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eedara, B.B.; Nyavanandi, D.; Narala, S.; Veerareddy, P.R.; Bandari, S. Improved Dissolution Rate and Intestinal Absorption of Fexofenadine Hydrochloride by the Preparation of Solid Dispersions: In Vitro and In Situ Evaluation. Pharmaceutics 2021, 13, 310. https://doi.org/10.3390/pharmaceutics13030310
Eedara BB, Nyavanandi D, Narala S, Veerareddy PR, Bandari S. Improved Dissolution Rate and Intestinal Absorption of Fexofenadine Hydrochloride by the Preparation of Solid Dispersions: In Vitro and In Situ Evaluation. Pharmaceutics. 2021; 13(3):310. https://doi.org/10.3390/pharmaceutics13030310
Chicago/Turabian StyleEedara, Basanth Babu, Dinesh Nyavanandi, Sagar Narala, Prabhakar Reddy Veerareddy, and Suresh Bandari. 2021. "Improved Dissolution Rate and Intestinal Absorption of Fexofenadine Hydrochloride by the Preparation of Solid Dispersions: In Vitro and In Situ Evaluation" Pharmaceutics 13, no. 3: 310. https://doi.org/10.3390/pharmaceutics13030310
APA StyleEedara, B. B., Nyavanandi, D., Narala, S., Veerareddy, P. R., & Bandari, S. (2021). Improved Dissolution Rate and Intestinal Absorption of Fexofenadine Hydrochloride by the Preparation of Solid Dispersions: In Vitro and In Situ Evaluation. Pharmaceutics, 13(3), 310. https://doi.org/10.3390/pharmaceutics13030310