
Therapeutic Potential of Injectable Nano-Mupirocin Liposomes for Infections Involving Multidrug-Resistant Bacteria

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Nano-Mupirocin Production
2.2.2. Quantification of Mupirocin
2.2.3. Antimicrobial Susceptibility Testing
2.2.4. Resistance Selection
Single-Step Antimicrobial Resistance Selection
Multi-Step Antimicrobial Resistance Selection
2.2.5. Nano-Mupirocin Pharmacokinetics (PK) in Rats
2.2.6. PK and Vaginal Biodistribution (BD) of Nano-Mupirocin
2.2.7. Bioanalytical Methods
2.2.8. Pharmacokinetic Analysis
2.2.9. Necrotizing Fasciitis, Dose Response Study
3. Results
3.1. Activity against Gram-Positive Bacteria
3.2. Resistance Selection
3.2.1. Gram-Positive Isolates
3.2.2. Resistance Selection with N. gonorrhoeae
3.3. Pharmacokinetic (PK) Study
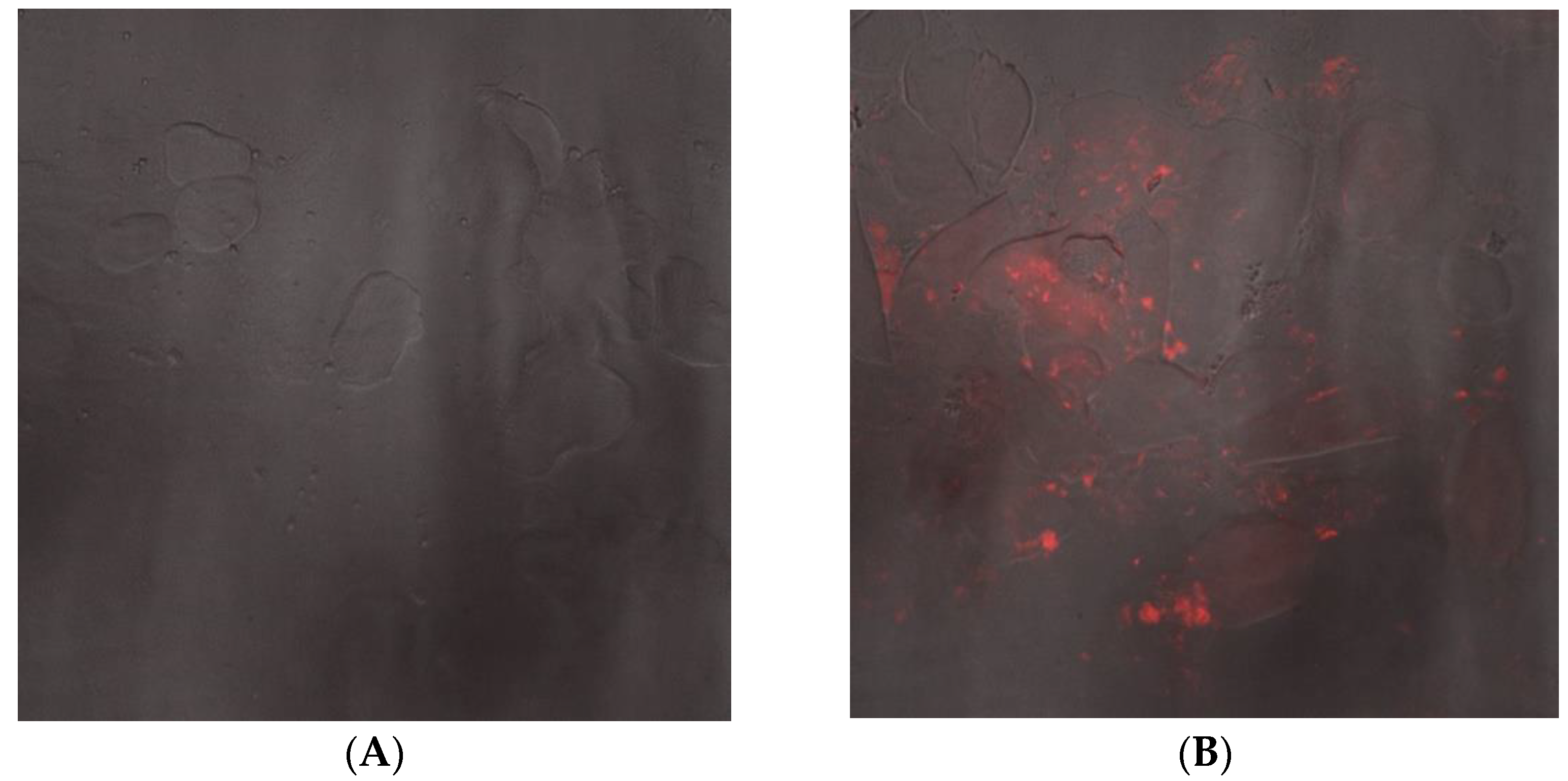
3.4. Nano-mupirocin Biodistribution into Murine Vaginal Secretions
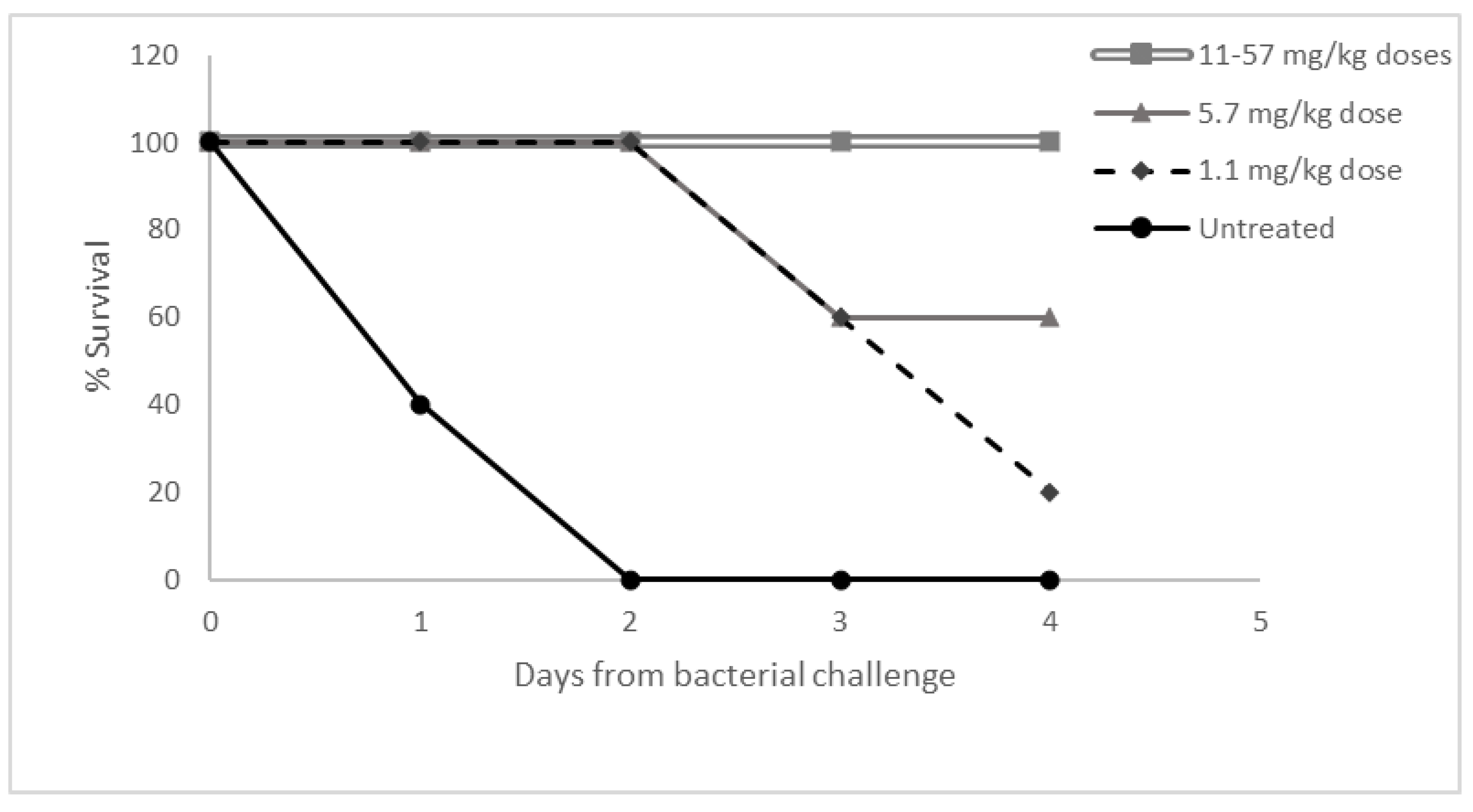
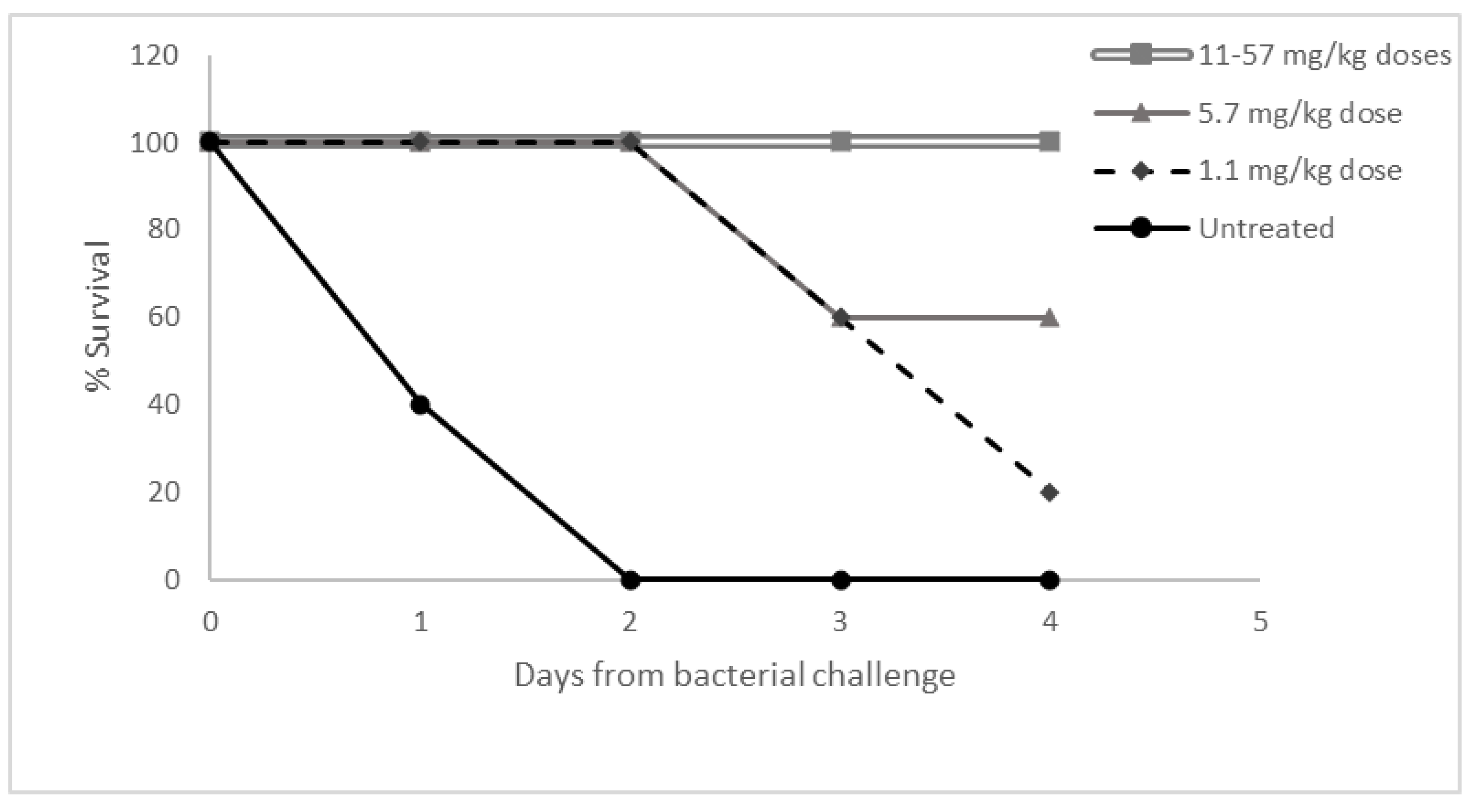
3.5. Dose Response Study in Necrotizing Fasciitis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hughes, J.; Mellows, G. Interaction of pseudomonic acid A with Escherichia coli B isoleucyl-tRNA synthetase. Biochem. J. 1980, 191, 209–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pappa, K. The clinical development of mupirocin. J. Am. Acad. Dermatol. 1990, 22, 873–879. [Google Scholar] [CrossRef]
- Fuller, A.; Mellows, G.; Wollford, M.; Banks, G.; Barrow, K.; Chain, E. Pseudomonic acid: An antibiotic produced by Pseudomonas fluorescens. Nature 1971, 234, 416–417. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, R.; Boon, R.J.; Griffin, K.E.; Masters, P.J.; Slocombe, B.; White, A. Antibacterial activity of mupirocin (pseudomonic acid), a new antibiotic for topical use. Antimicrob. Agents Chemother. 1985, 27, 495–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cern, A.; Golbraikh, A.; Sedykh, A.; Tropsha, A.; Barenholz, Y.; Goldblum, A. Quantitative structure-property relationship modeling of remote liposome loading of drugs. J. Control. Release 2012, 160, 147–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cern, A.; Barenholz, Y.; Tropsha, A.; Goldblum, A. Computer-aided design of liposomal drugs: In silico prediction and experimental validation of drug candidates for liposomal remote loading. J. Control. Release 2014, 173, 123–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cern, A.; Marcus, D.; Tropsha, A.; Barenholz, Y.; Goldblum, A. New drug candidates for liposomal delivery identified by computer modeling of liposomes, remote loading and leakage. J. Control. Release 2017, 252, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Azzopardi, E.A.; Ferguson, E.L.; Thomas, D.W. The enhanced permeability retention effect: A new paradigm for drug targeting in infection. J. Antimicrob. Chemother. 2013, 68, 257–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cern, A.; Michael-Gayego, A.; Bavli, Y.; Koren, E.; Goldblum, A.; Moses, A.E.; Xiong, Y.Q.; Barenholz, Y. Nano-mupirocin: Enabling the parenteral activity of mupirocin. Eur. J. Nanomed. 2016, 8, 139–149. [Google Scholar] [CrossRef]
- Goldman, O.; Cern, A.; Muesken, M.; Rohde, M.; Weiss, W.; Barenholz, Y.; Medina, E. Liposomal mupirocin holds promise for systemic treatment of invasive Staphylococcus aureus infections. J. Control. Release 2019, 316, 292–301. [Google Scholar] [CrossRef] [PubMed]
- CLSI. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard-Eleventh Edition M07-A11; Clinical and Laboratory Standards Institute (CLSI): Wayne, PA, USA, 2018. [Google Scholar]
- CLSI. Performance Standards for Antimicrobial Susceptibility Testing; Informational Supplement-Twenty-Eight Edition M100S; Clinical and Laboratory Standards Institute (CLSI): Wayne, PA, USA, 2018. [Google Scholar]
- CLSI. Methods for Determining Bactericidal Activity of Antimicrobial Agents; Approved Guideline M26-A; CLSI: Wayne, PA, USA, 1999. [Google Scholar]
- Hidalgo-Grass, C.; Dan-goor, M.; Maly, A.; Eran, Y.; Kwinn, L.A.; Nizet, V.; Ravins, M.; Jaffe, J.; Peyser, A.; Moses, A.E.; et al. Effect of a bacterial pheromone peptide on host chemokine degradation in group A streptococcal necrotising soft-tissue infections. Mech. Dis. 2004, 363, 696–703. [Google Scholar] [CrossRef]
- Cern, A.; Connolly, K.L.; Jerse, A.E.; Barenholz, Y. In vitro susceptibility of Neisseria gonorrhoeae strains to mupirocin. An antibiotic reformulated to parenteral nano-liposomal antibiotic. Antimicrob. Agents Chemother. 2018, 62, e02377-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, A.R.; Beale, A.S.; Boon, R.J.; Griffin, K.E.; Masters, P.J.; Sutherland, R. Antibacterial activity of mupirocin, an antibiotic produced by Pseudomonas fluorescens. In Mupirocin a Novel Topical Antibiotic; Royal Society of Medicine: London, UK, 1984; pp. 42–55. [Google Scholar]
- Cern, A.; Nativ-Roth, E.; Goldblum, A.; Barenholz, Y. Effect of solubilizing agents on mupirocin loading into and release from PEGylated nanoliposomes. J. Pharm. Sci. 2014, 103, 2131–2138. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.B.; Gorwitz, R.J.; Jernigan, J.A. Mupirocin resistance. Clin. Infect. Dis. 2009, 49, 935–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- CDER. Guidance for Industry: Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers; CDER: Rockville, MD, USA, 2005; Volume 25.
- Theuretzbacher, U.; Barbee, L.; Connolly, K.; Drusano, G.; Fernandes, P.; Hook, E.; Jerse, A.; O’Donnell, J.; Unemo, M.; Van Bambeke, F.; et al. Pharmacokinetic/pharmacodynamic considerations for new and current therapeutic drugs for uncomplicated gonorrhoea—challenges and opportunities. Clin. Microbiol. Infect. 2020, 26, 1630–1635. [Google Scholar] [CrossRef] [PubMed]
- Jerse, A.E. Experimental gonococcal genital tract infection and opacity protein expression in estradiol-treated mice. Infect. Immun. 1999, 67, 5699–5708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.Y.; Chang, H.; Burgess, M.; Mcmillan, N.A.J. Vaginal delivery of siRNA using a novel PEGylated lipoplex-entrapped alginate scaffold system. J. Control. Release 2011, 155, 418–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alirol, E.; Wi, T.E.; Bala, M.; Bazzo, M.L.; Chen, X.S.; Deal, C.; Dillon, J.A.R.; Kularatne, R.; Heim, J.; Hooft van Huijsduijnen, R.; et al. Multidrug-resistant gonorrhea: A research and development roadmap to discover new medicines. PLoS Med. 2017, 14, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manhart, L.E.; Jensen, J.S.; Bradshaw, C.S.; Golden, M.R.; Martin, D.H. Efficacy of antimicrobial therapy for Mycoplasma genitalium infections. Clin. Infect. Dis. 2015, 61, S802–S817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barenholz, Y. Doxil®-The first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef] [PubMed]
- Baines, P.; Jackson, D.; Mellows, G.; Swaisland, A.; Tasker, T. Mupirocin: Its chemistry and metabolism. In Mupirocin a Novel Topical Antibiotic; Wilkinson, D., Price, J., Eds.; Royal Society of Medicine: London, UK, 1984; pp. 13–22. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. Isolates with Indicated Mupirocin MIC (MSSA, MRSA) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. isolates with indicated Nano-mupirocin MIC (MSSA, MRSA) | µg/mL | 0.12 | 0.25 | 0.5 | 1 | 2 | 4 | 8 | 16 | 32 | 64 | >64 |
| 0.12 | 1, 3 | |||||||||||
| 0.25 | 6, 5 | 7, 1 | ||||||||||
| 0.5 | 7, 6 | 28, 18 | 0, 1 | |||||||||
| 1 | 1, 7 | 0, 1 | ||||||||||
| 2 | ||||||||||||
| 4 | ||||||||||||
| 8 | ||||||||||||
| 16 | 0, 1 | |||||||||||
| 32 | 0, 2 | |||||||||||
| 64 | 0, 1 | |||||||||||
| >64 | 0, 1 | 0, 1 | 1, 3 | |||||||||
| No. Isolates with Indicated Mupirocin MIC (S. pneumoniae, S. pyogenes) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| ≤ 0.06 | 0.06 | 0.12 | 0.25 | 0.5 | 1 | 2 | 4 | ||
| No. isolates with indicated Nano-mupirocin MIC (S. pneumoniae, S. pyogenes) | ≤0.06 | ||||||||
| 0.06 | 0, 1 | 0, 1 | |||||||
| 0.12 | 0, 13 | 0, 3 | |||||||
| 0.25 | 0, 3 | 3, 3 | |||||||
| 0.5 | 3, 1 | 1, 0 | 1, 0 | ||||||
| 1 | 1, 0 | 6, 0 | |||||||
| 2 | 1, 0 | ||||||||
| 4 | 1, 1 | 2, 0 | |||||||
| 8 | 2, 0 | 4, 0 | |||||||
| No. Isolates with MIC (µg/mL) | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.125 | 0.25 | 0.5 | 1 | 2 | 4 | 8 | 16 | 32 | 64 | 128 | 256 | ≥512 | |
| E. faecium VanR group (n = 115) | |||||||||||||
| Mupirocin | 7 | 73 | 9 | 25 | 1 | ||||||||
| Vancomycin | 1 | 2 | 112 | ||||||||||
| Linezolid | 67 | 32 | 6 | 8 | 2 | ||||||||
| E. faecalis VanR group (n = 101) | |||||||||||||
| Mupirocin | 1 | 12 | 42 | 42 | 3 | 1 | |||||||
| Vancomycin | 5 | 1 | 5 | 2 | 3 | 1 | 84 | ||||||
| Linezolid | 83 | 17 | 1 | ||||||||||
| MIC (µg/mL) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Organism No. | Mupirocin | Nano-Mupirocin | Vancomycin | Linezolid | Daptomycin | Penicillin | Erythromycin | Tetracycline | Levofloxacin | Trimethoprim/ |
| Sulfamethoxazole | ||||||||||
| E. faecium | ||||||||||
| 1146992 | 1 | 4 | >16 | >8 | 2 | >16 | >4 | 4 | >4 | >2 |
| 1533772 | 0.5 | 2 | >16 | >8 | 4 | >16 | 2 | 32 | >4 | >2 |
| 1765156 | 1 | 2 | >16 | 8 | 4 | >16 | >4 | 0.5 | >4 | >2 |
| 1766256 | 1 | 2 | >16 | 4 | 4 | >16 | >4 | 16 | >4 | >2 |
| 1602010 | 1 | 2 | 1 | 2 | 4 | >16 | 2 | >32 | >4 | >2 |
| 1602013 | 1 | 2 | 1 | 2 | 4 | >16 | 2 | >32 | >4 | >2 |
| 1765227 | 0.5 | 1 | 2 | 2 | 0.5 | 0.12 | >4 | >32 | 2 | ≤0.06 |
| E. faecalis | ||||||||||
| 862935 | 64 | >64 | 1 | 2 | 1 | 2 | 0.25 | >32 | 1 | ≤0.06 |
| 1569172 | >64 | >64 | 1 | 2 | 1 | 2 | 2 | >32 | 1 | ≤0.06 |
| 1606748 | 32 | >64 | 1 | 2 | 2 | 2 | 0.25 | 16 | 2 | ≤0.06 |
| 1765036 | 64 | >64 | 0.5 | 1 | 1 | 2 | 0.5 | 0.5 | 1 | ≤0.06 |
| 860769 | 32 | >64 | 1 | 2 | 2 | 2 | >4 | 32 | 1 | >2 |
| 1766601 | 64 | >64 | >16 | 2 | 0.5 | 2 | >4 | >32 | >4 | >2 |
| 1766602 | 64 | >64 | >16 | 2 | 2 | 8 | >4 | >32 | >4 | >2 |
| MIC (µg/mL) | MBC (µg/mL) | |||||
|---|---|---|---|---|---|---|
| Organism | Organism No. | Resistance | Mupirocin | Nano-Mupirocin | Mupirocin | Nano-Mupirocin |
| S. aureus | ATCC 29213 | NA | 0.12 | 0.5 | 32 | 32 |
| S. aureus | ATCC 29213 | NA | 0.25 | 0.5 | 16 | 16 |
| MRSA | 649380 | NA | 0.12 | 0.5 | 0.12 | 1 |
| MRSA | 649390 | NA | 0.25 | 1 | 0.25 | 2 |
| MRSA | 1308254 | Daptomycin non-susceptible | 0.12 | 0.5 | 0.25 | 2 |
| MRSA | 672231 | Vancomycin resistant | 0.06 | 0.25 | 0.12 | 1 |
| MRSA | 672233 | Vancomycin resistant | 0.06 | 0.5 | 0.12 | 1 |
| MRSA | 672232 | Vancomycin resistant | 0.12 | 0.5 | 0.5 | 4 |
| S. pneumoniae | ATCC 49619 | NA | 0.12 | 0.25 | 0.25 | 1 |
| S. pneumoniae | ATCC 49619 | NA | 0.25 | 0.5 | 0.5 | 1 |
| S. pyogenes | 1262561 | Macrolide resistant | 0.25 | 0.5 | 16 | 32 |
| S. pyogenes | 1426536 | Macrolide resistant | 0.03 | 0.12 | 8 | 8 |
| S. pyogenes | 1440834 | Macrolide resistant | 0.12 | 0.12 | 4 | 4 |
| T1/2 | Tmax | Cmax | C0 | AUC0_Tlast | AUCINF | Vz | Cl | |
|---|---|---|---|---|---|---|---|---|
| (h) | (h) | (µg/mL) | (µg/mL) | (h × µg/mL) | (h × µg/mL) | (mL/kg) | (mL/h/kg) | |
| Day 1 | ||||||||
| Male | ||||||||
| 10 mg/kg | 9.06 | 1.00 | 161 | 160 | 788 | 820 | 160 | 12.20 |
| 30 mg/kg | 8.33 | 0.08 | 551 | 596 | 2617 | 2745 | 131 | 10.93 |
| 100 mg/kg | 9.78 | 0.08 | 2087 | 2246 | 10,010 | 10,596 | 133 | 9.44 |
| Female | ||||||||
| 10 mg/kg | 9.04 | 0.08 | 216 | 265 | 761 | 808 | 161 | 12.38 |
| 30 mg/kg | 6.76 | 0.08 | 582 | 639 | 2784 | 2917 | 100 | 10.29 |
| 100 mg/kg | 8.89 | 0.08 | 2400 | 2610 | 10,218 | 10,848 | 118 | 9.22 |
| Day 14 | ||||||||
| Male | ||||||||
| 10 mg/kg | 9.87 | 0.08 | 248 | 266 | 1208 | 1234 | 115 | 8.11 |
| 30 mg/kg | 12.59 | 0.08 | 746 | 787 | 3714 | 3863 | 141 | 7.77 |
| 100 mg/kg | 12.41 | 0.08 | 2233 | 2381 | 13,554 | 14,213 | 126 | 7.04 |
| Female | ||||||||
| 10 mg/kg | 9.13 | 0.08 | 263 | 288 | 1036 | 1053 | 125 | 9.50 |
| 30 mg/kg | 8.72 | 0.08 | 636 | 664 | 3089 | 3143 | 120 | 9.55 |
| 100 mg/kg | 9.54 | 0.08 | 2227 | 2401 | 11,980 | 12,273 | 112 | 8.15 |
| T1/2 | Tmax | Cmax | AUC0_Tlast | AUCINF | %F a | |
|---|---|---|---|---|---|---|
| (h) | (h) | (µg/mL) | (h × µg/mL) | (h × µg/mL) | ||
| Day 1 | ||||||
| Male | 18.52 | 1.00 | 2.62 | 63.96 | 88.78 | 8.12 |
| Female | 13.45 | 8.00 | 4.79 | 105.68 | 129.75 | 13.89 |
| Day 14 | ||||||
| Male | 10.68 | 2.00 | 4.58 | 61.22 | 64.79 | 5.07 |
| Female | 9.01 | 4.00 | 4.39 | 77.19 | 80.30 | 7.45 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cern, A.; Bavli, Y.; Hod, A.; Zilbersheid, D.; Mushtaq, S.; Michael-Gayego, A.; Barasch, D.; Feinstein Rotkopf, Y.; Moses, A.E.; Livermore, D.M.; et al. Therapeutic Potential of Injectable Nano-Mupirocin Liposomes for Infections Involving Multidrug-Resistant Bacteria. Pharmaceutics 2021, 13, 2186. https://doi.org/10.3390/pharmaceutics13122186
Cern A, Bavli Y, Hod A, Zilbersheid D, Mushtaq S, Michael-Gayego A, Barasch D, Feinstein Rotkopf Y, Moses AE, Livermore DM, et al. Therapeutic Potential of Injectable Nano-Mupirocin Liposomes for Infections Involving Multidrug-Resistant Bacteria. Pharmaceutics. 2021; 13(12):2186. https://doi.org/10.3390/pharmaceutics13122186
Chicago/Turabian StyleCern, Ahuva, Yaelle Bavli, Atara Hod, Daniel Zilbersheid, Shazad Mushtaq, Ayelet Michael-Gayego, Dinorah Barasch, Yael Feinstein Rotkopf, Allon E. Moses, David M. Livermore, and et al. 2021. "Therapeutic Potential of Injectable Nano-Mupirocin Liposomes for Infections Involving Multidrug-Resistant Bacteria" Pharmaceutics 13, no. 12: 2186. https://doi.org/10.3390/pharmaceutics13122186
APA StyleCern, A., Bavli, Y., Hod, A., Zilbersheid, D., Mushtaq, S., Michael-Gayego, A., Barasch, D., Feinstein Rotkopf, Y., Moses, A. E., Livermore, D. M., & Barenholz, Y. (2021). Therapeutic Potential of Injectable Nano-Mupirocin Liposomes for Infections Involving Multidrug-Resistant Bacteria. Pharmaceutics, 13(12), 2186. https://doi.org/10.3390/pharmaceutics13122186