
In Silico Simulation of the Systemic Drug Exposure Following the Topical Application of Opioid Analgesics in Patients with Cutaneous Lesions


Abstract
1. Introduction
2. Materials and Methods
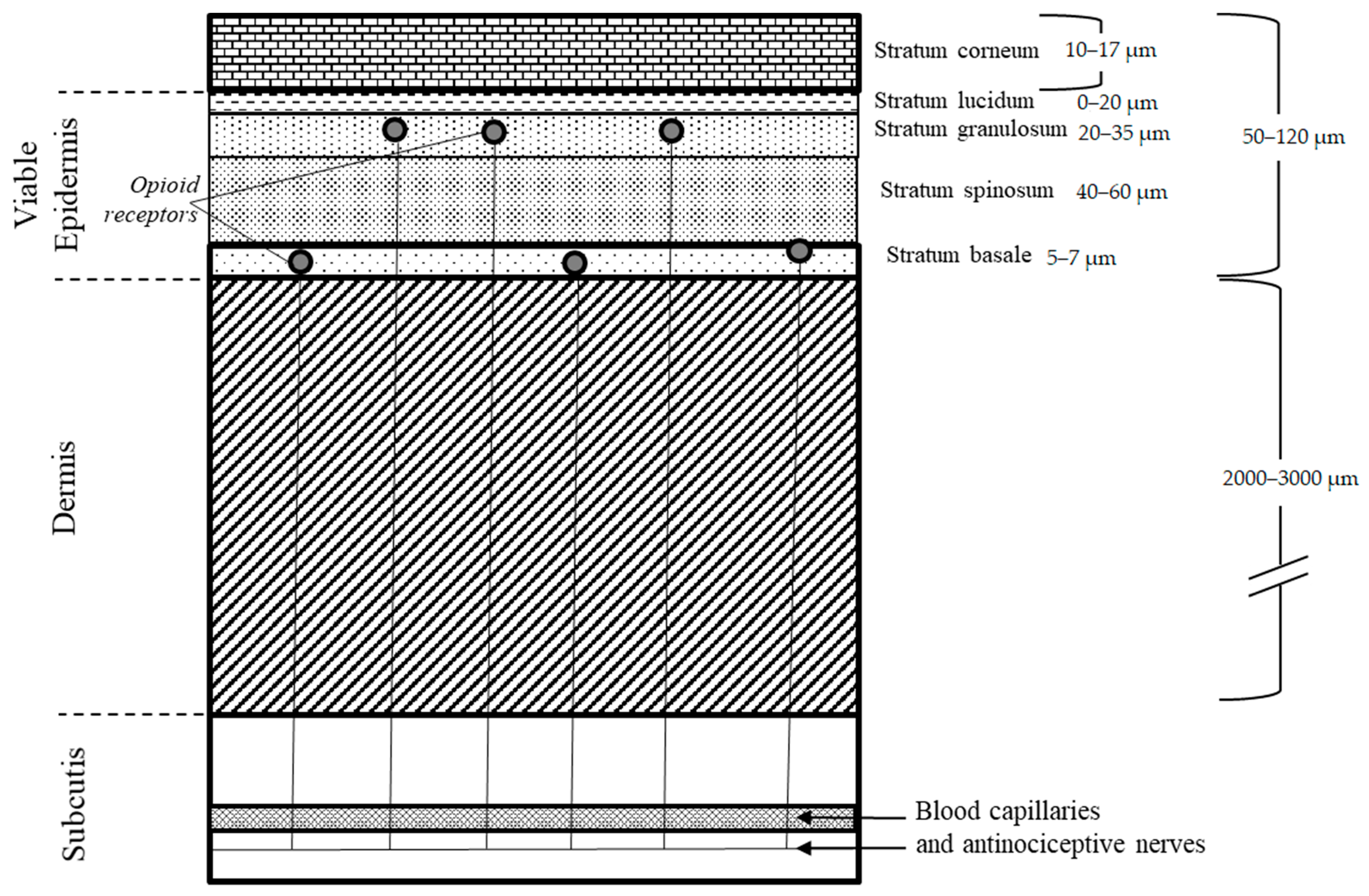
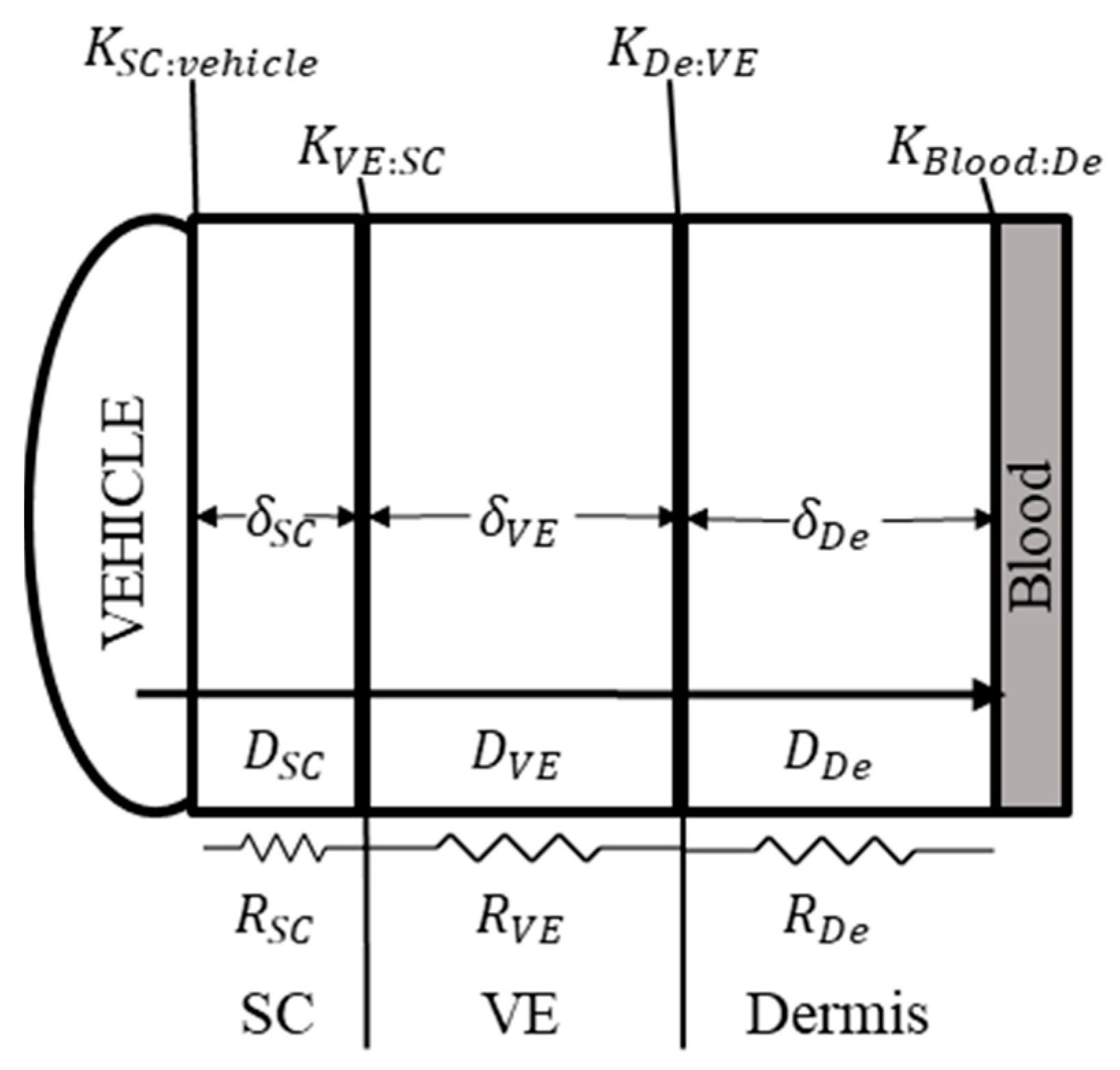
2.1. BIOiSIM Model Expansion
2.1.1. Intra-Skin Permeability Prediction
2.1.2. Calculation of Intra-Skin Concentration
2.2. Test Dataset and Data Curation
2.3. Subjects
2.4. Statistical Analysis
3. Results
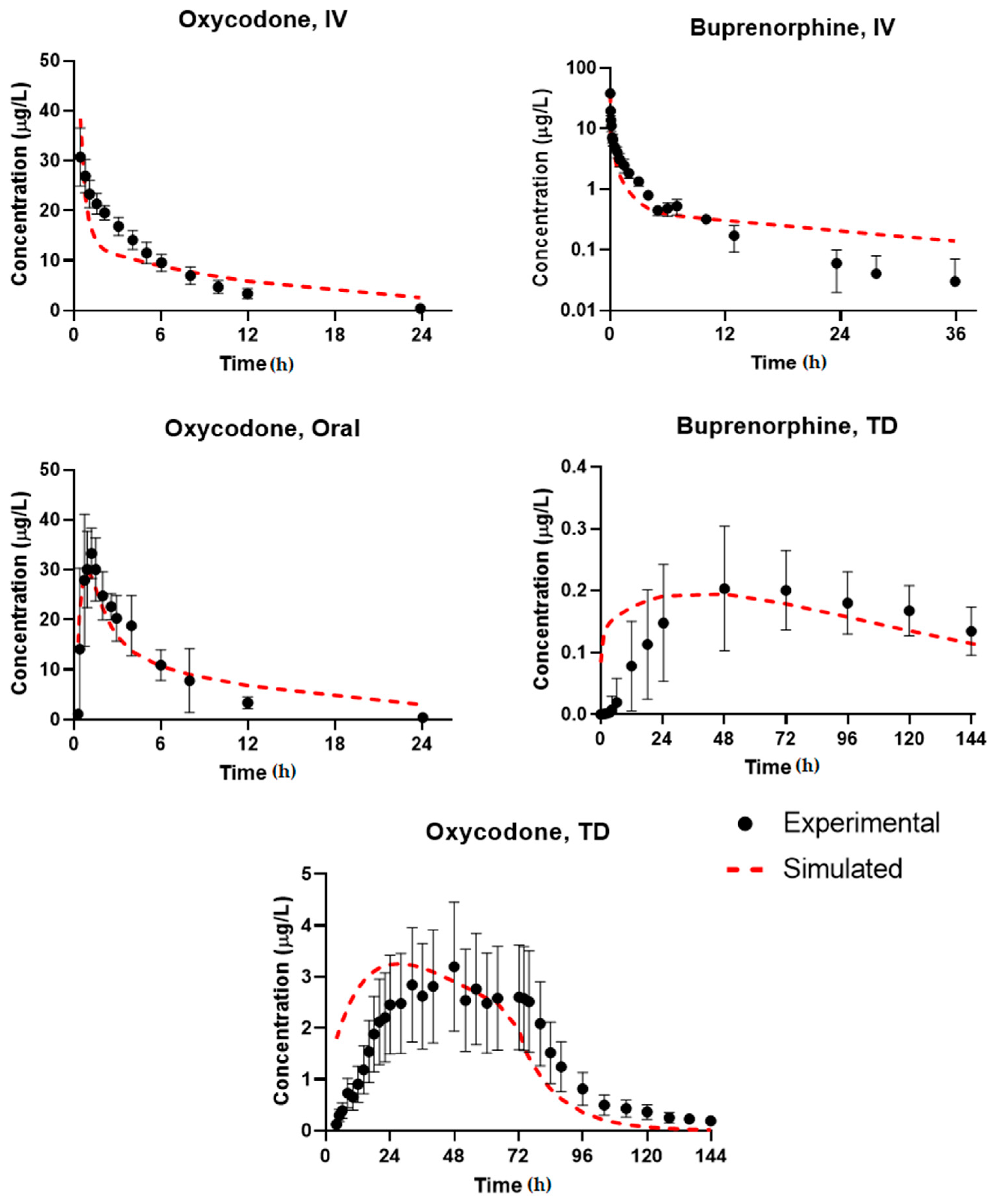
3.1. Simulation Accuracy
3.2. Predicted Skin Model Parameters
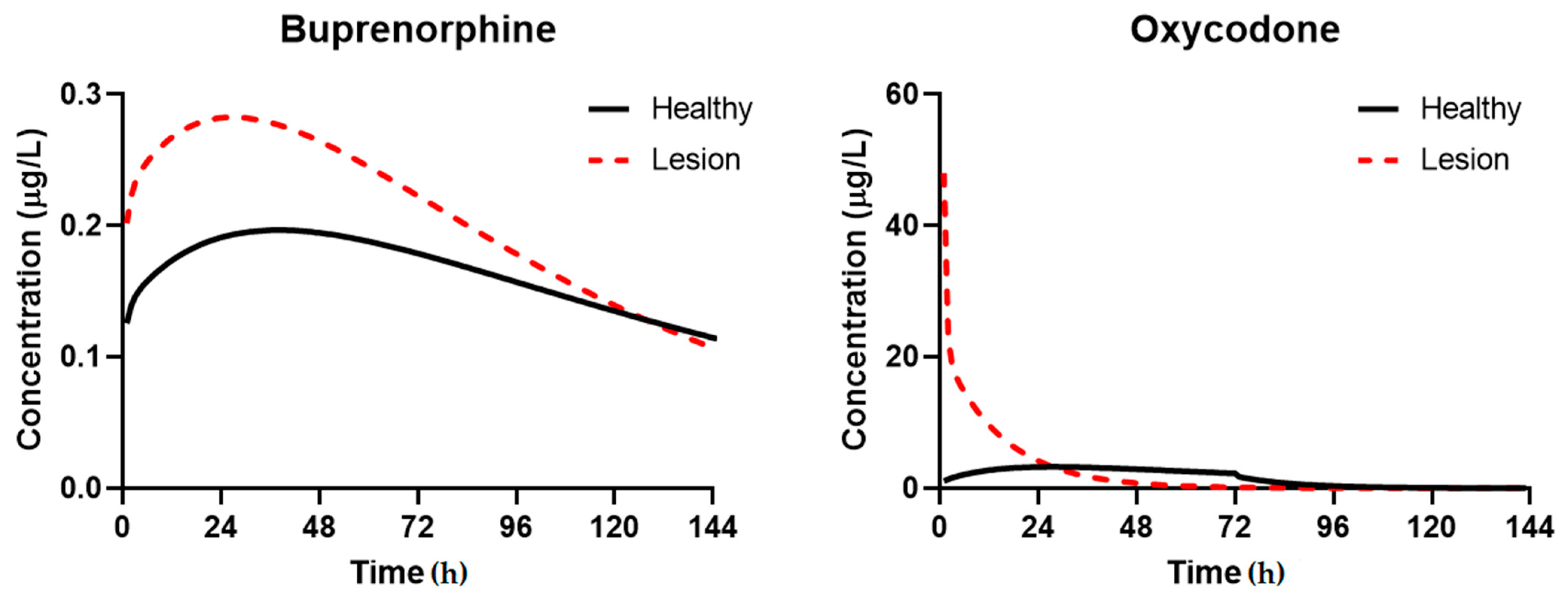
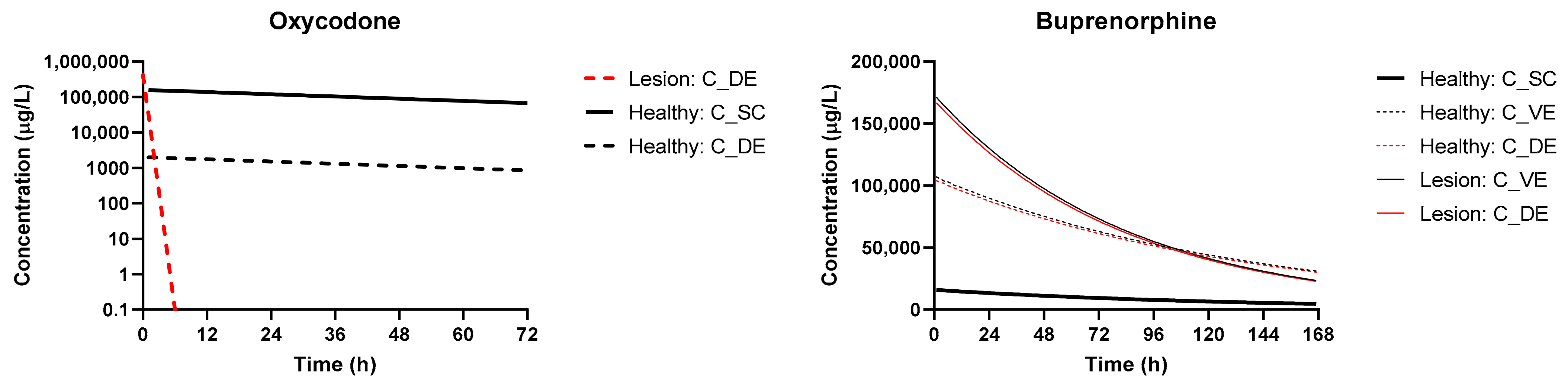
3.3. Simulation of Disposition in Patients with Lesions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Leppert, W.; Malec-Milewska, M.; Zajaczkowska, R.; Wordliczek, J. Transdermal and Topical Drug Administration in the Treatment of Pain. Molecules 2018, 23, 681. [Google Scholar] [CrossRef]
- Flores, M.P.; de Castro, A.P.C.R.; dos Santos Nascimento, J. Topical analgesics. Rev. Bras. Anestesiol. 2012, 7094, 70122–70128. [Google Scholar] [CrossRef]
- Bigliardi-Qi, M.; Gaveriaux-Ruff, C.; Hohl, D.; Bigliardi, P.L. Opioid receptors in skin—Link between stress and skin disease? Exp. Derm. 2006, 15, 643–648. [Google Scholar] [CrossRef]
- Ständer, S.; Gunzer, M.; Metze, D.; Luger, T.; Steinhoff, M. Localization of μ-opioid receptor 1A on sensory nerve fibers in human skin. Regul. Pept. 2002, 110, 75–83. [Google Scholar] [CrossRef]
- Wei, J.C.J.; Edwards, G.A.; Martin, D.J.; Huang, H.; Crichton, M.L.; Kendall, M.A.F. Allometric scaling of skin thickness, elasticity, viscoelasticity to mass for micro-medical device translation: From mice, rats, rabbits, pigs to humans. Sci. Rep. 2017, 7, 15885. [Google Scholar] [CrossRef] [PubMed]
- James, D.L.; Jowza, M. Principles of Burn Pain Management. Clin. Plast. Surg. 2017, 44, 737–747. [Google Scholar] [CrossRef]
- Hansen, S.; Lehr, C.M.; Schaefer, U.F. Improved input parameters for diffusion models of skin absorption. Adv. Drug Deliv. Rev. 2013, 65, 251–264. [Google Scholar] [CrossRef]
- Musazzi, U.M.; Matera, C.; Dallanoce, C.; Vacondio, F.; De Amici, M.; Vistoli, G.; Cilurzo, F.; Minghetti, P. On the selection of an opioid for local skin analgesia: Structure-skin permeability relationships. Int. J. Pharm. 2015, 489, 177–185. [Google Scholar] [CrossRef]
- Lachiewicz, A.M.; Williams, F.N.; Carson, S.S.; Trexler, J.M.; Nielsen, C.A.; van Duin, D.; Weber, D.J.; Williams, S.D.; Jones, S.W.; Cairns, B.A. Improving Research Enrollment of Severe Burn Patients. J. Burn Care Res. 2017, 38, e807–e813. [Google Scholar] [CrossRef]
- Todo, H. Transdermal Permeation of Drugs in Various Animal Species. Pharmaceutics 2017, 9, 33. [Google Scholar] [CrossRef] [PubMed]
- Maharao, N.; Antontsev, V.; Hou, H.; Walsh, J.; Varshney, J. Scalable in silico Simulation of Transdermal Drug Permeability: Application of BIOiSIM Platform. Drug Des. Devel. 2020, 14, 2307–2317. [Google Scholar] [CrossRef] [PubMed]
- Pecoraro, B.; Tutone, M.; Hoffman, E.; Hutter, V.; Almerico, A.M.; Traynor, M. Predicting Skin Permeability by Means of Computational Approaches: Reliability and Caveats in Pharmaceutical Studies. J. Chem. Inf. Model 2019, 59, 1759–1771. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.J.; Lian, G.P.; Han, L.J. Prediction of human skin permeability using artificial neural network (ANN) modeling. Acta Pharm. Sin. 2007, 28, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Ellison, C.A.; Tankersley, K.O.; Obringer, C.M.; Carr, G.J.; Manwaring, J.; Rothe, H.; Duplan, H.; Genies, C.; Gregoire, S.; Hewitt, N.J.; et al. Partition coefficient and diffusion coefficient determinations of 50 compounds in human intact skin, isolated skin layers and isolated stratum corneum lipids. Toxicol. Vitr. 2020, 69, 104990. [Google Scholar] [CrossRef]
- Mitragotri, S.; Anissimov, Y.G.; Bunge, A.L.; Frasch, H.F.; Guy, R.H.; Hadgraft, J.; Kasting, G.B.; Lane, M.E.; Roberts, M.S. Mathematical models of skin permeability: An overview. Int. J. Pharm. 2011, 418, 115–129. [Google Scholar] [CrossRef]
- Obata, Y.; Takayama, K. Related Topic: Determination of Partition Coefficient from Vehicle to Skin. In Skin Permeation and Disposition of Therapeutic and Cosmeceutical Compounds; Sugibayashi, K., Ed.; Springer: Tokyo, Japan, 2017; pp. 385–389. [Google Scholar] [CrossRef]
- Varrassi, G.; Marinangeli, F.; Ciccozzi, A.; Iovinelli, G.; Facchetti, G.; Ciccone, A. Intra-articular buprenorphine after knee arthroscopy. A randomised, prospective, double-blind study. Acta Anaesthesiol. Scand. 1999, 43, 51–55. [Google Scholar] [CrossRef]
- Ballas, S.K. Treatment of painful sickle cell leg ulcers with topical opioids. Blood 2002, 99, 1096. [Google Scholar] [CrossRef]
- Pergolizzi, J.V., Jr.; Mercadante, S.; Echaburu, A.V.; Van den Eynden, B.; Fragoso, R.M.; Mordarski, S.; Lybaert, W.; Beniak, J.; Oronska, A.; Slama, O.; et al. The role of transdermal buprenorphine in the treatment of cancer pain: An expert panel consensus. Curr. Med. Res. Opin. 2009, 25, 1517–1528. [Google Scholar] [CrossRef]
- Chakravarty, K.; Antontsev, V.; Jagarapu, A.; Bundey, Y.; Hou, H.; Maharao, N.; Varshney, J. Accelerated repurposing and drug development of pulmonary hypertension therapies for COVID-19 treatment using an AI-integrated biosimulation platform. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Maharao, N.; Antontsev, V.; Wright, M.; Varshney, J. Entering the era of computationally driven drug development. Drug Metab. Rev. 2020, 52, 283–298. [Google Scholar] [CrossRef] [PubMed]
- Potts, R.O.; Guy, R.H. Predicting skin permeability. Pharm. Res. 1992, 9, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Shatkin, J.A.; Brown, H.S. Pharmacokinetics of the dermal route of exposure to volatile organic chemicals in water: A computer simulation model. Environ. Res. 1991, 56, 90–108. [Google Scholar] [CrossRef]
- Kretsos, K.; Miller, M.A.; Zamora-Estrada, G.; Kasting, G.B. Partitioning, diffusivity and clearance of skin permeants in mammalian dermis. Int. J. Pharm. 2008, 346, 64–79. [Google Scholar] [CrossRef] [PubMed]
- Polak, S.; Ghobadi, C.; Mishra, H.; Ahamadi, M.; Patel, N.; Jamei, M.; Rostami-Hodjegan, A. Prediction of concentration-time profile and its inter-individual variability following the dermal drug absorption. J. Pharm. Sci. 2012, 101, 2584–2595. [Google Scholar] [CrossRef]
- Nitsche, J.M.; Wang, T.F.; Kasting, G.B. A two-phase analysis of solute partitioning into the stratum corneum. J. Pharm. Sci. 2006, 95, 649–666. [Google Scholar] [CrossRef] [PubMed]
- Harding, C.R. The stratum corneum: Structure and function in health and disease. Dermatol. Ther. 2004, 17 (Suppl. 1), 6–15. [Google Scholar] [CrossRef]
- Scott, R.C.; Corrigan, M.A.; Smith, F.; Mason, H. The influence of skin structure on permeability: An intersite and interspecies comparison with hydrophilic penetrants. J. Investig. Derm. 1991, 96, 921–925. [Google Scholar] [CrossRef]
- Lombardo, F.; Berellini, G.; Obach, R.S. Trend Analysis of a Database of Intravenous Pharmacokinetic Parameters in Humans for 1352 Drug Compounds. Drug Metab. Dispos. 2018, 46, 1466–1477. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, H.V.; Zhang, H.; Caritis, S.N.; Venkataramanan, R. A physiologically based pharmacokinetic modelling approach to predict buprenorphine pharmacokinetics following intravenous and sublingual administration. Br. J. Clin. Pharm. 2017, 83, 2458–2473. [Google Scholar] [CrossRef]
- Purdue Pharma L.P. Butrans® (Buprenorphine) Transdermal System; NDA 021306/S-027; FDA: Silver Spring, MD, USA, 2017.
- Benet, L.Z.; Broccatelli, F.; Oprea, T.I. BDDCS applied to over 900 drugs. AAPS J. 2011, 13, 519–547. [Google Scholar] [CrossRef]
- Boger, E.; Evans, N.; Chappell, M.; Lundqvist, A.; Ewing, P.; Wigenborg, A.; Friden, M. Systems Pharmacology Approach for Prediction of Pulmonary and Systemic Pharmacokinetics and Receptor Occupancy of Inhaled Drugs. CPT Pharmacomet. Syst. Pharm. 2016, 5, 201–210. [Google Scholar] [CrossRef]
- Campbell, J.L.; Andersen, M.E.; Clewell, H.J. A hybrid CFD-PBPK model for naphthalene in rat and human with IVIVE for nasal tissue metabolism and cross-species dosimetry. Inhal. Toxicol. 2014, 26, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Jamei, M.; Bajot, F.; Neuhoff, S.; Barter, Z.; Yang, J.; Rostami-Hodjegan, A.; Rowland-Yeo, K. A mechanistic framework for in vitro-in vivo extrapolation of liver membrane transporters: Prediction of drug-drug interaction between rosuvastatin and cyclosporine. Clin. Pharm. 2014, 53, 73–87. [Google Scholar] [CrossRef]
- Jorge, L.L.; Feres, C.C.; Teles, V.E. Topical preparations for pain relief: Efficacy and patient adherence. J. Pain Res. 2010, 4, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Graham, T.; Grocott, P.; Probst, S.; Wanklyn, S.; Dawson, J.; Gethin, G. How are topical opioids used to manage painful cutaneous lesions in palliative care? A critical review. Pain 2013, 154, 1920–1928. [Google Scholar] [CrossRef] [PubMed]
- Stein, C.; Lang, L.J. Peripheral mechanisms of opioid analgesia. Curr. Opin. Pharm. 2009, 9, 3–8. [Google Scholar] [CrossRef]
- Faymonville, M.E.; Libbrecht, D. Transdermal buprenorphine: A current overview of pharmacological and clinical data. Rev. Med. Liege 2008, 63, 671–676. [Google Scholar]
- Bhattacharya, S.; Mishra, R.K. Pressure ulcers: Current understanding and newer modalities of treatment. Indian J. Plast. Surg. 2015, 48, 4–16. [Google Scholar] [CrossRef]
- Soriano-Ruiz, J.L.; Calpena-Campmany, A.C.; Silva-Abreu, M.; Halbout-Bellowa, L.; Bozal-de Febrer, N.; Rodriguez-Lagunas, M.J.; Clares-Naveros, B. Design and evaluation of a multifunctional thermosensitive poloxamer-chitosan-hyaluronic acid gel for the treatment of skin burns. Int. J. Biol. Macromol. 2020, 142, 412–422. [Google Scholar] [CrossRef]
- Loden, M. Biophysical properties of dry atopic and normal skin with special reference to effects of skin care products. Acta Derm. Venereol. Suppl. (Stockh.) 1995, 192, 1–48. [Google Scholar] [CrossRef]
- Chast, F.; Bardin, C.; Sauvageon-Martre, H.; Callaert, S.; Chaumeil, J.C. Systemic morphine pharmacokinetics after ocular administration. J. Pharm. Sci. 1991, 80, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Avdeef, A.; Barrett, D.A.; Shaw, P.N.; Knaggs, R.D.; Davis, S.S. Octanol-, chloroform-, and propylene glycol dipelargonat-water partitioning of morphine-6-glucuronide and other related opiates. J. Med. Chem. 1996, 39, 4377–4381. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Property | Oxycodone | Buprenorphine |
|---|---|---|
| Chemical class | Weak base | Weak base |
| Behavior | Full opioid agonist | Opioid agonist-antagonist |
| Plasma protein fraction unbound (fu,p) | 0.55 | 0.04 [29] |
| Blood:plasma binding coefficient (B:P) | 1.0465 * | 0.55 [30] |
| Clearance [L/h] | 28.3 * | 85.5 * |
| Tissue:plasma partition coefficient (Kp) | 5.29 * | 13.3464 |
| Transdermal permeability (kperm) [cm2/h] | 0.0001 * | 0.000593 * |
| Bioavailability (Oral) | 0.735 | N/A |
| Bioavailability (TD) | 0.3312 * | 0.15 [31] |
| First-order absorption rate constant (ka) | 0.8281 * | N/A |
| logP | 0.255 [32] | 4.98 [32] |
| pKa acid | 13.56 | 7.5 |
| pKa base | 8.21 | 12.54 |
| Drug Name | ROA | Dosage | Experimental Setup |
|---|---|---|---|
| Oxycodone | IV | 0.1 mg/kg | 12 healthy subjects were given 0.1 mg/kg of intravenous oxycodone after pre-treatments with placebo. |
| Oral | 15 mg | 15 mg oral dose of the drug was administered to 12 healthy volunteers. | |
| Transdermal | 23.4 mg, Patch | Subjects received a single 3-day (72 h) application of three 40 cm2 solid matrix oxycodone transdermal patches containing 6.7 mg tocopheryl phosphate mixture and 23.4 mg of oxycodone per patch. | |
| Buprenorphine | IV | 1.2 mg | Buprenophine in a single dose 1.2 mg was administered to six healthy male volunteers. |
| Transdermal | 1.68 mg | A buprenorphine transdermal system delivering 10 mcg/hour was applied to healthy volunteers. |
| Compounds | Buprenorphine | Oxycodone | ||||
|---|---|---|---|---|---|---|
| Output Metric | ROA | IV | TD | IV | Oral | TD |
| Dose (mg) | 1.2 | 1.68 | 6.75 | 39 | 23.4 | |
| AUC(0–t) (µg*h/L) | Observed * | NA | NA | NA | 180.00 | 209.05 |
| Calculated | 17.45 | 25.99 | 153.10 | 180.29 | 214.01 | |
| Predicted | 14.83 | 26.27 | 161.28 | 208.86 | 220.04 | |
| AAFE | 1.18 | 1.01 | 1.05 | 1.16 | 1.03 | |
| AFE | 1.18 | 0.99 | 0.95 | 0.86 | 0.97 | |
| Cmax (µg/L) | Observed * | 37.52 | NA | NA | 38.00 | 3.40 |
| Calculated | 38.16 | 0.20 | 30.73 | 33.26 | 3.20 | |
| Predicted | 36.76 | 0.20 | 38.41 | 29.95 | 3.26 | |
| AAFE | 1.04 | 1.03 | 1.25 | 1.11 | 1.02 | |
| AFE | 1.04 | 1.03 | 0.80 | 1.11 | 0.98 | |
| Tmax (h) | Observed * | 0.04 | NA | NA | 1.08 | 49.30 |
| Calculated | 0.04 | 48.02 | 0.43 | 1.19 | 47.99 | |
| Predicted | 0.04 | 37.99 | 0.43 | 0.86 | 27.25 | |
| AAFE | 1.00 | 1.26 | 1.00 | 1.39 | 1.76 | |
| AFE | 1.00 | 1.26 | 1.00 | 1.39 | 1.76 | |
| Statistics | GMFE | 1.07 | 1.10 | 1.10 | 1.21 | 1.23 |
| p-value | 0.4474 | 0.0112 | 0.7468 | 0.1666 | 0.8696 | |
| Significant difference? (p-value < 0.05) | No | Yes | No | No | No | |
| Parameter Calculated | Oxycodone | Buprenorphine | |
|---|---|---|---|
| Partition Coefficients (K) | SC/w | 3.73 | 5.29 |
| De/w | 10.29 | 57.29 | |
| VE/SC | 2.76 | 10.83 | |
| Permeability (kperm) [cm/h] | Total | 1.000 × 10−4 | 5.93 × 10−4 |
| SC | 1.005 × 10−4 | 15.6 × 10−4 | |
| VE | 0.82 | 0.04 | |
| Dermis | 220 × 10−4 | 9.88 × 10−4 | |
| Resistance (R) [h/cm] | Total | 10,000 | 1686.82 |
| SC | 9952.99 | 641.39 | |
| VE | 1.21 | 26.98 | |
| Dermis | 45.49 | 1011.71 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khotimchenko, M.; Antontsev, V.; Chakravarty, K.; Hou, H.; Varshney, J. In Silico Simulation of the Systemic Drug Exposure Following the Topical Application of Opioid Analgesics in Patients with Cutaneous Lesions. Pharmaceutics 2021, 13, 284. https://doi.org/10.3390/pharmaceutics13020284
Khotimchenko M, Antontsev V, Chakravarty K, Hou H, Varshney J. In Silico Simulation of the Systemic Drug Exposure Following the Topical Application of Opioid Analgesics in Patients with Cutaneous Lesions. Pharmaceutics. 2021; 13(2):284. https://doi.org/10.3390/pharmaceutics13020284
Chicago/Turabian StyleKhotimchenko, Maksim, Victor Antontsev, Kaushik Chakravarty, Hypatia Hou, and Jyotika Varshney. 2021. "In Silico Simulation of the Systemic Drug Exposure Following the Topical Application of Opioid Analgesics in Patients with Cutaneous Lesions" Pharmaceutics 13, no. 2: 284. https://doi.org/10.3390/pharmaceutics13020284
APA StyleKhotimchenko, M., Antontsev, V., Chakravarty, K., Hou, H., & Varshney, J. (2021). In Silico Simulation of the Systemic Drug Exposure Following the Topical Application of Opioid Analgesics in Patients with Cutaneous Lesions. Pharmaceutics, 13(2), 284. https://doi.org/10.3390/pharmaceutics13020284