
The Artemiside-Artemisox-Artemisone-M1 Tetrad: Efficacies against Blood Stage P. falciparum Parasites, DMPK Properties, and the Case for Artemiside

 , , ,
, , ,  ,
,  and
and
Abstract
1. Introduction
2. Results
2.1. Artemisox Efficacy
2.1.1. Asexual Blood Stage Parasites
2.1.2. Blood Stage Gametocytes
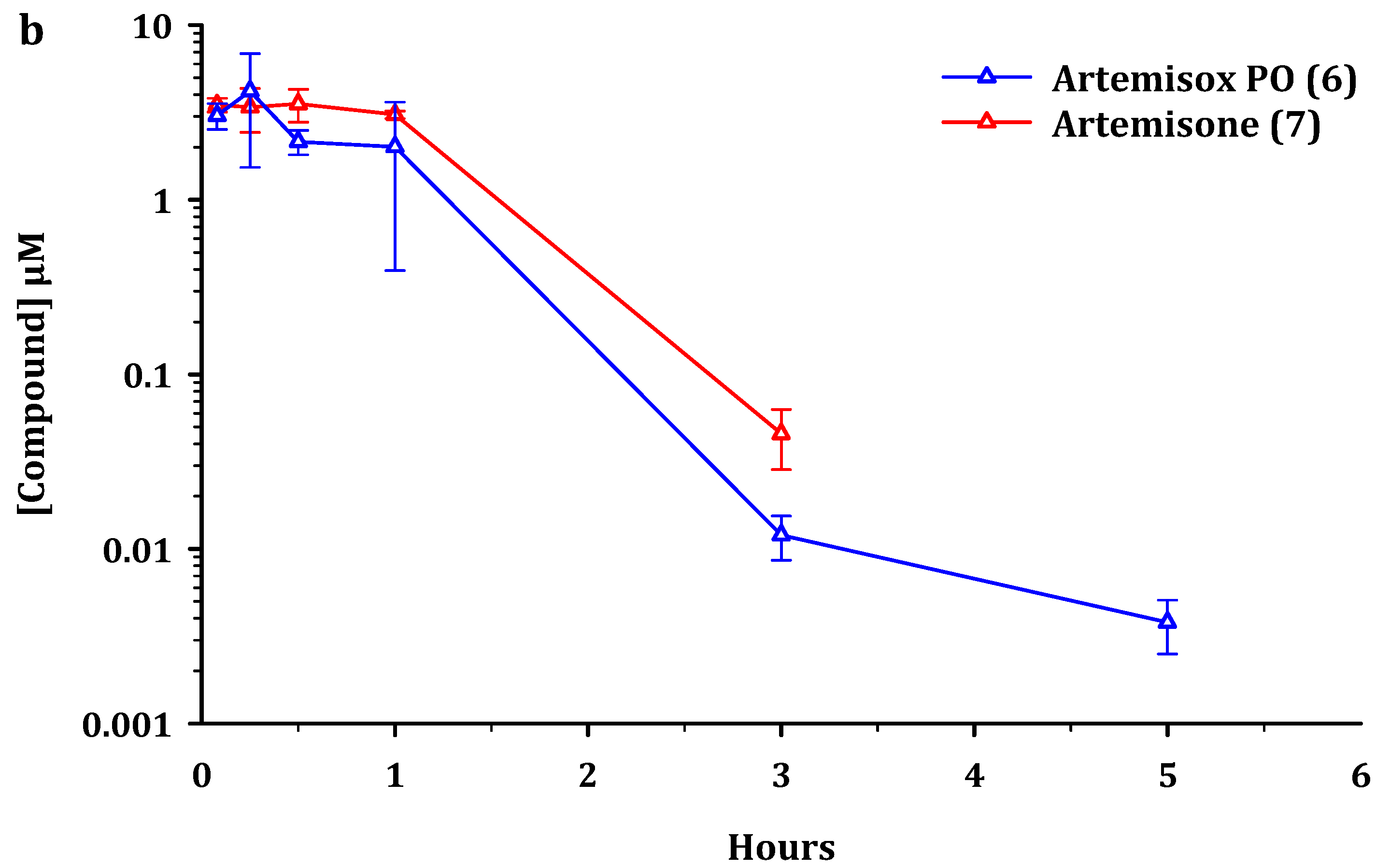
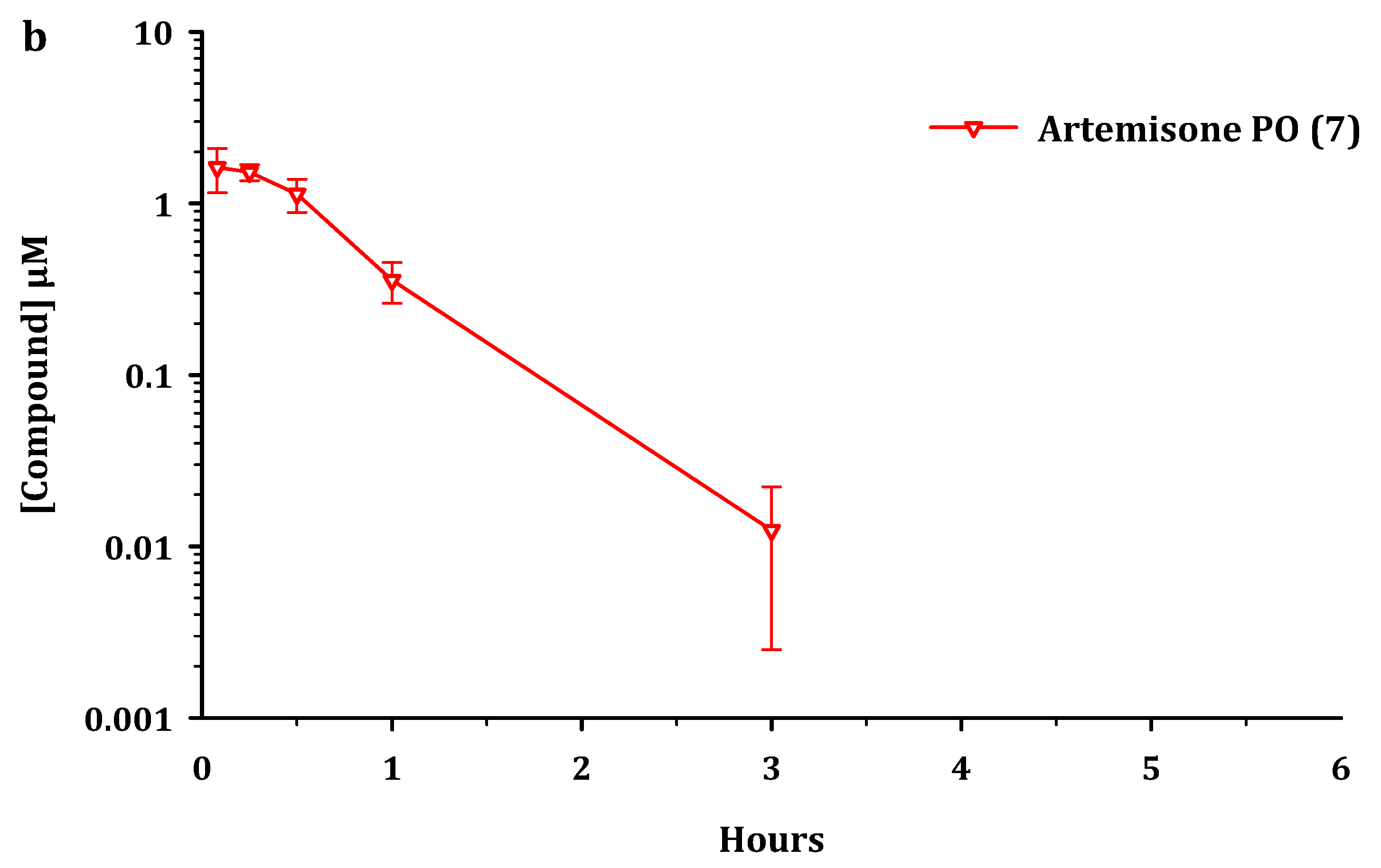
2.2. Metabolism and Pharmacokinetics of Artemiside, Artemisox and Artemisone
2.3. Pharmacokinetic Parameters
3. Discussion
3.1. Metabolism of Artemiside 5
3.2. Pharmacokinetics
3.3. Comparison of Efficacies of Artemiside 5 and Artemisone 7
3.4. Comparative Toxicities of Artemiside 5 and Artemisone 7
4. Materials and Methods
4.1. Efficacy
4.2. Cytotoxicity
4.3. Pharmacokinetics and Metabolism
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Disclaimer
References
- World Health Organization. World Malaria Report; World Health Organization: Geneva, Switzerland, 2020; ISBN 978-92-4-001579-1. [Google Scholar]
- Hamilton, W.L.; Amato, R.; van der Pluijm, R.W.; Jacob, C.G.; Quang, H.H.; Thuy-Nhien, N.T.; Hien, T.T.; Hongvanthong, B.; Chindavongsa, K.; Mayxay, M.; et al. Evolution and expansion of multidrug resistant malaria in Southeast Asia: A genomic epidemiology study. Lancet Infect. Dis. 2019, 19, 943–951. [Google Scholar] [CrossRef]
- Witmer, K.; Dahalan, F.A.; Delves, M.J.; Yahiya, S.; Watson, O.J.; Straschil, U.; Chiwcharoen, D.; Sornboon, B.; Pukrittayakamee, S.; Pearson, R.D.; et al. Transmission of artemisinin-resistant malaria parasites to mosquitoes under antimalarial drug pressure. Antimicrob. Agents Chemother. 2020, 65, e00898-20. [Google Scholar] [CrossRef] [PubMed]
- Ippolito, M.M.; Moser, K.A.; Kabuya, J.-B.B.; Cunningham, C.; Juliano, J.J. Antimalarial drug resistance and implications for the WHO global technical strategy. Curr. Epidemiol. Rep. 2021, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Balikagala, B.; Fukuda, N.; Ikeda, M.; Katuro, O.T.; Tachibana, S.-I.; Yamauchi, M.; Opio, W.; Emoto, S.; Anywar, D.A.; Kimura, E.; et al. Evidence of artemisinin-resistant malaria in Africa. N. Engl. J. Med. 2021, 385, 1163–1171. [Google Scholar] [CrossRef]
- Coertzen, D.; Reader, J.; van der Watt, M.; Nondaba, S.H.; Gibhard, L.; Wiesner, L.; Smith, P.; D’Alessandro, S.; Taramelli, D.; Wong, H.N.; et al. Artemisone and artemiside-potent pan-reactive antimalarial agents that also synergize redox imbalance in P. falciparum transmissible gametocyte stages. Antimicrob. Agents Chemother. 2018, 62, e02214-17. [Google Scholar] [CrossRef]
- Wong, H.N.; Padín-Irizarry, V.; van der Watt, M.E.; Reader, J.; Liebenberg, W.; Wiesner, L.; Smith, P.; Eribez, K.; Winzeler, E.A.; Kyle, D.E.; et al. Optimal 10-aminoartemisinins with potent transmission-blocking capabilities for new artemisinin combination therapies—Activities against blood stage P. falciparum Including PfKI3 C580Y mutants and liver stage P. berghei parasites. Front. Chem. 2020, 7, 901. [Google Scholar] [CrossRef] [PubMed]
- Beteck, R.M.; Seldon, R.; Coertzen, D.; van der Watt, M.E.; Reader, J.; Mackenzie, J.S.; Lamprecht, D.A.; Abraham, M.; Eribez, K.; Müller, J.; et al. Accessible and distinct decoquinate derivatives active against Mycobacterium tuberculosis and apicomplexan parasites. Commun. Chem. 2018, 1, 62. [Google Scholar] [CrossRef]
- Liu, J.M.; Ni, M.Y.; Fan, J.F.; Tu, Y.Y.; Wu, Z.H.; Wu, Y.L.; Chui, W.S. Structure and reactions of arteannuin. Acta Chim. Sin. 1979, 37, 129–141. [Google Scholar]
- Li, Y.; Yu, P.-L.; Chen, Y.-X.; Li, L.-Q.; Gai, Y.-Z.; Wang, D.-S.; Zheng, Y.-P. Synthesis of some derivatives of artemisinin. Kexue Tongbao 1979, 24, 667–669. [Google Scholar]
- Liu, X. Study on artemisinin derivatives. Yao Xue Tong Bao 1980, 15, 183. [Google Scholar]
- Zhang, J.F. A Detailed Chronological Record of Project 523 and the Discovery and Development of Qinghaosu (Artemisinin); Yangcheng Evening News Publisher: Guangzhou, China, 2006. [Google Scholar]
- Guo, Z. Artemisinin anti-malarial drugs in China. Acta Pharm. Sin. B 2016, 6, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Van Agtmael, M.; Gupta, V.; Van der Wösten, T.H.; Rutten, J.P.B.; Van Boxtel, C.J. Grapefruit juice increases the bioavailability of artemether. Eur. J. Clin. Pharmacol. 1999, 55, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Lefèvre, G.; Thomsen, M.S. Clinical pharmacokinetics of artemether and lumefantrine (Riamet®). Clin. Drug Investig. 1999, 18, 467–480. [Google Scholar] [CrossRef]
- Batty, K.T.; Ilett, K.F.; Powell, S.M.; Martin, J.; Davis, T.M.E. Relative bioavailability of artesunate and dihydroartemisinin: Investigations in the isolated perfused rat liver and in healthy Caucasian volunteers. Am. J. Trop. Med. Hyg. 2002, 66, 130–136. [Google Scholar] [CrossRef]
- Li, Q.; Xie, L.; Melendez, V.; Weina, P. Bioequivalence of two intravenous artesunate products with its active metabolite following single and multiple injections. Pharmaceuticals 2011, 4, 138–153. [Google Scholar] [CrossRef]
- Morris, C.A.; Duparc, S.; Borghini-Fuhrer, I.; Jung, D.; Shin, C.-S.; Fleckenstein, L. Review of the clinical pharmacokinetics of artesunate and its active metabolite dihydroartemisinin following intravenous, intramuscular, oral or rectal administration. Malar. J. 2011, 10, 263. [Google Scholar] [CrossRef]
- Kouakou, Y.I.; Tod, M.; Leboucher, G.; Lavoignat, A.; Bonnot, G.; Bienvenu, A.-L.; Picot, S. Systematic review of artesunate pharmacokinetics: Implication for treatment of resistant malaria. Int. J. Infect. Dis. 2019, 89, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Anh, C.X.; Chavchich, M.; Birrell, G.W.; Van Breda, K.; Travers, T.; Rowcliffe, K.; Lord, A.R.; Shanks, G.D.; Edstein, M.D. Pharmacokinetics and ex vivo antimalarial activity of artesunate-amodiaquine plus methylene blue in healthy volunteers. Antimicrob. Agents Chemother. 2020, 64, e01441-19. [Google Scholar] [CrossRef] [PubMed]
- Haynes, R.K.; Chan, H.W.; Lung, C.M.; Ng, N.C.; Wong, H.N.; Shek, L.Y.; Williams, I.D.; Gomes, M.F.; Cartwright, A. Artesunate and dihydroartemisinin (DHA): Unusual decomposition products formed under mild conditions and comments on the fitness of DHA as an antimalarial drug. ChemMedChem. 2007, 2, 1448–1463. [Google Scholar] [CrossRef]
- Jansen, F.H. The pharmaceutical death-ride of dihydroartemisinin. Malar. J. 2010, 9, 212. [Google Scholar] [CrossRef]
- Parapini, S.; Olliaro, P.; Navaratnam, V.; Taramelli, D.; Basilico, N. Stability of the antimalarial drug dihydroartemisinin under physiologically-relevant conditions: Implications for clinical treatment, pharmacokinetic and in vitro assays. Antimicrob. Agents. Chemother. 2015, 59, 4046–4052. [Google Scholar] [CrossRef] [PubMed]
- Haynes, R.K.; Fugmann, B.; Stetter, J.; Rieckmann, K.; Heilmann, H.-D.; Chan, H.-W.; Cheung, M.-K.; Lam, W.-L.; Wong, H.-N.; Croft, S.L.; et al. Artemisone—A highly active antimalarial drug of the artemisinin class. Angew. Chem. Int. Ed. 2006, 45, 2082–2088. [Google Scholar] [CrossRef] [PubMed]
- Watson, D.J.; Laing, L.; Gibhard, L.; Wong, H.N.; Haynes, R.K.; Wiesner, L. Towards new transmission-blocking combination therapies—Pharmacokinetics of 10-amino-artemisinins and 11-aza-artemisinin, and comparison with DHA and artemether. Antimicrob. Agents Chemother. 2021, 65, e00990-21. [Google Scholar] [CrossRef] [PubMed]
- Mbengue, A.; Bhattacharjee, S.; Pandharkar, T.; Liu, H.; Estiu, G.; Stahelin, R.V.; Rizk, S.S.; Njimoh, D.L.; Ryan, Y.; Chotivanich, K.; et al. A molecular mechanism of artemisinin resistance in Plasmodium falciparum malaria. Nature 2015, 520, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Van Hook, A.M. Antimalarial drugs inhibit PI3P production. Sci. Signal. 2015, 8, ec118. [Google Scholar] [CrossRef]
- Haynes, R.K.; Schmuck, G. Establishment of an in vitro screening model for neurodegeneration induced by antimalarial drugs of the artemisinin-type. Neurotox. Res. 2000, 2, 37–49. [Google Scholar] [CrossRef]
- Ramos-Martín, V.; González-Martínez, C.; Mackenzie, I.; Schmutzhard, J.; Pace, C.; Lalloo, D.G.; Terlouw, D.J. Neuroauditory toxicity of artemisinin combination therapies—Have safety concerns been addressed? Am. J. Trop. Med. Hyg. 2014, 91, 62–73. [Google Scholar] [CrossRef]
- Dalrymple, D.G. Artemisia annua, Artemisinin, ACTs & Malaria Control in Africa. In Tradition, Science and Public Policy; Politics & Prose: Washington, DC, USA, 2012; ISBN 978-0-615-61599-8. See Comments in Support of DHA by T.N.C Wells, Medicines for Malaria Venture (MMV), Geneva, on p. 49. [Google Scholar]
- Kümpornsin, K.; Loesbanluechai, D.; de Cozar, C.; Kotanan, N.; Chotivanich, K.; White, N.J.; Wilairat, P.; Gomez-Lorenzo, M.G.; Gamo, F.J.; Sanz, L.M.; et al. Lumefantrine attenuates Plasmodium falciparum artemisinin resistance during the early ring stage. Int. J. Parasitol. Drugs Drug Resist. 2021, 17, 186–190. [Google Scholar] [CrossRef]
- Manh, N.D.; Thanh, N.V.; Quang, H.H.; Van, N.T.T.; San, N.N.; Phong, N.C.; Birrell, G.W.; Edstein, M.D.; Edgel, K.A.; Martin, N.J.; et al. Pyronaridine-artesunate (Pyramax®) for the treatment of artemisinin and piperaquine-resistant Plasmodium falciparum in the central highlands of Vietnam. Antimicrob. Agents Chemother. 2021. [Google Scholar] [CrossRef]
- Haynes, R.K. From artemisinin to new artemisinin antimalarials: Biosynthesis, extraction, old and new derivatives, stereochemistry and medicinal chemistry requirements. Curr. Top. Med. Chem. 2006, 6, 509–537. [Google Scholar] [CrossRef]
- Haynes, R.K.; Ho, W.-Y.; Chan, H.-W.; Fugmann, B.; Stetter, J.; Croft, S.L.; Vivas, L.; Peters, W.; Robinson, B.L. Highly antimalaria-active artemisinin derivatives: Biological activity does not correlate with chemical reactivity. Angew. Chem. Int. Ed. 2004, 43, 1381–1385. [Google Scholar] [CrossRef]
- Haynes, R.K.; Wong, H.N.; Wu, Y.; Wu, W.K.; Cheu, K.W.; Williams, I.D.; Krishna, S.; Slavic, K.; Gravett, A.M.; Liu, W.M. Methylene homologues of artemisone: An unexpected structure–activity relationship and a possible implication for the design of C10-substituted artemisinins. ChemMedChem 2016, 11, 1469–1479. [Google Scholar] [CrossRef]
- Chan, W.C.; Chan, D.H.W.; Lee, K.W.; Tin, W.S.; Wong, H.N.; Haynes, R.K. Evaluation and optimization of synthetic routes from dihydroartemisinin to the alkylamino-artemisinins artemiside and artemisone: A test of N-glycosylation methodologies on a lipophilic peroxide. Tetrahedron 2018, 74, 5156–5171. [Google Scholar] [CrossRef]
- Schmuck, G.; Temerowski, M.; Haynes, R.K.; Fugmann, B. Identification of non-neurotoxic arte misinin derivatives in vivo and in vitro. Res. Adv. Antimicrob. Agents Chemother. 2003, 3 (Suppl. 2), 35–47. [Google Scholar]
- D’Alessandro, S.; Gelati, M.; Basilico, N.; Parati, E.A.; Haynes, R.K.; Taramelli, D. Differential effects on angiogenesis of two antimalarial compounds, dihydroartemisinin and artemisone: Implications for embryotoxicity. Toxicology 2007, 241, 66–74. [Google Scholar] [CrossRef]
- Nagelschmitz, J.; Voith, B.; Wensing, G.; Roemer, A.; Fugmann, B.; Haynes, R.K.; Kotecka, B.M.; Rieckmann, K.H.; Edstein, M.D. First assessment in humans of the safety, tolerability, pharmaco-kinetics, and ex vivo pharmacodynamic antimalarial activity of the new artemisinin derivative artemisone. Antimicrob. Agents Chemother. 2008, 52, 3085–3091. [Google Scholar] [CrossRef]
- Grobler, L.; Chavchich, M.; Haynes, R.K.; Edstein, M.D.; Grobler, A.F. Assessment of the induction of dormant ring stages in Plasmodium falciparum parasites by artemisone and artemisone entrapped in Pheroid vesicles in vitro. Antimicrob. Agents Chemother. 2014, 58, 7579–7582. [Google Scholar] [CrossRef][Green Version]
- Stuchlíková, L.R.; Matoušková, P.; Vokřál, I.; Lamka, J.; Szotáková, B.; Sečkařová, A.; Dimunová, D.; Nguyen, L.T.; Várady, M.; Skálová, L. Metabolism of albendazole, ricobendazole and flubendazole in Haemonchus contortus adults: Sex differences, resistance-related differences and the identification of new metabolites. Int. J. Parasitol. Drugs Drug Resist. 2018, 8, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, A.Y.; Wyllie, S.; Patterson, S.; Oza, S.L.; Read, K.D.; Fairlamb, A.H. Cross-resistance to nitro drugs and implications for treatment of human African trypanosomiasis. Antimicrob. Agents Chemother. 2010, 54, 2893–2900. [Google Scholar] [CrossRef] [PubMed]
- Wyllie, S.; Patterson, S.; Stojanovski, L.; Simeons, F.R.C.; Norval, S.; Kime, R.; Read, K.D.; Fairlamb, A.H. The anti-trypanosome drug fexinidazole shows potential for treating visceral leishmaniasis. Sci. Transl. Med. 2012, 4, 119re1. [Google Scholar] [CrossRef]
- Lelièvre, J.; Almela, M.J.; Lozano, S.; Miguel, C.; Franco, V.; Leroy, D.; Herreros, E. Activity of clinically relevant antimalarial drugs on Plasmodium falciparum mature gametocytes in an ATP bioluminescence “transmission blocking” assay. PLoS ONE 2012, 7, e35019. [Google Scholar] [CrossRef]
- Siciliano, G.; Santha Kumar, T.R.; Bona, R.; Camarda, G.; Calabretta, M.M.; Cevenini, L.; Davioud-Charvet, E.; Becker, K.; Cara, A.; Fidock, D.A.; et al. A high susceptibility to redox imbalance of the transmissible stages of Plasmodium falciparum revealed with a luciferase-based mature gametocyte assay. Mol. Microbiol. 2017, 104, 306–331. [Google Scholar] [CrossRef] [PubMed]
- Bowman, C.M.; Benet, L.Z. In vitro-in vivo extrapolation and hepatic clearance-dependent underprediction. J. Pharm. Sci. 2019, 108, 2500–2504. [Google Scholar] [CrossRef] [PubMed]
- Vogt, W. Oxidation of methionyl residues in proteins: Tools, targets, and reversal. Free Rad. Biol. Med. 1995, 18, 93–105. [Google Scholar] [CrossRef]
- Liang, X.; Kaya, A.; Zhang, Y.; Le, D.T.; Hua, D.; Gladyshev, V.N. Characterization of methionine oxidation and methionine sulfoxide reduction using methionine-rich cysteine-free proteins. BMC Biochem. 2012, 13, 21. [Google Scholar] [CrossRef]
- Kehm, R.; Baldensperger, T.; Raupbach, J.; Höhn, A. Protein oxidation-formation mechanisms, detection and relevance as biomarkers in human diseases. Redox Biol. 2021, 42, 101901. [Google Scholar] [CrossRef]
- Regueiro-Ren, A. “Cyclic Sulfoxides and Sulfones in Drug Design” in “Applications of Heterocycles in the Design of Drugs and Agricultural Products”. In Advances in Heterocyclic Chemistry; Meanwell, N.A., Lolli, M.L., Eds.; Elsevier: Amsterdam, The Netherlands, 2021; Volume 134, pp. 2–320. [Google Scholar] [CrossRef]
- Bakshi, R.P.; Nenortas, E.; Tripathi, A.K.; Sullivan, D.J.; Shapiro, T.A. Model system to define pharmacokinetic requirements for antimalarial drug efficacy. Sci. Transl. Med. 2013, 5, 205ra135. [Google Scholar] [CrossRef]
- Batty, K.T.; Gibbons, P.L.; Davis, T.M.E.; Ilett, K.F. Short Report: Pharmacokinetics of dihydroartemisinin in a murine malaria model. Am. J. Trop. Med. Hyg. 2008, 78, 641–642. [Google Scholar] [CrossRef]
- Xie, L.H.; Li, Q.; Zhang, J.; Weina, P.J. Pharmacokinetics, tissue distribution and mass balance of radiolabeled dihydroartemisinin in male rats. Malar. J. 2009, 8, 112. [Google Scholar] [CrossRef]
- Xing, J.; Bai, K.H.; Liu, T.; Wang, R.L.; Zhang, L.F.; Zhang, S.Q. The multiple-dosing pharmacokinetics of artemether, artesunate, and their metabolite dihydroartemisinin in rats. Xenobiotica 2011, 41, 252–258. [Google Scholar] [CrossRef]
- Arey, R.; Reisfeld, B. Predicting the disposition of the antimalarial drug artesunate and its active metabolite dihydroartemisinin using physiologically based pharmacokinetic modeling. Antimicrob. Agents Chemother. 2021, 65, e02280-20. [Google Scholar] [CrossRef]
- Lanteri, C.A.; Chaorattanakawee, S.; Lon, C.; Saunders, D.L.; Rutvisuttinunt, W.; Yingyuen, K.; Bathurst, I.; Ding, X.C.; Tyner, S.D. Ex vivo activity of endoperoxide antimalarials, including artemisone and arterolane, against multidrug-resistant Plasmodium falciparum isolates from Cambodia. Antimicrob. Agents Chemother. 2014, 58, 5831–5840. [Google Scholar] [CrossRef][Green Version]
- Sissoko, A.; Vásquez-Ocmín, P.; Maciuk, A.; Barbieri, D.; Neveu, G.; Rondepierre, L.; Grougnet, R.; Leproux, P.; Blaud, M.; Hammad, K.; et al. A chemically stable fluorescent mimic of dihydro artemisinin, artemether, and arteether with conserved bioactivity and specificity shows high pharmacological relevance to the antimalarial drugs. ACS Infect. Dis. 2020, 6, 1532–1547. [Google Scholar] [CrossRef]
- Adjalley, S.H.; Johnston, G.L.; Li, T.; Eastman, R.T.; Ekland, E.H.; Eappen, A.G.; Richman, A.; Sim, B.K.; Lee, M.C.; Hoffman, S.L.; et al. Quantitative assessment of Plasmodium falciparum sexual development reveals potent transmission-blocking activity by methylene blue. Proc. Natl. Acad. Sci. USA 2011, 108, 1214–1223. [Google Scholar] [CrossRef]
- Lucantoni, L.; Fidock, D.A.; Avery, V.M. Luciferase-Based, high-throughput assay for screening and profiling transmission-blocking compounds against Plasmodium falciparum gametocytes. Antimicrob. Agents Chemother. 2016, 60, 2097–2107. [Google Scholar] [CrossRef] [PubMed]
- D'Alessandro, S.; Camarda, G.; Corbett, Y.; Siciliano, G.; Parapini, S.; Cevenini, L.; Michelini, E.; Aldo Roda, A.; Leroy, D.; Taramelli, D.; et al. A chemical susceptibility profile of the Plasmodium falciparum transmission stages by complementary cell-based gametocyte assays. J. Antimicrob. Chemother. 2016, 71, 1148–1158. [Google Scholar] [CrossRef]
- Plouffe, D.M.; Wree, M.; Du, A.Y.; Meister, S.; Li, F.; Patra, K.; Lubar, A.; Okitsu, S.L.; Flannery, E.L.; Kato, N.; et al. High-throughput assay and discovery of small molecules that interrupt malaria transmission. Cell Host Microbe 2016, 19, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Reader, J.; Botha, M.; Theron, A.; Lauterbach, S.B.; Rossouw, C.; Engelbrecht, D.; Wepener, M.; Smit, A.; Leroy, D.; Mancama, D.; et al. Nowhere to hide: Interrogating different metabolic parameters of Plasmodium falciparum gametocytes in a transmission blocking drug discovery pipeline towards malaria elimination. Malar. J. 2015, 14, 213. [Google Scholar] [CrossRef]
- Ma, N.; Hunt, N.H.; Madigan, M.C.; Chan-Ling, T. Correlation between enhanced vascular permeability, up-regulation of cellular adhesion molecules and monocyte adhesion to the endothelium in the retina during the development of fatal murine cerebral malaria. Am. J. Pathol. 1996, 149, 1745–1762. [Google Scholar] [PubMed]
- Lackner, P.; Beer, R.; Helbok, R.; Broessner, G.; Engelhardt, K.; Brenneis, C.; Schmutzhard, E.; Pfaller, K. Scanning electron microscopy of the neuropathology of murine cerebral malaria. Malar. J. 2006, 5, 116. [Google Scholar] [CrossRef]
- Waknine-Grinberg, J.H.; Hunt, N.; Bentura-Marciano, A.; McQuillan, J.A.; Chan, H.W.; Chan, W.C.; Barenholz, Y.; Haynes, R.K.; Golenser, J. Artemisone effective against murine cerebral malaria. Malar. J. 2010, 9, 1–15. [Google Scholar] [CrossRef]
- Guo, J.; Guiguemde, A.W.; Bentura-Marciano, A.; Clark, J.; Haynes, R.K.; Chan, W.C.; Wong, H.-N.; Hunt, N.H.; Guy, R.K.; Golenser, J. Synthesis of artemiside and its effects in combination with conventional drugs against severe murine malaria. Antimicrob. Agents Chemother. 2012, 56, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Dunay, I.R.; Chan, W.C.; Haynes, R.K.; Sibley, L.D. Artemisone and artemiside control acute and reactivated toxoplasmosis in a murine model. Antimicrob. Agents Chemother. 2009, 53, 4450–4456. [Google Scholar] [CrossRef]
- Bhattacharjee, A.K.; Karle, J.M. Stereoelectronic properties of antimalarial artemisinin analogues in relation to neurotoxicity. Chem. Res. Toxicol. 1999, 12, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Schmuck, G.; Roehrdanz, E.; Haynes, R.K.; Kahl, R. Neurotoxic mode of action of artemisinin. Antimicrob. Agents Chemother. 2002, 46, 821–827. [Google Scholar] [CrossRef]
- Nontprasert, A.; Pukrittayakamee, S.; Prakongpan, S.; Supanaranond, W.; Looareesuwan, S.; White, N.J. Assessment of the neurotoxicity of oral dihydroartemisinin in mice. Trans. R. Soc. Trop. Med. Hyg. 2002, 96, 99–101. [Google Scholar] [CrossRef]
- Verlinden, B.K.; Niemand, J.; Snyman, J.; Sharma, S.K.; Beattie, R.J.; Woster, P.M.; Birkholtz, L. Discovery of novel alkylated (bis)urea and (bis)thiourea polyamine analogues with potent antimalarial activities. J. Med. Chem. 2011, 54, 6624–6633. [Google Scholar] [CrossRef]
- Smilkstein, M.; Sriwilaijaroen, N.; Kelly, J.X.; Wilairat, P.; Riscoe, M. Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob. Agents Chemother. 2004, 48, 1803–1806. [Google Scholar] [CrossRef] [PubMed]
- South African Bureau of Standards. SANS 10386:2008, Edition 1. South African National Standard: The Care and Use of Animals for Scientific Purposes; SABS Standards Division: Pretoria, South Africa, 2008. [Google Scholar]
- Department of Health. Ethics in Health Research: Principles, Processes and Structures, 2nd ed; Department of Health, Republic of South Africa: Pretoria, South Africa, 2015; Available online: https://ahrecs.com/resources/ethics-health-research-principles-processes-structures-2nd-ed-south-africa/ (accessed on 30 March 2021).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug a | IC50 nM b | IC50 µM e | ||||
|---|---|---|---|---|---|---|
| NF54 | K1 | RI c | W2 | RI d | HepG2 | |
| Chloroquine f | 10.0 ± 3.0 | 154 ± 14 | 15.4 | 233 ± 49 | 23.3 | 58.4 |
| Methylene Blue f | 5.0 ± 0.8 | 6.45 ± 0.30 | 1.29 | 5.13 ± 0.31 | 1.03 | - |
| DHA 2 f | 2.51 ± 0.19 | 1.51 ± 0.33 | 0.6 | 1.74 ± 0.22 | 0.7 | - |
| Artemether 3 f | 1.86 ± 0.17 | 9 ± 2 | 4.8 | 7 ± 1 | 3.8 | - |
| Artesunate 4 f | 3.00± 0.29 | 4 ± 1 | 1.3 | 2.4 ± 0.4 | 0.8 | - |
| Artemiside 5 f | 1.11 ± 0.17 | 1.6 ± 0.4 | 1.47 | 1.75 ± 0.27 | 1.58 | >50 |
| Artemisox 6 | 1.95 ± 0.25 | 1.5 ± 0.5 | 0.8 | 1.5 ± 0.4 | 0.7 | >50 |
| Artemisone 7 f | 1.2 ± 0.4 | 1.01 ± 0.19 | 0.85 | 1.6 ± 0.4 | 1.36 | >50 |
| M1 8 | 2.63 ± 0.24 | 1.50 ± 0.23 | 0.57 | 2.26 ± 0.08 | 0.86 | >50 |
| Compound a | IC50 nM b | |
|---|---|---|
| EG | LG | |
| Methylene Blue c | 95.0 ± 11.3 | 143.0 ± 16.7 |
| DHA 2 c | 43.0 ± 3.9 | 33.66 ± 1.98 |
| Artemether 3 c | 37.7 ± 2.0 | 136.2 ± 85.9 |
| Artesunate 4 c | 62.8 ± 3.0 | 259.4 ± 80 |
| Artemiside 5 c | 16.4 ± 1.0 | 1.5 ± 0.5 d |
| Artemisox 6 | 18.94 ± 0.98 | 2.72 ± 0.09 d |
| Artemisone 7 c | 1.94 ± 0.11 | 42.4 ± 3.3 d |
| M1 8 | 13.4 ± 2.7 | ND e |
| Drug iv | t½ h | MRT0-last h | CL L/h/kg | Vss L/kg | AUC0-last µmol.h/L | AUC0-∞ µmol.h/L |
|---|---|---|---|---|---|---|
| 5 | 1.2 ± 0.1 | 1.0 ± 0.1 | 3.5 ± 0.4 | 4.1 ± 0.5 | 3.8 ± 0.5 | 3.9 ± 0.4 |
| 6 from 5 | - | - | - | - | 1.9 ± 0.5 | - |
| 7 from 5 | - | - | - | - | 0.32 ± 0.12 | - |
| 6 | 0.43 ± 0.12 | 0.57 ± 0.04 | 6.9 ± 2.0 | 4.0 ± 1.2 | 2.0 ± 0.7 | 2.0 ± 0.7 |
| 7 from 6 | - | - | - | - | 1.1 ± 0.2 | - |
| 7 | 0.39 ± 0.08 | 0.56 ± 0.07 | 5.3 ± 1.5 | 3.0 ± 0.5 | 2.4 ± 0.6 | 2.5 ± 0.6 |
| Drug po | t½ h | Cmax µM | MRT0-last h | MAT h | AUC0-last µmol.h/L | F % | Ratio AUC0-last Metabolite/Parent |
|---|---|---|---|---|---|---|---|
| 5 | 1.40 ± 0.04 | 0.13 ± 0.01 | 1.9 ± 0.1 | 0.92 ± 0.20 | 0.36 ± 0.06 | 1.0 ± 0.3 | - |
| 6 from 5 | - | 2.5 ± 0.3 | - | - | 4.0 ± 0.8 | - | 11.1 ± 1.5 |
| 7 from 5 | - | 1.8 ± 0.1 | - | - | 2.8 ± 0.3 | - | 7.8 ± 0.7 |
| 6 | 0.54 ± 0.10 | 4.5 ± 2.3 | 0.69 ± 0.09 | 0.13 ± 0.07 | 3.3 ± 1.4 | 16 ± 2 | - |
| 7 from 6 | - | 3.8 ± 0.6 | - | - | 4.7 ± 0.6 | - | 1.5 ± 0.4 |
| 7 | 0.39 ± 0.05 | 1.7 ± 0.4 | 0.54 ± 0.13 | 0 | 1.1 ± 0.1 | 4.9 ± 1.7 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gibhard, L.; Coertzen, D.; Reader, J.; van der Watt, M.E.; Birkholtz, L.-M.; Wong, H.N.; Batty, K.T.; Haynes, R.K.; Wiesner, L. The Artemiside-Artemisox-Artemisone-M1 Tetrad: Efficacies against Blood Stage P. falciparum Parasites, DMPK Properties, and the Case for Artemiside. Pharmaceutics 2021, 13, 2066. https://doi.org/10.3390/pharmaceutics13122066
Gibhard L, Coertzen D, Reader J, van der Watt ME, Birkholtz L-M, Wong HN, Batty KT, Haynes RK, Wiesner L. The Artemiside-Artemisox-Artemisone-M1 Tetrad: Efficacies against Blood Stage P. falciparum Parasites, DMPK Properties, and the Case for Artemiside. Pharmaceutics. 2021; 13(12):2066. https://doi.org/10.3390/pharmaceutics13122066
Chicago/Turabian StyleGibhard, Liezl, Dina Coertzen, Janette Reader, Mariëtte E. van der Watt, Lyn-Marie Birkholtz, Ho Ning Wong, Kevin T. Batty, Richard K. Haynes, and Lubbe Wiesner. 2021. "The Artemiside-Artemisox-Artemisone-M1 Tetrad: Efficacies against Blood Stage P. falciparum Parasites, DMPK Properties, and the Case for Artemiside" Pharmaceutics 13, no. 12: 2066. https://doi.org/10.3390/pharmaceutics13122066
APA StyleGibhard, L., Coertzen, D., Reader, J., van der Watt, M. E., Birkholtz, L.-M., Wong, H. N., Batty, K. T., Haynes, R. K., & Wiesner, L. (2021). The Artemiside-Artemisox-Artemisone-M1 Tetrad: Efficacies against Blood Stage P. falciparum Parasites, DMPK Properties, and the Case for Artemiside. Pharmaceutics, 13(12), 2066. https://doi.org/10.3390/pharmaceutics13122066